Synthesis, Crystal Structure, Vibration Spectral, and DFT Studies of 4-Aminoantipyrine and Its Derivatives

Abstract
:1. Introduction
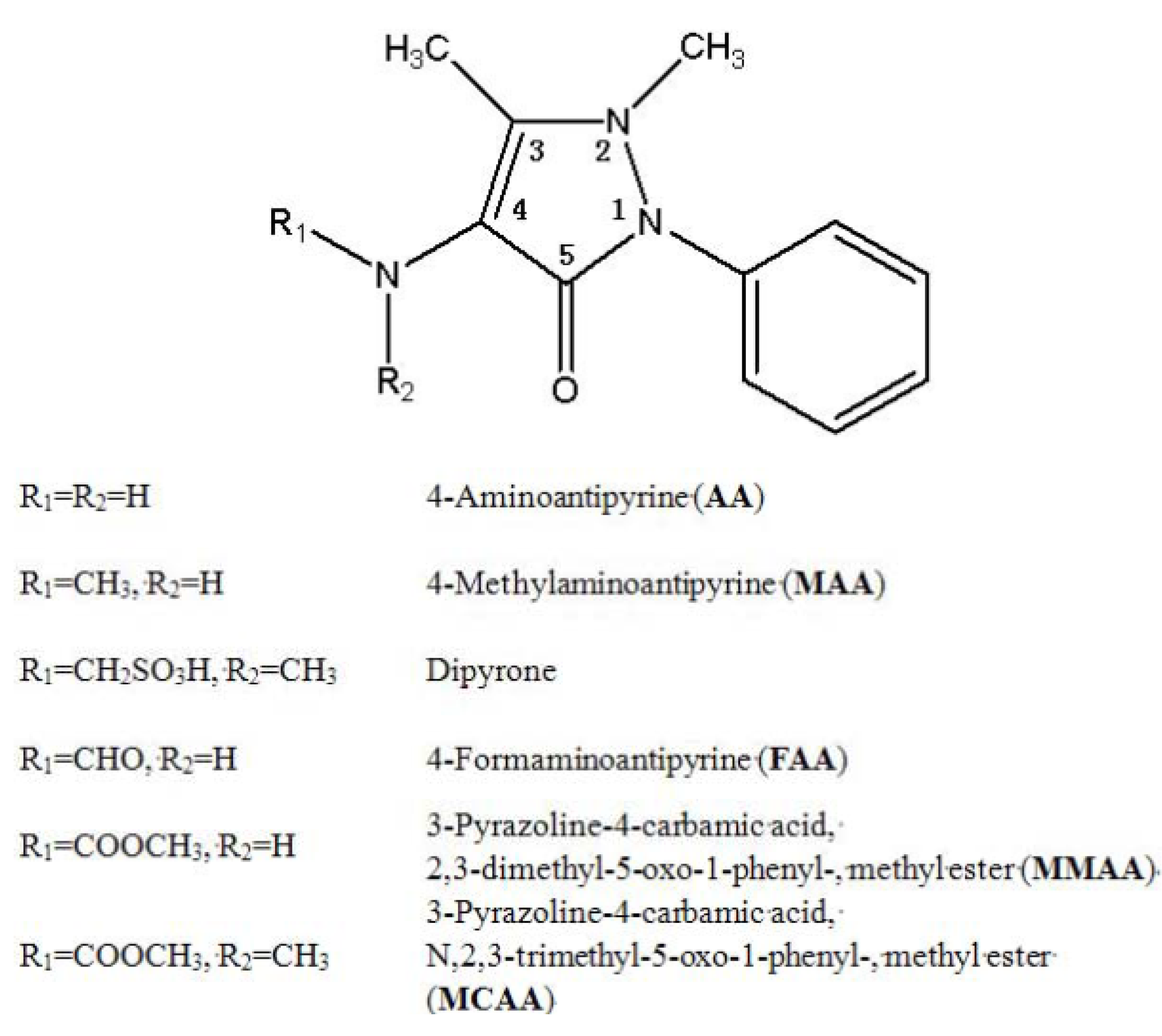
2. Results and Discussion
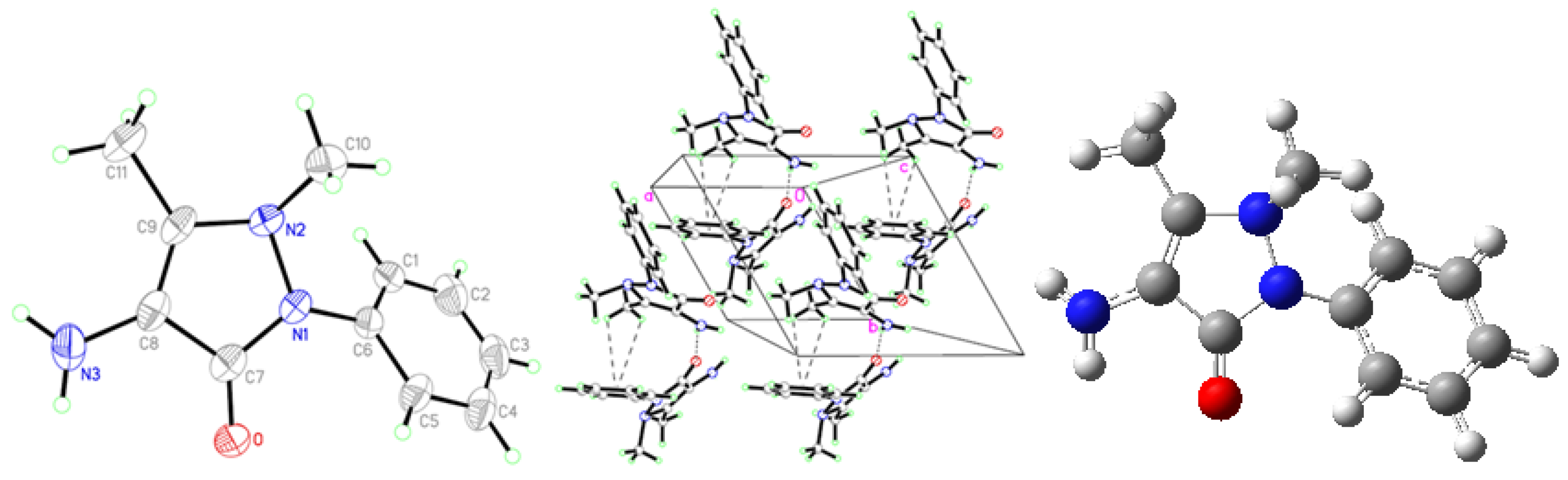
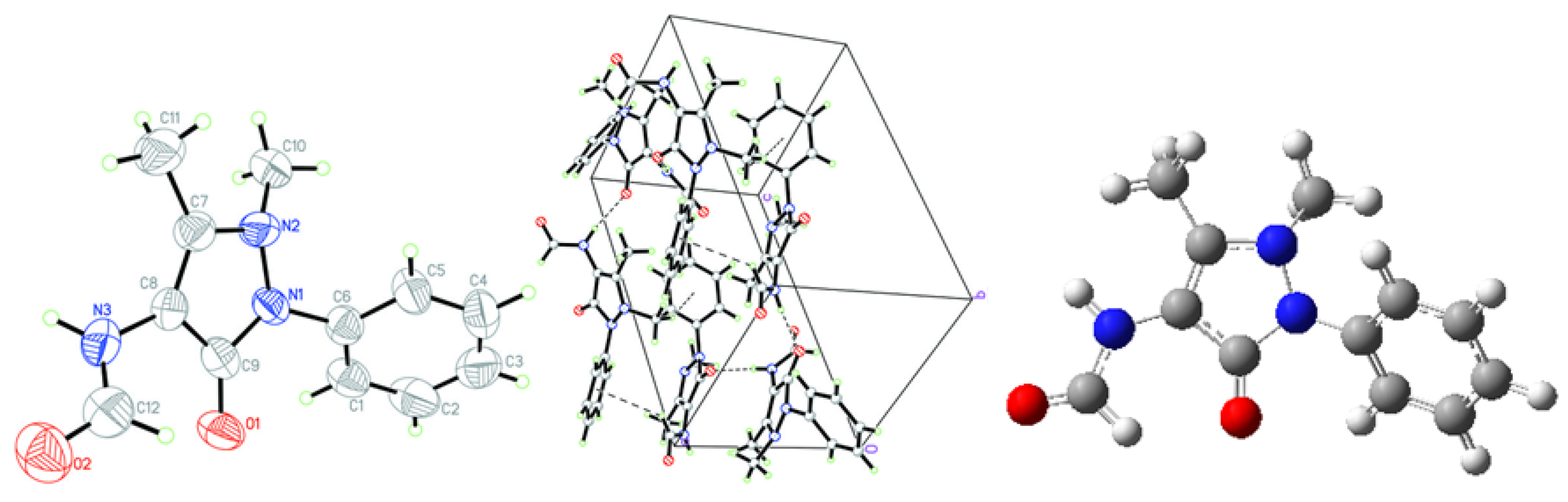
2.1. Crystallographic Results

{kind=link}
{kind=link}
{kind=link}
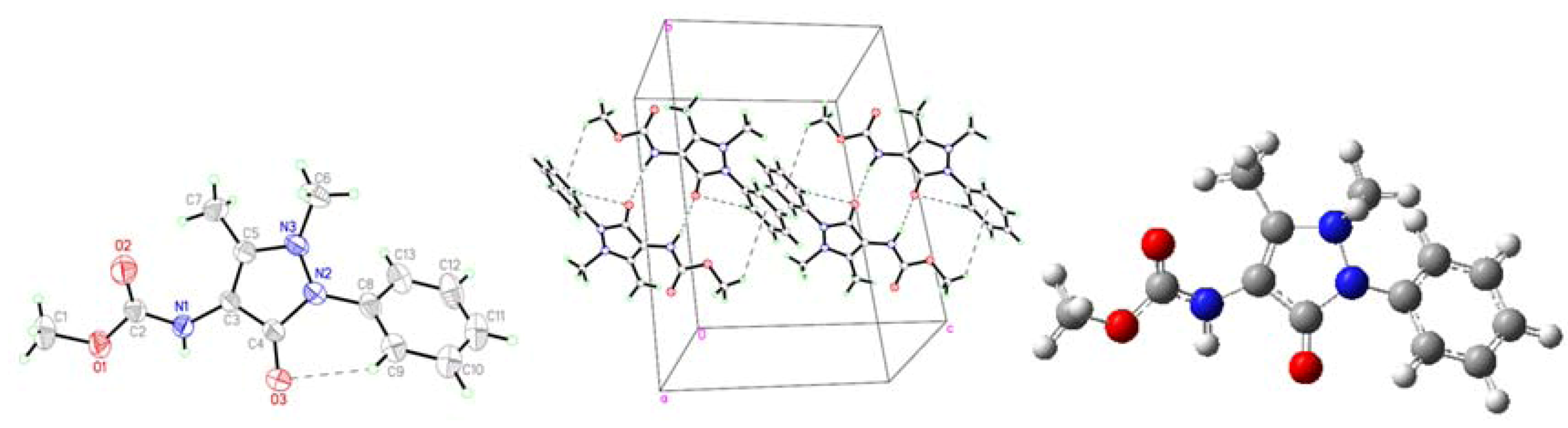{kind=link}
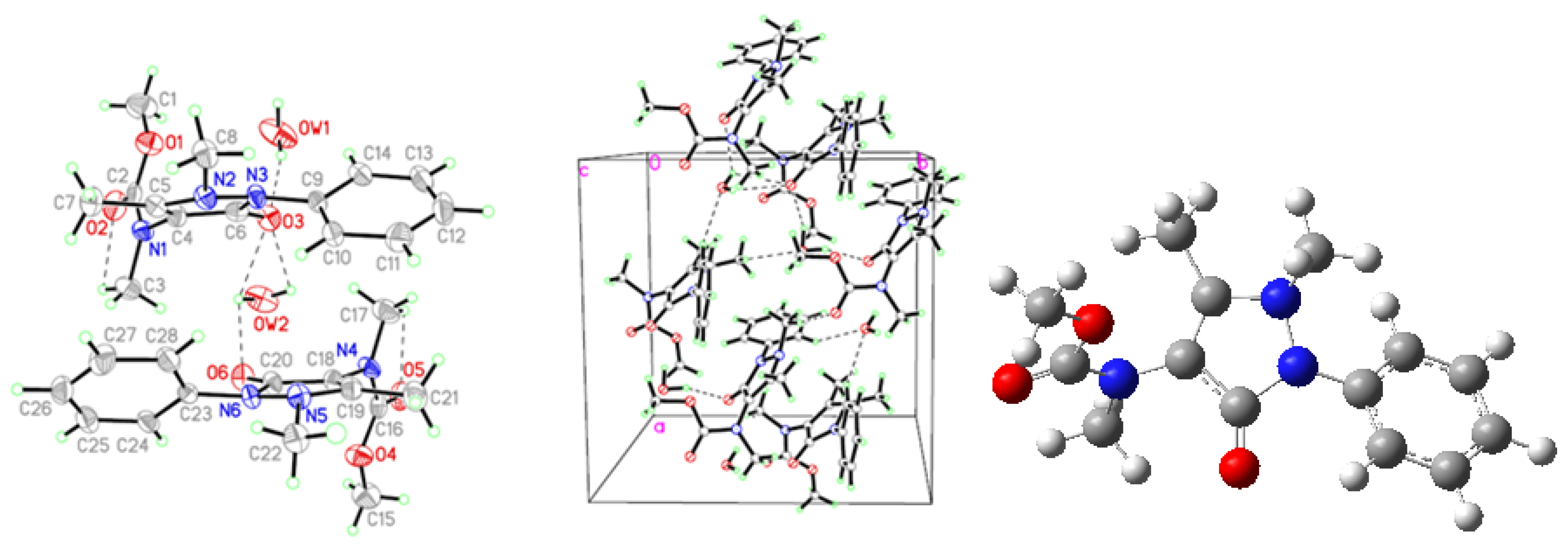{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AA | MMAA | MCAA | |
|---|---|---|---|
| empirical formula | C11H13N3O | C13H15N3O3 | C28H38N6O8 |
| formula weight | 203.24 | 261.28 | 586.64 |
| temperature [K] | 293 (2) | 293 (2) | 293 (2) |
| wavelength [Å] | 0.71073 | 0.71073 | 0.71073 |
| crystal system, | hexagonal | monoclinic | monoclinic |
| space group | P65 | P21/c | Cc |
| unit cell dimensions | |||
| a [Å] | 7.5160 (11) | 6.7180 (13) | 12.044 (2) |
| b [Å] | 7.5160 (11) | 17.305 (4) | 11.961 (2) |
| c [Å] | 32.005 (6) | 11.455 (2) | 20.724 (4) |
| α [º] | 90.00 | 90.00 | 90.00 |
| β [º] | 90.00 | 97.33 (3) | 97.47 (3) |
| γ [º] | 120.00 | 90.00 | 90.00 |
| volume [Å3] | 1565.7 (5) | 1320.8 (5) | 2960.1 (10) |
| Z | 6 | 4 | 4 |
| ρcalcd [g cm−3] | 1.293 | 1.314 | 1.316 |
| μ [mm−1] | 0.087 | 0.095 | 0.098 |
| F (000) | 648 | 552 | 1248 |
| crystal size [mm3] | 0.05 × 0.10 × 0.20 | 0.10 × 0.10 × 0.20 | 0.10 × 0.10 × 0.20 |
| θ range [º] for data collection | 3.13 to 25.31 | 2.14 to 25.27 | 1.98 to 25.27 |
| index ranges | 0 ≤ h ≤ 7 | 0 ≤ h ≤ 8 | 0 ≤ h ≤ 14 |
