Photochemical Synthesis and Properties of 1,6- and 1,8-Naphthalenophanes

Abstract
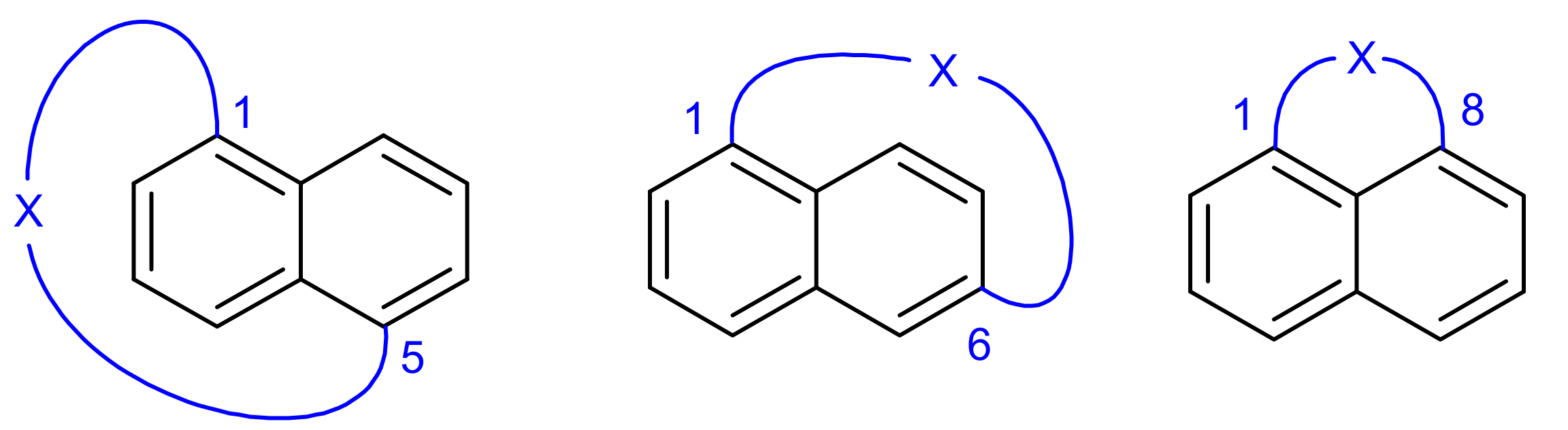
:1. Introduction
2. Results and Discussion
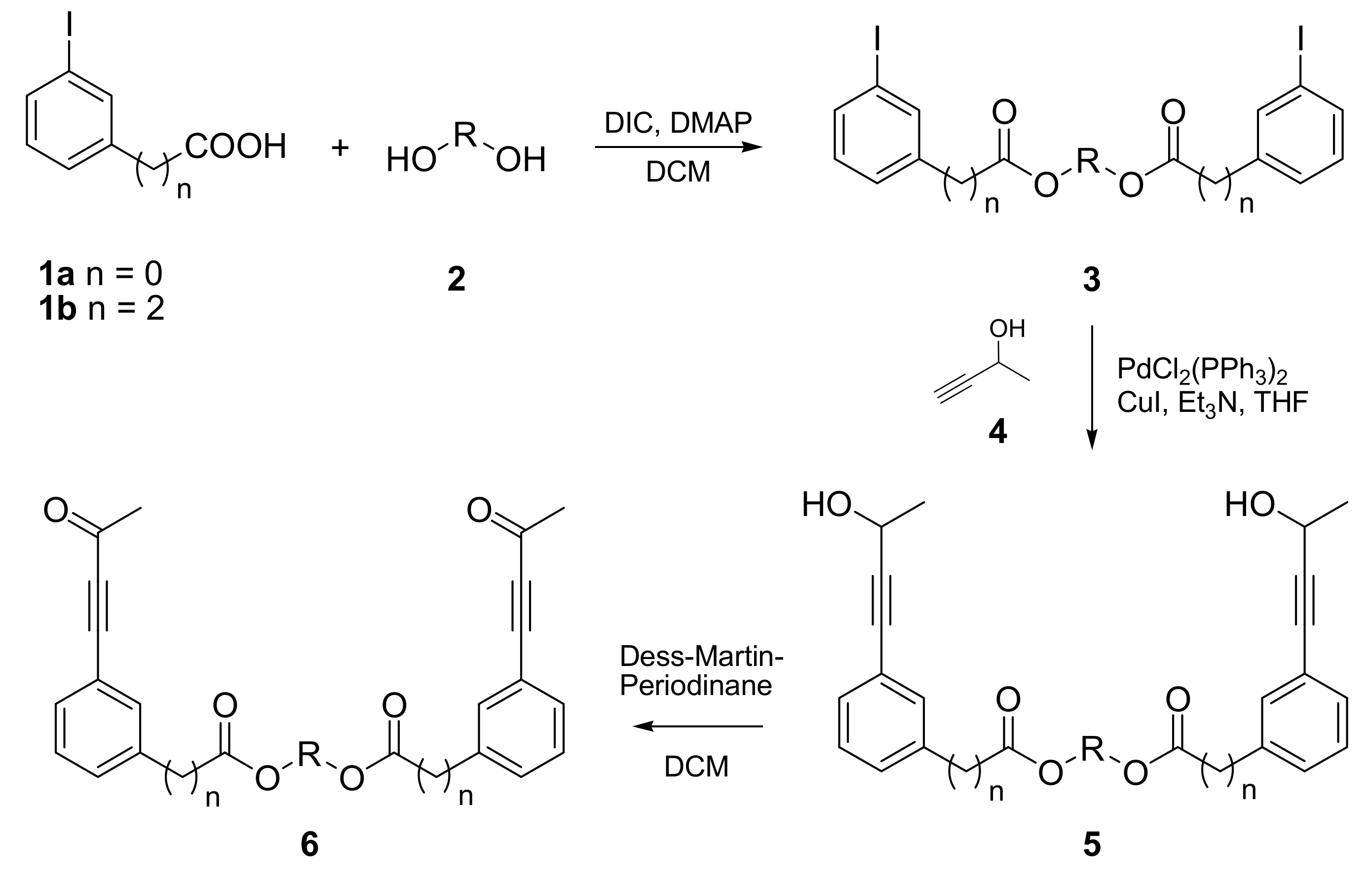