| 0 ≤ k ≤ 7 | 0 ≤ k ≤ 20 | 0 ≤ k ≤ 14 | |
| −38 ≤ l ≤ 38 | −13 ≤ l ≤ 13 | −24 ≤ l ≤ 24 | |
| reflections collected | 2,331 | 2,603 | 2,956 |
| independent reflections | 1898 [ Rint = 0.097] | 2392 [ Rint = 0.023] | 2818 [ Rint = 0.084] |
| max. and min. transmission | 0.9957/0.9829 | 0.9905/0.9812 | 0.9903/0.9807 |
| data/restraints/parameters | 1898/1/136 | 2392/0/173 | 2818/3/387 |
| goodness-of-fit on F2 | 1.005 | 1.000 | 1.009 |
| final R indices [I > 2σ (I)]; R1, wR2 | 0.0638, 0.1667 | 0.0552, 0.1488 | 0.0651, 0.1526 |
| R1, wR2 (all data) | 0.0992, 0.1901 | 0.0886, 0.1704 | 0.0969, 0.1716 |
| largest diff. peak and hole [e·Å−3] | 0.153 and −0.160 | 0.239 and −0.188 | 0.224 and −0.273 |
| Parameters | AA | |
|---|---|---|
| Bond lengths (Å) | Experimental | B3LYP/6-31G (d) |
| O1-C7 | 1.229 (6) | 1.228 |
| N1-C7 | 1.379 (6) | 1.400 |
| N1-C6 | 1.418 (6) | 1.417 |
| N1-N2 | 1.431 (5) | 1.420 |
| N2-C9 | 1.402 (6) | 1.421 |
| N2-C10 | 1.459 (6) | 1.475 |
| N3-C8 | 1.365 (7) | 1.394 |
| Bond angles (°) | ||
| C7-N1-C6 | 126.9 (4) | 125.11 |
| C6-N1-N2 | 119.3 (4) | 119.28 |
| C9-N2-C10 | 117.9 (4) | 115.26 |
| N1-N2-C10 | 110.3 (4) | 111.49 |
| C1-C6-N1 | 122.7 (5) | 120.76 |
| C5-C6-N1 | 117.2 (4) | 119.23 |
| O-C7-N1 | 125.1 (4) | 126.98 |
| O-C7-C8 | 129.2 (5) | 127.79 |
| N1-C7-C8 | 105.7 (4) | 105.17 |
| C9-C8-N3 | 129.9 (5) | 132.14 |
| N3-C8-C7 | 121.4 (5) | 119.44 |
| N2-C9-C11 | 119.3 (4) | 119.85 |
| Parameters | FAA | |
|---|---|---|
| Bond lengths (Å) | Experimental | B3LYP/6-31G (d) |
| O1-C9 | 1.248 (5) | 1.226 |
| N1-C9 | 1.384 (5) | 1.405 |
| N1-C6 | 1.424 (5) | 1.420 |
| N1-N2 | 1.412 (5) | 1.413 |
| N2-C7 | 1.359 (5) | 1.405 |
| N2-C10 | 1.463 (5) | 1.474 |
| N3-C8 | 1.419 (5) | 1.396 |
| N3-C12 | 1.305 (6) | 1.382 |
| O2-C12 | 1.228 (5) | 1.217 |
| C9-N1-C6 | 124.3 (3) | 124.92 |
| C6-N1-N2 | 118.3 (3) | 119.36 |
| C7-N2-C10 | 123.0 (4) | 116.78 |
| N1-N2-C10 | 117.4 (4) | 112.61 |
| C1-C6-N1 | 119.4 (4) | 119.25 |
| C5-C6-N1 | 120.4 (4) | 120.57 |
| O1-C9-N1 | 123.6 (4) | 125.40 |
| O1-C9-C8 | 131.7 (4) | 130.29 |
| N1-C9-C8 | 104.7 (4) | 104.27 |
| C7-C8-N3 | 127.8 (4) | 127.35 |
| N3-C8-C9 | 122.7 (4) | 124.17 |
| N2-C7-C11 | 120.4 (4) | 120.17 |
| C12-N3-C8 | 122.2 (4) | 127.67 |
| O2-C12-N3 | 124.7 (5) | 121.89 |
| Parameters | MMAA | |
|---|---|---|
| Bond lengths (Å) | Experimental | B3LYP/6-31G (d) |
| O3-C4 | 1.235 (3) | 1.228 |
| N2-C4 | 1.396 (3) | 1.400 |
| N2-C8 | 1.422 (3) | 1.418 |
| N2-N3 | 1.408 (3) | 1.415 |
| N3-C5 | 1.384 (3) | 1.411 |
| N3-C6 | 1.476 (3) | 1.474 |
| N1-C3 | 1.407 (3) | 1.397 |
| N1-C2 | 1.346 (3) | 1.374 |
| O2-C2 | 1.208 (3) | 1.217 |
| O1-C2 | 1.342 (3) | 1.357 |
| O1-C1 | 1.428 (4) | 1.433 |
| Bond angles (°) | ||
| C4-N2-C8 | 125.2 (2) | 125.32 |
| C8-N2-N3 | 118.9 (2) | 119.58 |
| C5-N3-C6 | 119.8 (2) | 116.43 |
| N2-N3-C6 | 114.1 (2) | 112.43 |
| C13-C8-N2 | 120.7 (3) | 120.77 |
| C9-C8-N2 | 119.2 (2) | 119.11 |
| O3-C4-N2 | 123.9 (2) | 127.11 |
| O3-C4-C3 | 130.6 (2) | 127.96 |
| N2-C4-C3 | 105.5 (2) | 104.86 |
| C5-C3-N1 | 129.6 (2) | 133.63 |
| N1-C3-C4 | 121.5 (2) | 117.07 |
| N3-C5-C7 | 120.3 (2) | 118.98 |
| C2-N1-C3 | 124.0 (2) | 125.31 |
| O2-C2-N1 | 126.1 (2) | 126.26 |
| O1-C2-N1 | 109.5 (2) | 109.11 |
| O2-C2-O1 | 124.4 (2) | 124.62 |
| C2-O1-C1 | 116.8 (2) | 114.74 |
| Parameters | MCAA | |
|---|---|---|
| Bond lengths (Å) | Experimental | B3LYP/6-31G(d) |
| O3-C6 | 1.240 (8) | 1.225 |
| N3-C6 | 1.386 (9) | 1.415 |
| N3-C9 | 1.420 (7) | 1.419 |
| N2-N3 | 1.420 (7) | 1.414 |
| N2-C5 | 1.373 (8) | 1.396 |
| N2-C8 | 1.459 (8) | 1.472 |
| N1-C4 | 1.412 (8) | 1.409 |
| N1-C2 | 1.367 (10) | 1.378 |
| O2-C2 | 1.215 (8) | 1.218 |
| O1-C2 | 1.351 (9) | 1.361 |
| O1-C1 | 1.411 (10) | 1.434 |
| N1-C3 | 1.456 (9) | 1.468 |
| Bond angles (°) | ||
| C6-N3-C9 | 126.3 (5) | 124.67 |
| C9-N3-N2 | 120.6 (5) | 119.17 |
| C5-N2-C8 | 123.9 (6) | 117.83 |
| N3-N2-C8 | 118.2 (5) | 113.26 |
| C10-C9-N3 | 120.6 (5) | 119.01 |
| C14-C9-N3 | 117.6 (6) | 120.91 |
| O3-C6-N3 | 123.1 (6) | 125.21 |
| O3-C6-C4 | 131.6 (6) | 130.54 |
| N3-C6-C4 | 105.2 (5) | 104.21 |
| C5-C4-N1 | 125.6 (6) | 128.24 |
| N1-C4-C6 | 125.0 (6) | 123.01 |
| N2-C5-C7 | 120.8 (6) | 120.67 |
| C2-N1-C4 | 120.7 (6) | 123.23 |
| O2-C2-N1 | 125.0 (7) | 124.89 |
| O1-C2-N1 | 111.2 (6) | 111.66 |
| O2-C2-O1 | 123.8 (8) | 123.45 |
| C2-O1-C1 | 114.9 (7) | 114.12 |
| C2-N1-C3 | 120.0 (6) | 118.43 |
| C4-N1-C3 | 118.5 (6) | 117.53 |
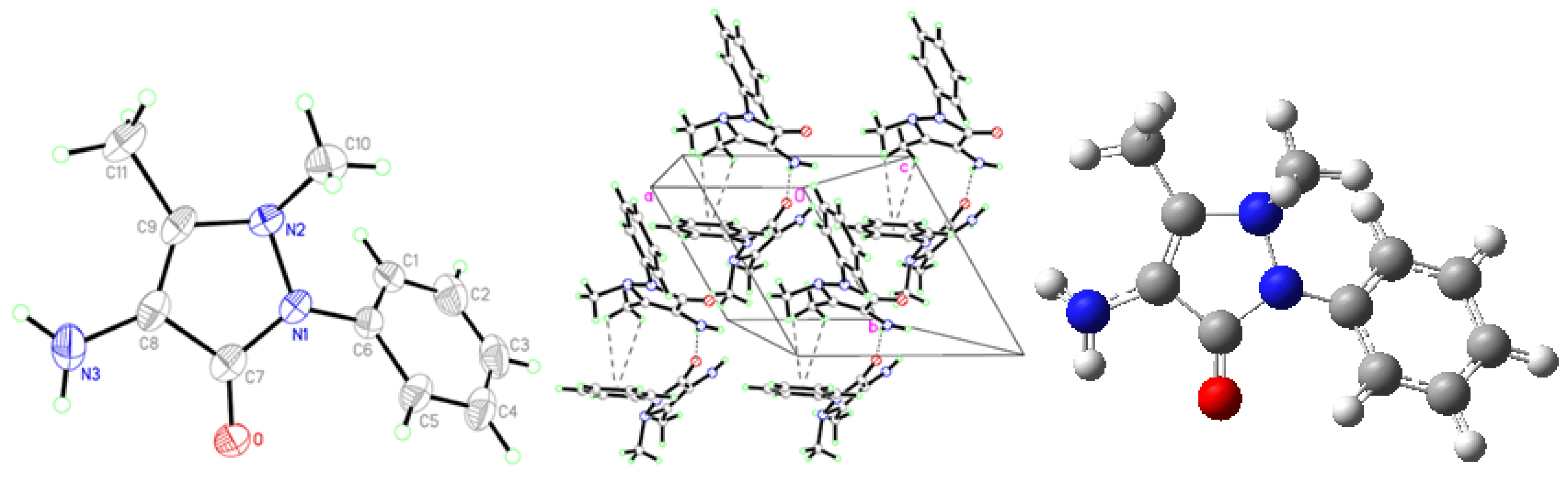
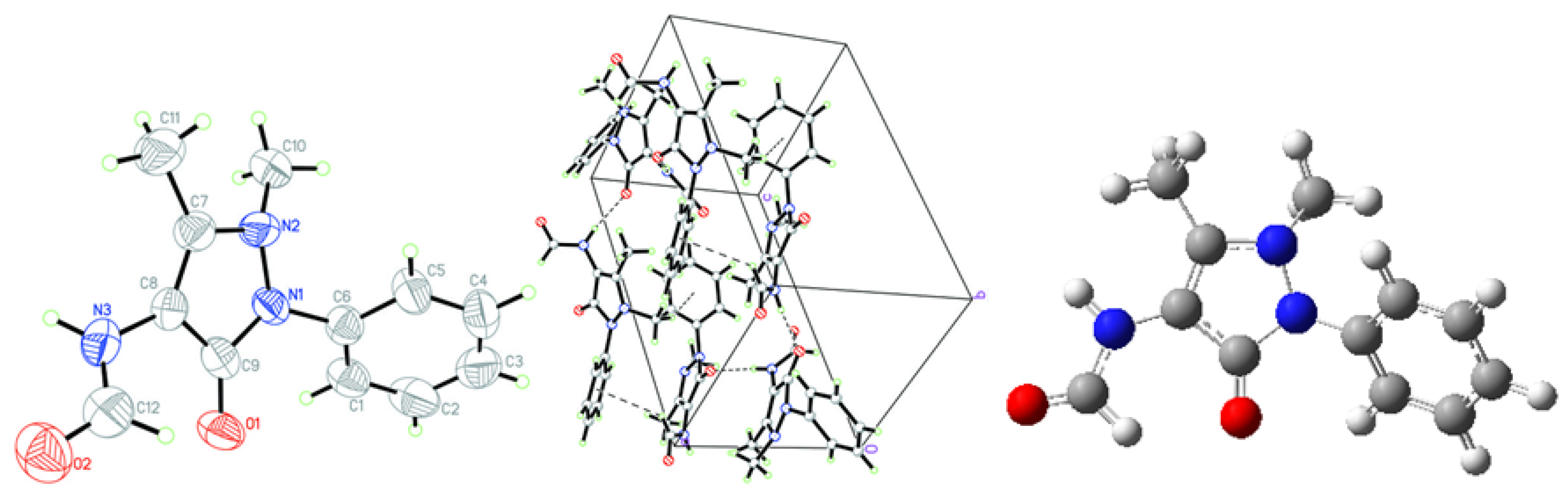
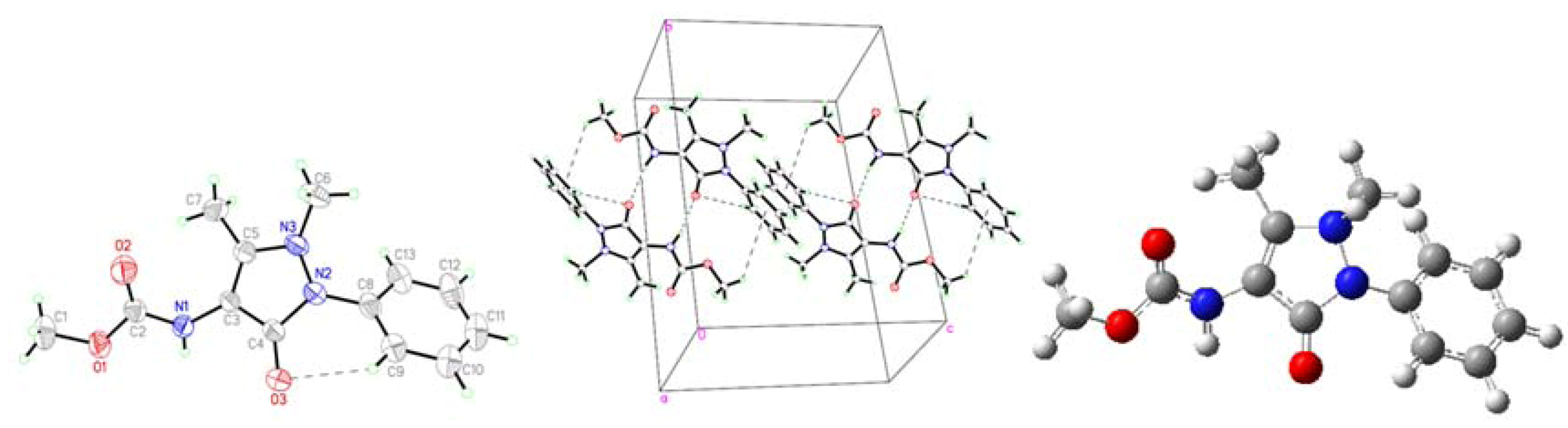
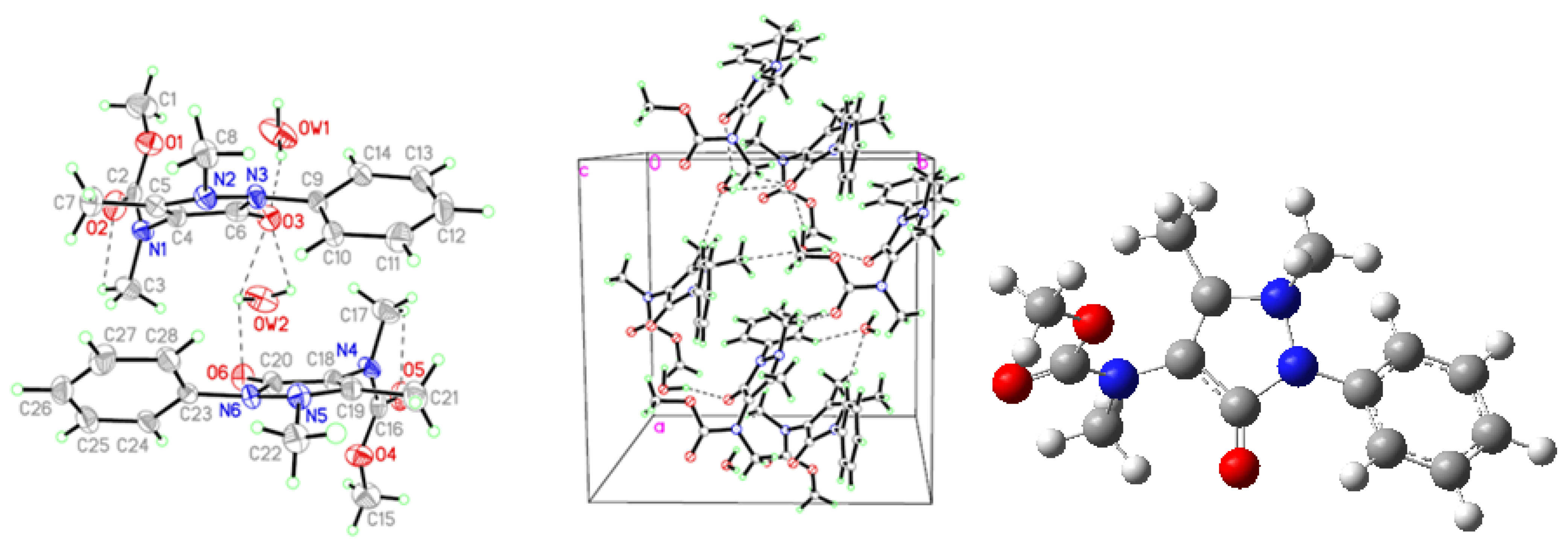
| D-H···A | D-H | H···A | D···A | D–H···A | Symmetry codes |
|---|---|---|---|---|---|
| AA | |||||
| N3-H3A···O | 0.86 | 2.23 | 3.039 (6) | 156.0 | x − y, −1 + x, −1/6 + z |
| C11-H11A···Cg1 | 0.96 | 3.21 | 3.476 (6) | 98.2 | x − y, −1 + x, −1/6 + z |
| C11-H11C···Cg1 | 0.96 | 2.99 | 3.476 (6) | 112.7 | x − y,−1 + x, −1/6 + z |
| FAA | |||||
| N3-H3A···O1 | 0.86 | 2.01 | 2.864 (5) | 172.0 | −x, y + 1/2,−z + 3/2 |
| C10-H10B···Cg1 | 0.96 | 2.85 | 3.733 (5) | 153.0 | −x − 1, y + 3/2, −z + 3/2 |
| C12-H12A···Cg1 | 0.93 | 3.03 | 3.647 (5) | 125.0 | −x, y+ 3/2, −z + 3/2 |
| MMAA | |||||
| N1-H1A···O3 | 0.86 | 2.01 | 2.850 (3) | 166.0 | 1 − x, 1 − y, 2 − z |
| C9-H9A···O3 | 0.93 | 2.52 | 2.960 (3) | 109.0 | |
| C1-H1C···Cg1 | 0.96 | 3.11 | 3.686 (4) | 120.1 | x, 1/2 − y, 1/2 + z |
| C7-H7B···Cg1 | 0.96 | 3.23 | 3.763 (3) | 116.7 | 2 − x, 1− y, 2− z |
| MCAA | |||||
| OW1-HW1B···O3 | 1.01 | 1.90 | 2.850 (9) | 156.0 | |
| C17-H17A···O5 | 0.96 | 2.34 | 2.766 (10) | 106.0 | |
| OW2-HW2B···O3 | 0.85 | 2.51 | 2.860 (7) | 106.0 | |
| OW2-HW2A···O3 | 0.85 | 2.55 | 2.860 (7) | 102.0 | |
| OW2-HW2A···O6 | 0.85 | 2.41 | 2.871 (8) | 114.0 | |
| C3-H3A···O2 | 0.96 | 2.43 | 2.806 (10) | 103.0 | |
| OW1-HW1A···O6#1 | 0.96 | 1.90 | 2.823 (9) | 159.0 | 1/2 + x, 1/2 + y, z |
| C8-H8C···OW1#2 | 0.96 | 2.49 | 3.056 (10) | 118.0 | −1/2 + x, 1/2 + y, z |
| C10-H10A···OW2#2 | 0.93 | 2.52 | 3.358 (9) | 151.0 | −1/2 + x, 1/2 + y, z |
| C22-H22A···O5#2 | 0.96 | 2.56 | 3.510 (8) | 171.0 | −1/2 + x, 1/2 + y, z |
| C28-H28A···OW2#2 | 0.93 | 2.54 | 3.384 (9) | 151.0 | −1/2 + x, 1/2 + y, z |
| C3-H3B···Cg1#3 | 0.96 | 3.20 | 4.038 (9) | 147.0 | x, y, z |
| C8-H8B···Cg1#1 | 0.96 | 2.80 | 3.737 (8) | 165.3 | 1/2 + x, 1/2 + y, z |
| C14-H14A···Cg2#1 | 0.93 | 3.11 | 3.582 (7) | 113.0 | 1/2 + x,1/2 + y, z |
| C17-H17B···Cg3#3 | 0.96 | 3.36 | 4.020 (9) | 127.4 | x, y, z |
| C22-H22B···Cg3#4 | 0.96 | 2.90 | 3.814 (8) | 160.5 | −1/2 + x,−1/2 + y, z |
| C24-H24A···Cg4#4 | 0.93 | 3.12 | 3.539 (8) | 109.4 | −1/2 + x, −1/2 + y, z |
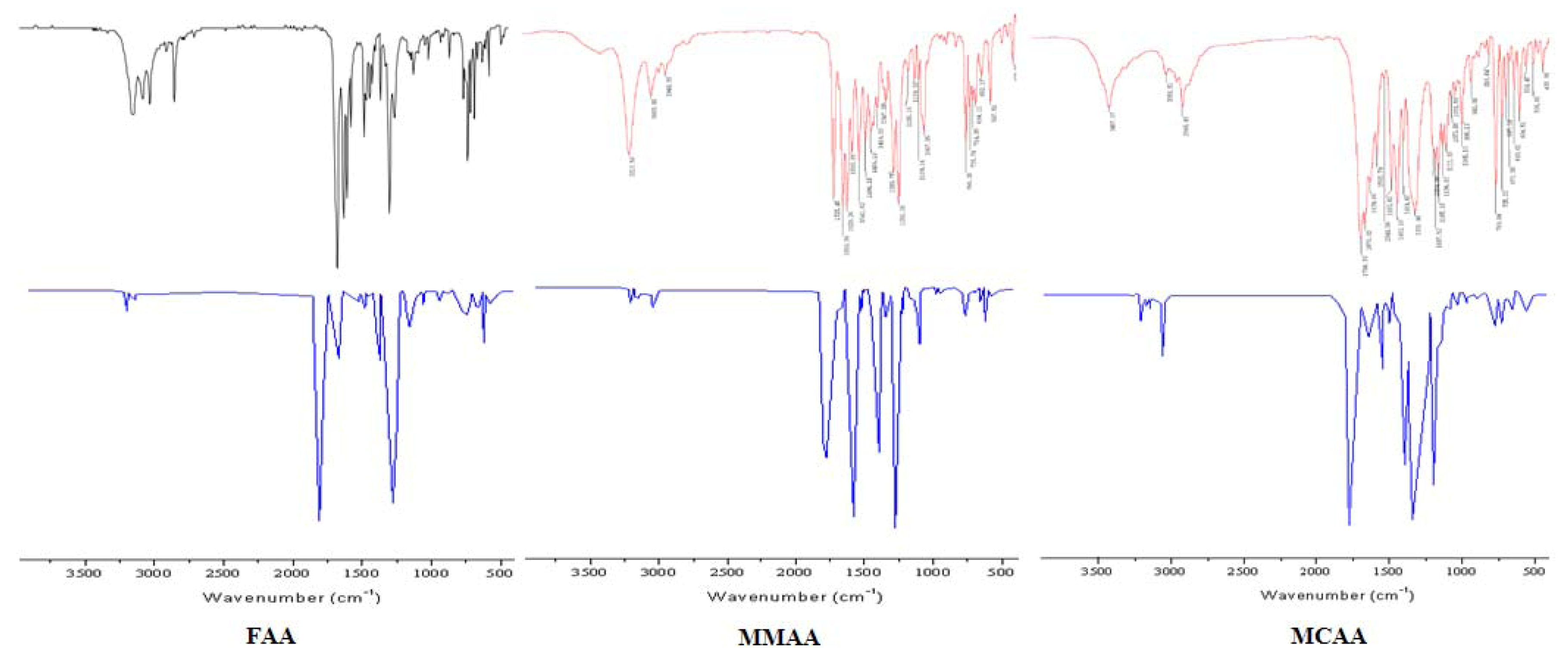
2.2. Vibration Spectra
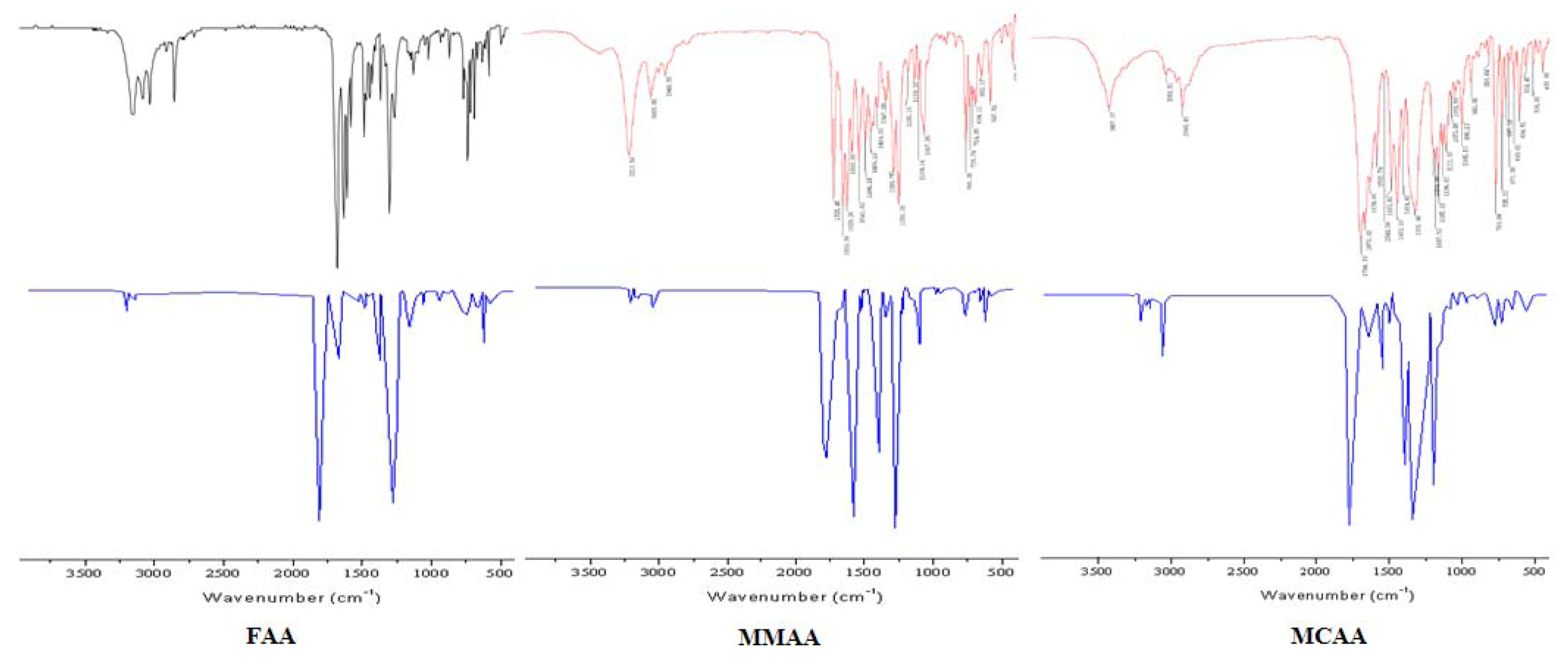
| FAA | MMAA | MCAA | ||||||
|---|---|---|---|---|---|---|---|---|
| Exp. | B3LYP/6-31G * | Vibrational assignments | Exp. | B3LYP/6-31G * | Vibrational assignments | Exp. | B3LYP/6-31G * | Vibrational assignments |
| 3190ms | 3187 | νN-H | 3213s | 3210 | νN-H | 3056w | 3055 | ν=C-H |
| 3049ms | 3044 | ν=C-H | 3053m | 3057 | ν=C-H | 2948m | 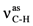 | |
| 2925ms | | 2948w | | 1700vs | 1690 | νC=O | ||
| 2878ms | 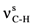 | 1725vs | 1727 | νC=O | 1676vs | 1659 | νC=O | |
| 1689vs | 1690 | νC=O | 1659vs | 1659 | νC=O | 1639s | 1643 | νC=C |
| 1636vs | 1643 | νC=O | 1629vs | νC=C | 1593ms | νC=C | ||
| 1545s | 1544 | 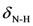 | 1593s | νC=C | 1544w | 1546 | ||
| 1490s | 1480 | νC=C | 1541s | 1538 | | 1493s | 1498 | νC=C |
| 1386ms | 1395 | 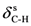 | 1494s | 1492 | νC=C | 1452vs | 1455 | νC=C |
| 1216ms | 1211 | νC-N | 1456ms | 1449 | νC=C | 1331vs | 1340 | |
| 1140m | 1129 | νC-N | 1347m | 1346 | | 1205ms | 1209 | νC-N |
| 1106w | 1113 | νC-N | 1290s | νC-N | 1187ms | 1188 | νC-N | |
| 1020w | 1017 | νC-N | 1253vs | νC-O | 1165ms | 1169 | νC-O | |
| 856w | 850 | 1185w | 1188 | νC-N | 1137ms | 1141 | νC-N | |
| 768ms | 774 | γ=C-H | 1139w | 1141 | νC-N | 1111ms | 1108 | νC-N |
| 698ms | 708 | γN-H | 1109w | 1117 | νC-N | 1073w | 1072 | νC-O |
| 666w | 659 | γ=C-H | 1067s | 1065 | νC-O | 1051w | 1045 | νC-N |
| 638w | 632 | 765s | 758 | γ=C-H | 996m | 991 | ||
| 735m | 740 | 769s | 773 | γ=C-H | ||||
| 714m | 709 | γN-H | 725m | 728 | ||||
| 694m | 703 | γ=C-H | ||||||
| 652w | 661 | |||||||
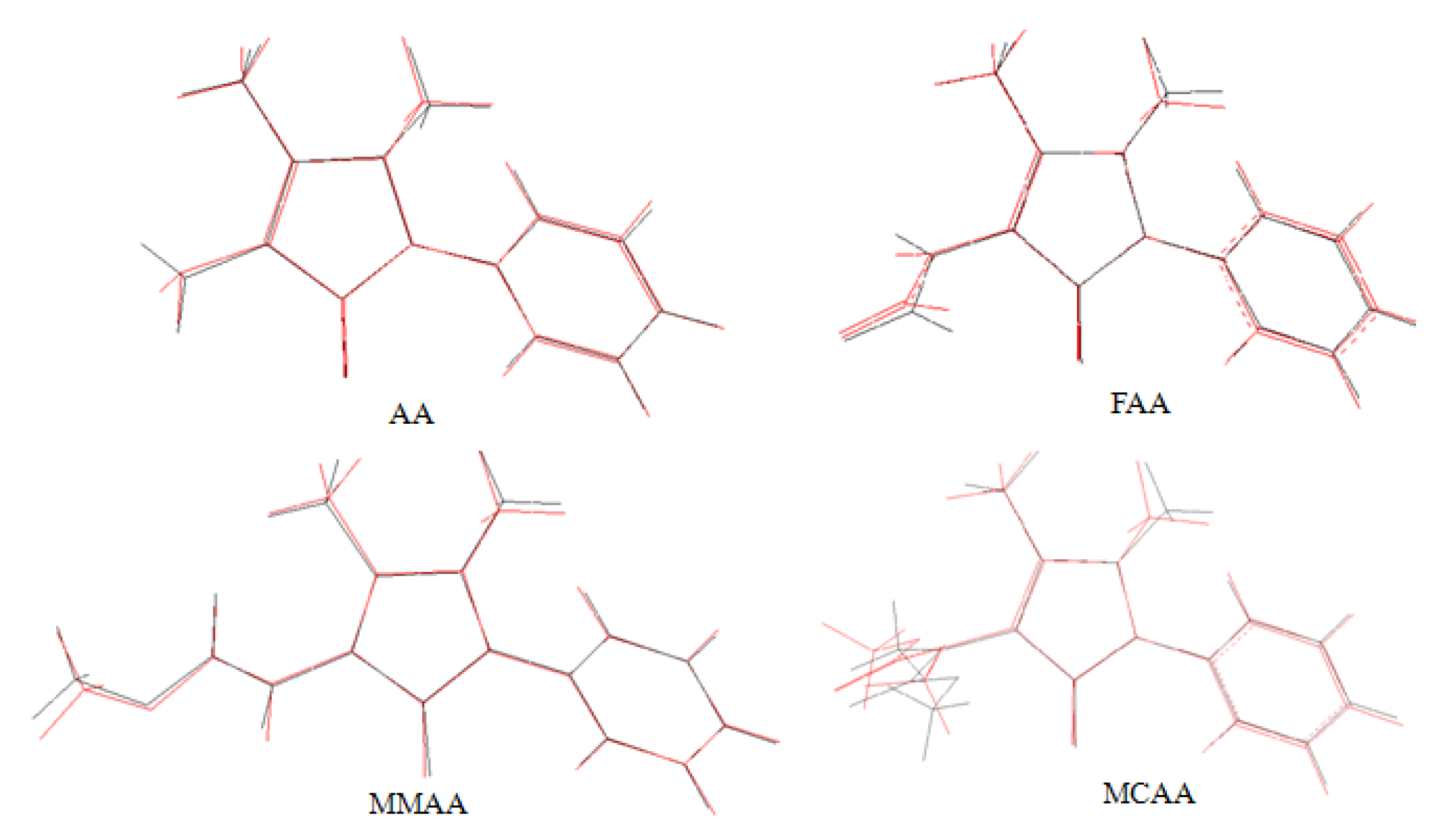
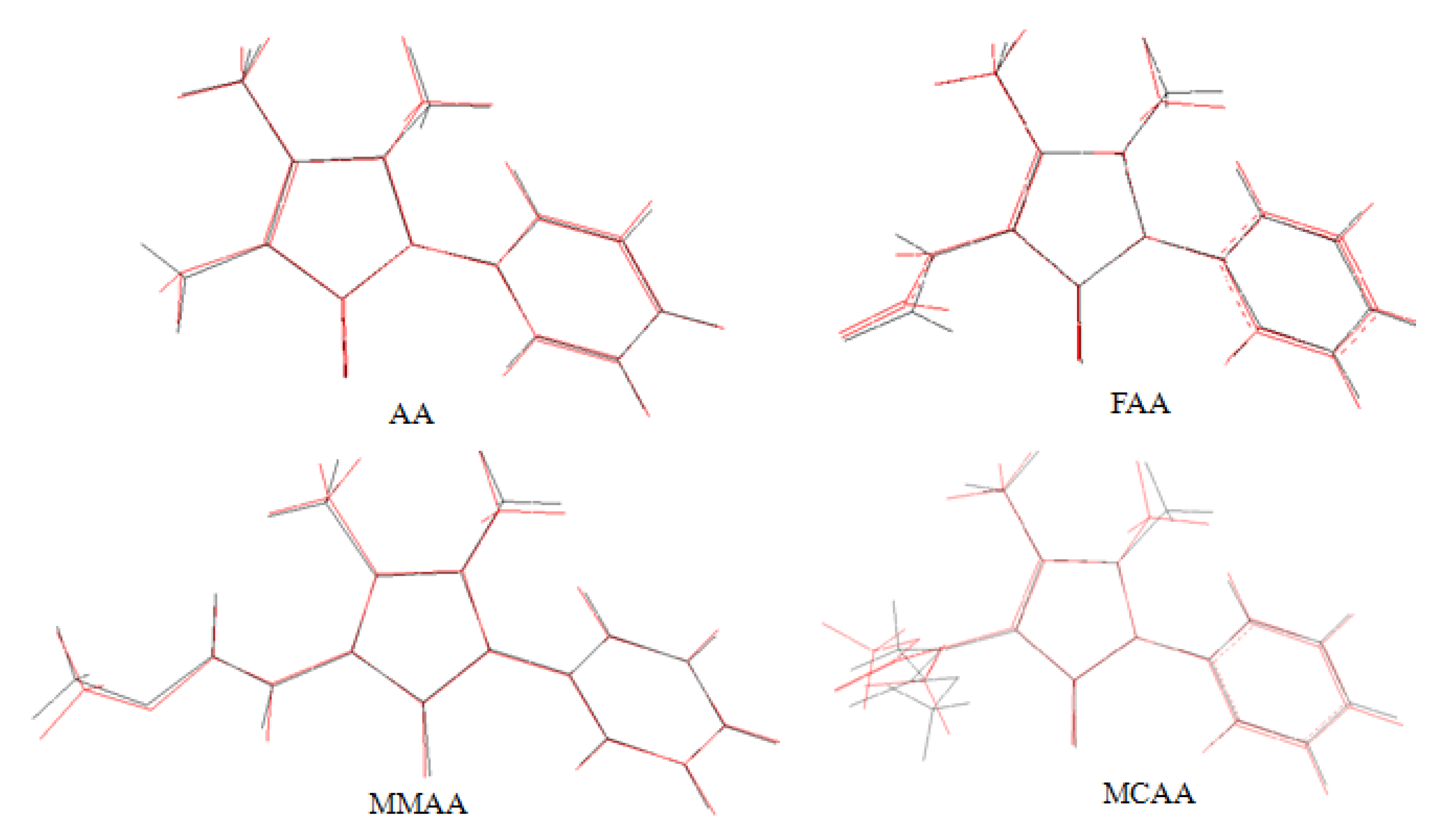
2.3. Theoretical Structures
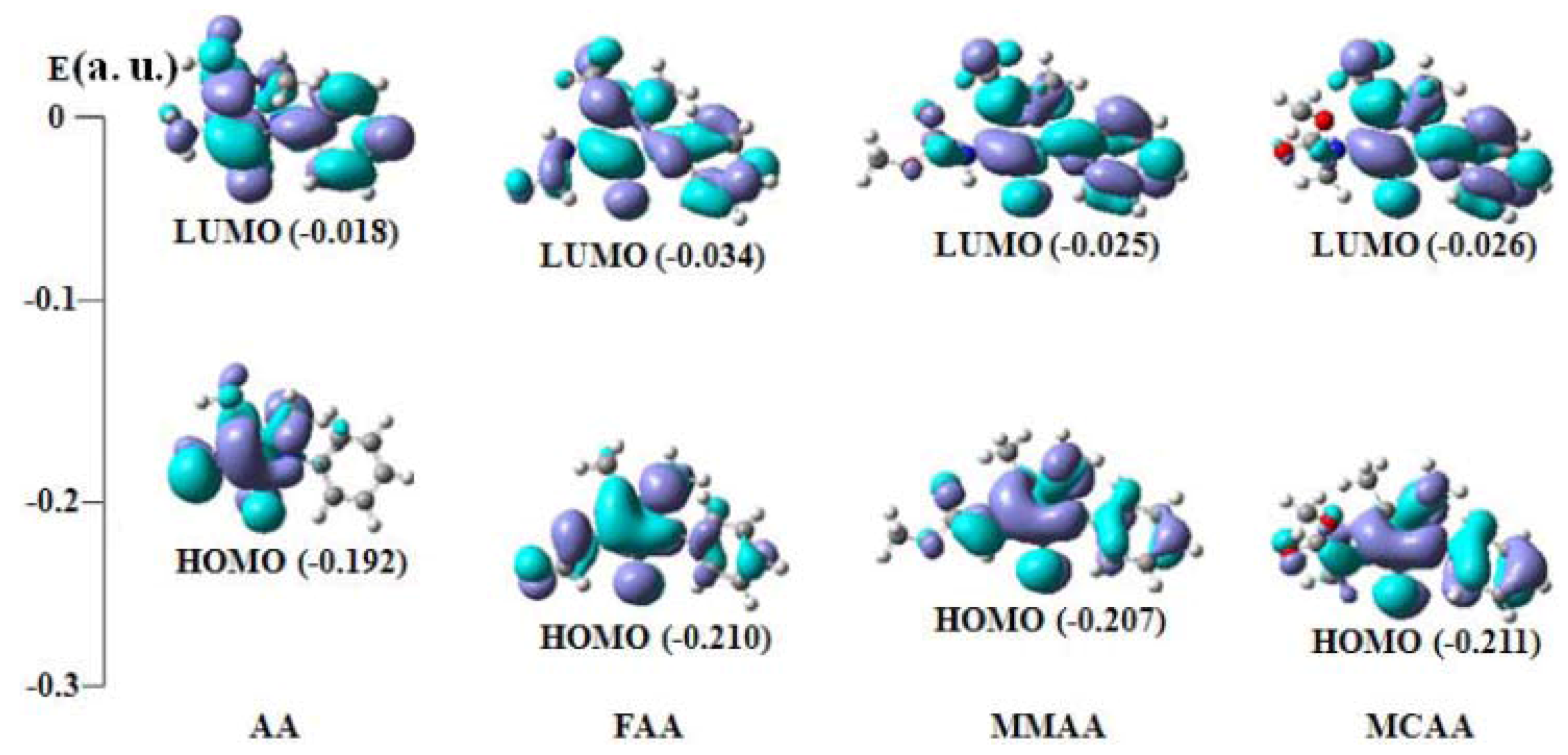
2.4. Frontier Molecular Orbital Analysis
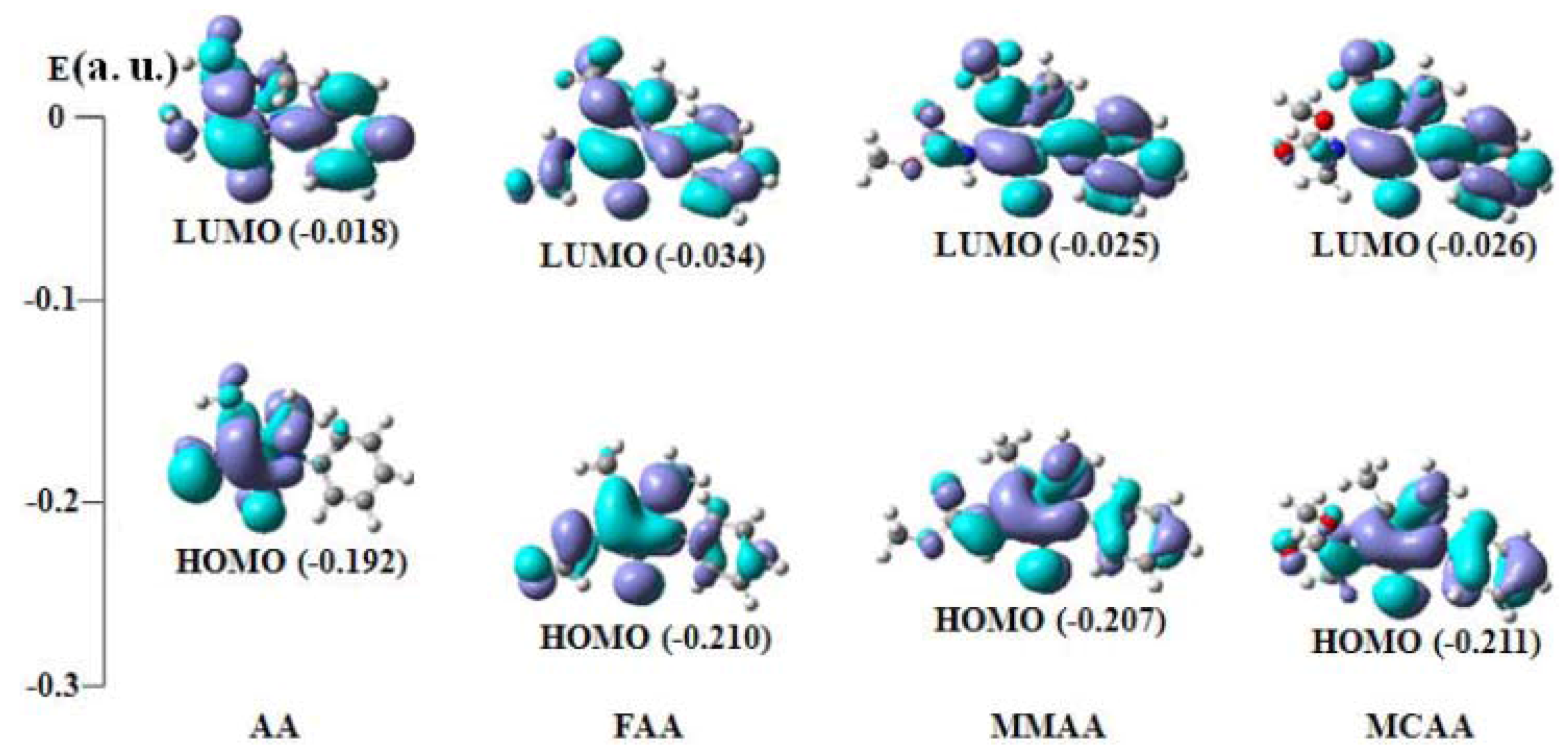
| Compound | E(HOMO) | E (LUMO) | ∆E (LUMO−HOMO) |
|---|---|---|---|
| AA | −0.192 (54a) | −0.018 (55a) | 0.174 |
| FAA | −0.210 (61a) | −0.034 (62a) | 0.176 |
| MMAA | −0.207 (69a) | −0.025 (70a) | 0.182 |
| MCAA | −0.211 (73a) | −0.026 (74a) | 0.185 |
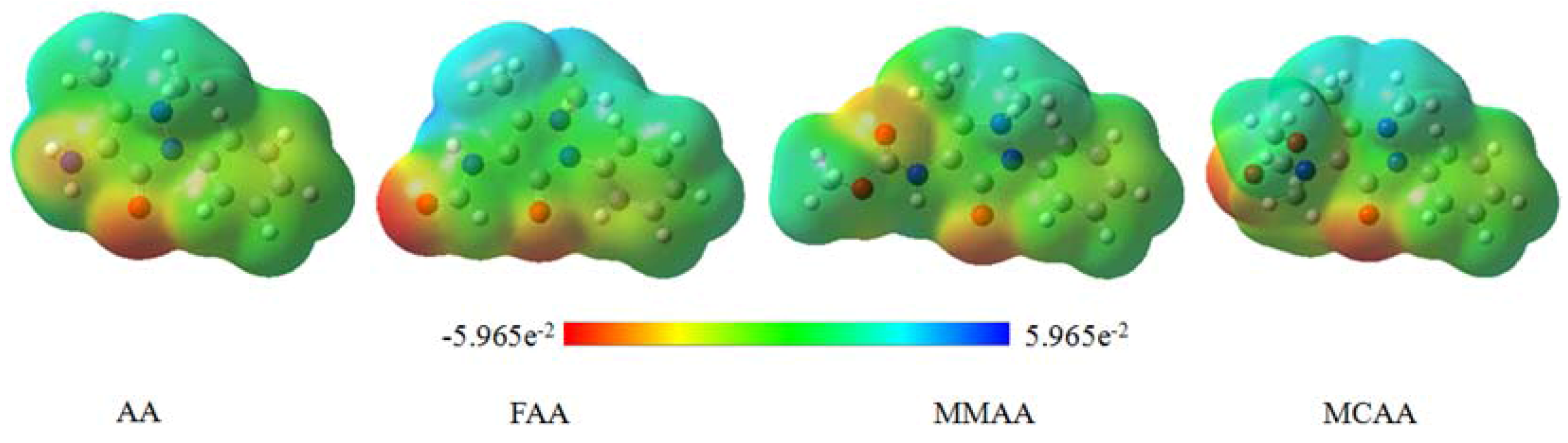
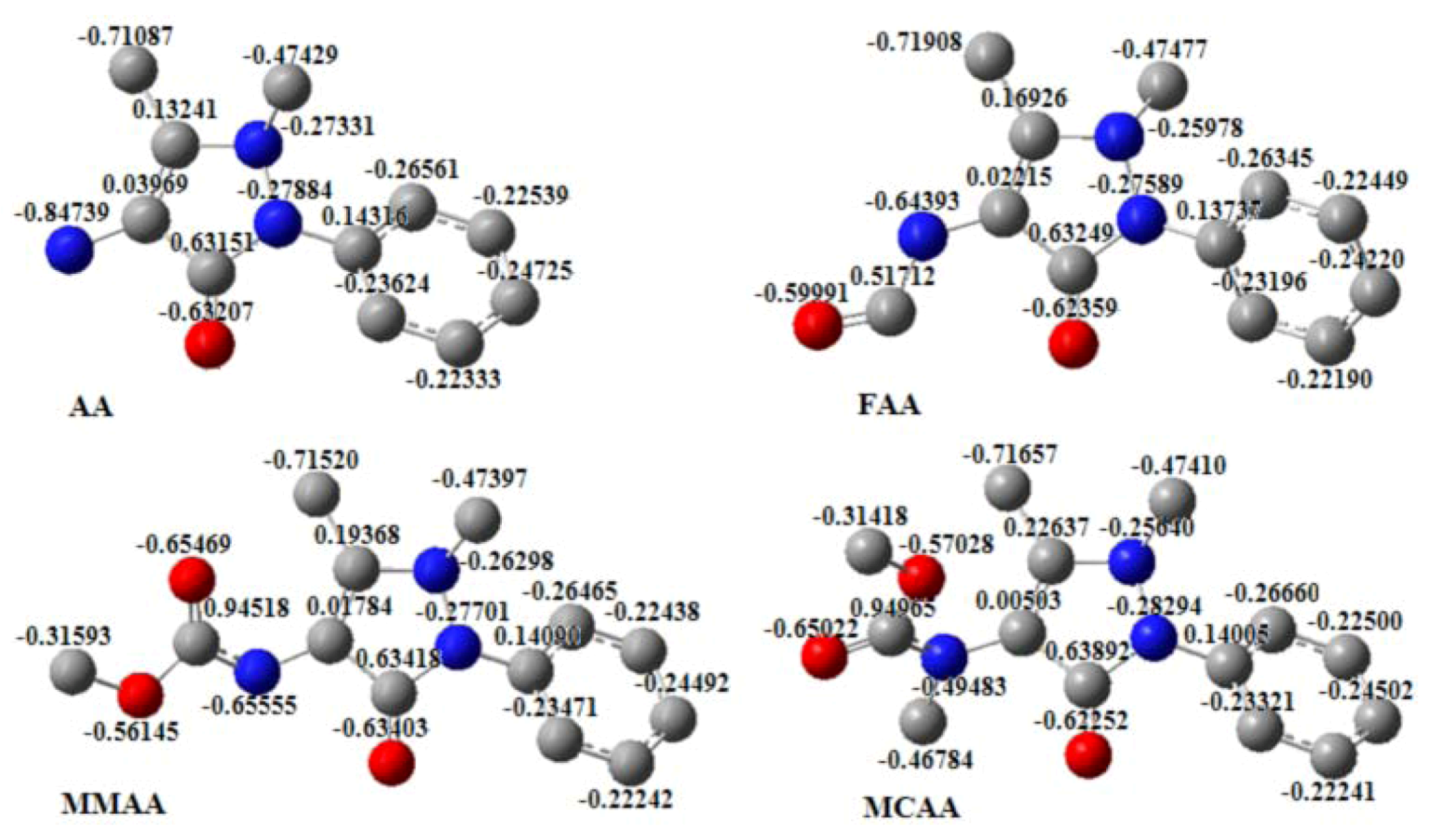
2.5. Molecular Electrostatic Potential
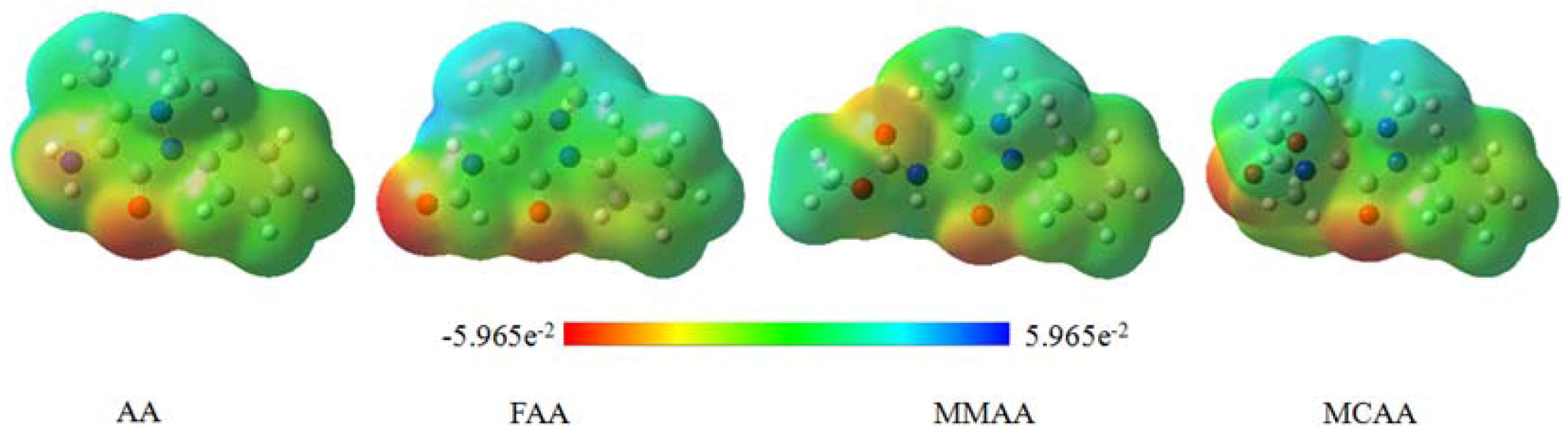

3. Experimental
3.1. General
3.2. Synthesis
3.3. Crystallography
3.4. Theoretical Calculation
4. Conclusions
Acknowledgments
- Sample Availability: Samples of the compounds, AA, FAA, MMAA, and MCAA, are available from the authors.
References
- Cechinel Filho, V.; Correa, R.; Vaz, Z.; Calixto, J.B.; Nunes, R.J.; Pinheiro, T.R.; Andricopulo, A.D.; Yunes, R.A. Further studies on analgesic activity of cyclic imides. Farmaco 1998, 53, 55–57. [Google Scholar] [CrossRef]
- Sondhi, S.M.; Sharma, V.K.; Verma, R.P.; Singhal, N.; Shukla, R.; Raghubir, R.; Dubey, M.P. Synthesis, anti-inflammatory, and analgesic activity evaluation of some mercapto pyrimidine and pyrimidobenzimidazole derivatives. Synthesis 1999, 1999, 878–884. [Google Scholar] [CrossRef]
- Mishra, A.P. Physicochemical and antimicrobial studies on nickel(II) and copper(II) Schiff base complexes derived from 2-furfuraldehyde. J. Indian Chem. Soc. 1999, 76, 35–37. [Google Scholar]
- Raman, N.; Kulandaisamy, A.; Jeyasubramanian, K. Synthesis, spectral, redox, and antimicrobial activity of Schiff base transition metal(II) complexes derived from 4-aminoantipyrine and benzil. Syn. React. Inorg. Met. Org. Chem. 2002, 32, 1583–1610. [Google Scholar] [CrossRef]
- Raman, N.; Kulandaisamy, A.; Jeyasubramanian, K. Synthesis, Structural Characterization, Redox, and Antibacterial Studies of 12-Membered Tetraaza Macrocyclic Cu(II), Ni(II), Co(II), Zn(II), and VO(IV) Complexes Derived from 1,2-bis(imino-4'-antipyrinyl)-1,2-diphenylethane and o-Phenylenediamine. Syn. React. Inorg. Met. Org. Chem. 2004, 34, 17–43. [Google Scholar]
- Sondhi, S.M.; Singhal, N.; Verma, R.P.; Arora, S.K.; Dastidar, S.G. Synthesis of hemin and porphyrin derivatives and their evaluation for anticancer activity. Indian J. Chem. Sect. B 2001, 40B, 113–119. [Google Scholar]
- Sutcliffe, J.A. Antibacterial Agents: Solutions for the Evolving Problems of Resistance. Bioorg. Med. Chem. Lett. 2003, 13, 4159–4161. [Google Scholar] [CrossRef]
- Ergun, H.; Frattarelli, D.A.C.; Aranda, J.V.J. Characterization of the role of physicochemical factors on the hydrolysis of dipyrone. J. Pharm. Biomed. Anal. 2004, 35, 479–487. [Google Scholar] [CrossRef]
- Hohlfeld, T.; Zimmermann, N.; Weber, A.A.; Jessen, G.; Weber, H.; Schroer, K.; Hoeltje, H.D.; Ebel, R. Pyrazolinone analgesics prevent the antiplatelet effect of aspirin and preserve human platelet thromboxane synthesis. J. Thromb. Haemost. 2008, 6, 166–173. [Google Scholar]
- Picot, D.; Loll, P.J.; Garavito, R.M. The x-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature 1994, 367, 243–249. [Google Scholar]
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; McDonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Gildehaus, D.; Miyashiro, J.M.; Penning, T.D.; Seibert, K.; et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar]
- Li, Y.; Zhang, H.B.; Liu, Y.Y.; Li, F.S.; Liu, X.N. Synthesis, characterization, and quantum chemical calculation studies on 3-(3-nitrophenylsulfonyl)aniline. J. Mol. Struct. 2011, 997, 110–116. [Google Scholar] [CrossRef]
- Wang, H.W.; Yang, M.M.; Lu, Q.S.; Li, F.S. N-(1,5-Dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl) formamide. Acta Crystallogr. Sect. E Struct. Rep. Online 2011, E67, o1310. [Google Scholar]
- Fukui, K.; Yonezawa, T.; Shingu, H.J. A molecular-orbital theory of reactivity in aromatic hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Padmaja, L.; Ravikumar, C.; Sajan, D.; Hubert Joe, I.; Jayakumar, V.S.; Pettit, G.R.; Neilsen, F.O. Density functional study on the structural conformations and intramolecular charge transfer from the vibrational spectra of the anticancer drug combretastatin-A2. J. Raman Spectrosc. 2009, 40, 419–428. [Google Scholar] [CrossRef]
- Ravikumar, C.; Hubert Joe, I.; Jayakumar, V.S. Charge transfer interactions and nonlinear optical properties of push-pull chromophore benzaldehyde phenylhydrazone: A vibrational approach. Chem. Phys. Lett. 2008, 460, 552–558. [Google Scholar] [CrossRef]
- Scrocco, E.; Tomasi, J. Electronic molecular structure, reactivity and intermolecular forces: An euristic interpretation by means of electrostatic molecular potentials. Adv. Quantum Chem. 1978, 11, 115–121. [Google Scholar] [CrossRef]
- Luque, F.J.; Lopez, J.M.; Orozco, M. Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects”. Theor. Chem. Acc. 2000, 103, 343–345. [Google Scholar]
- Okulik, N.; Jubert, A.H. Theoretical analysis of the reactive sites of non-steroidal anti-inflammatory drugs. Internet Electron. J. Mol. Des. 2005, 4, 17–30. [Google Scholar]
- Politzer, P.; Laurence, P.R.; Jayasuriya, K. Molecular electrostatic potentials: An effective tool for the elucidation of biochemical phenomena. Environ. Health Perspect. 1985, 61, 191–202. [Google Scholar] [CrossRef]
- Scrocco, E.; Tomasi, J. Topics in Current Chemistry; Springer: Berlin, Germany, 1973; Volume 7, p. 95. [Google Scholar]
- Politzer, P.; Truhlar, D.G. Chemical Applications of Atomic and Molecular Electrostatic Potentials; Basic Books: New York, NY, USA, 1981. [Google Scholar]
- Bocca, C.C.; Basso, E.A.; Gauze, G.F. Substituent effects on the reduction of 2-OMe, 2-SMe and 2-SeMe cyclohexanones by LiAlH4: An investigation of conformational equilibrium and transition states. Chem. Phys. Lett. 2005, 413, 434–439. [Google Scholar] [CrossRef]
- Politzer, K.C.D. The Force Concept in Chemistry; Van Nostrand Reinhold Publisher: New York, NY, USA, 1981; Chapter 6. [Google Scholar]
- Politzer, S.M. Reviews in Computational Chemistry; VCH Publishers: New York, NY, USA, 1991; Chapter 7. [Google Scholar]
- Naray-Szabo, G.; Ferenczy, G.G. Molecular Electrostatics. Chem. Rev. 1995, 95, 829–847. [Google Scholar] [CrossRef]
- Murray, S.; Politzer, P. Molecular Orbital Calculations for Biological Systems; Oxford University Press: New York, NY, USA, 1998; Chapter 3. [Google Scholar]
- Michaux, C. A new potential cyclooxygenase-2 inhibitor, pyridinic analogue of nimesulide. Eur. J. Med. Chem. 2005, 40, 1316–1324. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, Y.; Liu, Y.; Wang, H.; Xiong, X.; Wei, P.; Li, F. Synthesis, Crystal Structure, Vibration Spectral, and DFT Studies of 4-Aminoantipyrine and Its Derivatives. Molecules 2013, 18, 877-893. https://doi.org/10.3390/molecules18010877
Li Y, Liu Y, Wang H, Xiong X, Wei P, Li F. Synthesis, Crystal Structure, Vibration Spectral, and DFT Studies of 4-Aminoantipyrine and Its Derivatives. Molecules. 2013; 18(1):877-893. https://doi.org/10.3390/molecules18010877
Chicago/Turabian StyleLi, Yi, Yuanyuan Liu, Haowei Wang, Xiaohui Xiong, Ping Wei, and Fangshi Li. 2013. "Synthesis, Crystal Structure, Vibration Spectral, and DFT Studies of 4-Aminoantipyrine and Its Derivatives" Molecules 18, no. 1: 877-893. https://doi.org/10.3390/molecules18010877
APA StyleLi, Y., Liu, Y., Wang, H., Xiong, X., Wei, P., & Li, F. (2013). Synthesis, Crystal Structure, Vibration Spectral, and DFT Studies of 4-Aminoantipyrine and Its Derivatives. Molecules, 18(1), 877-893. https://doi.org/10.3390/molecules18010877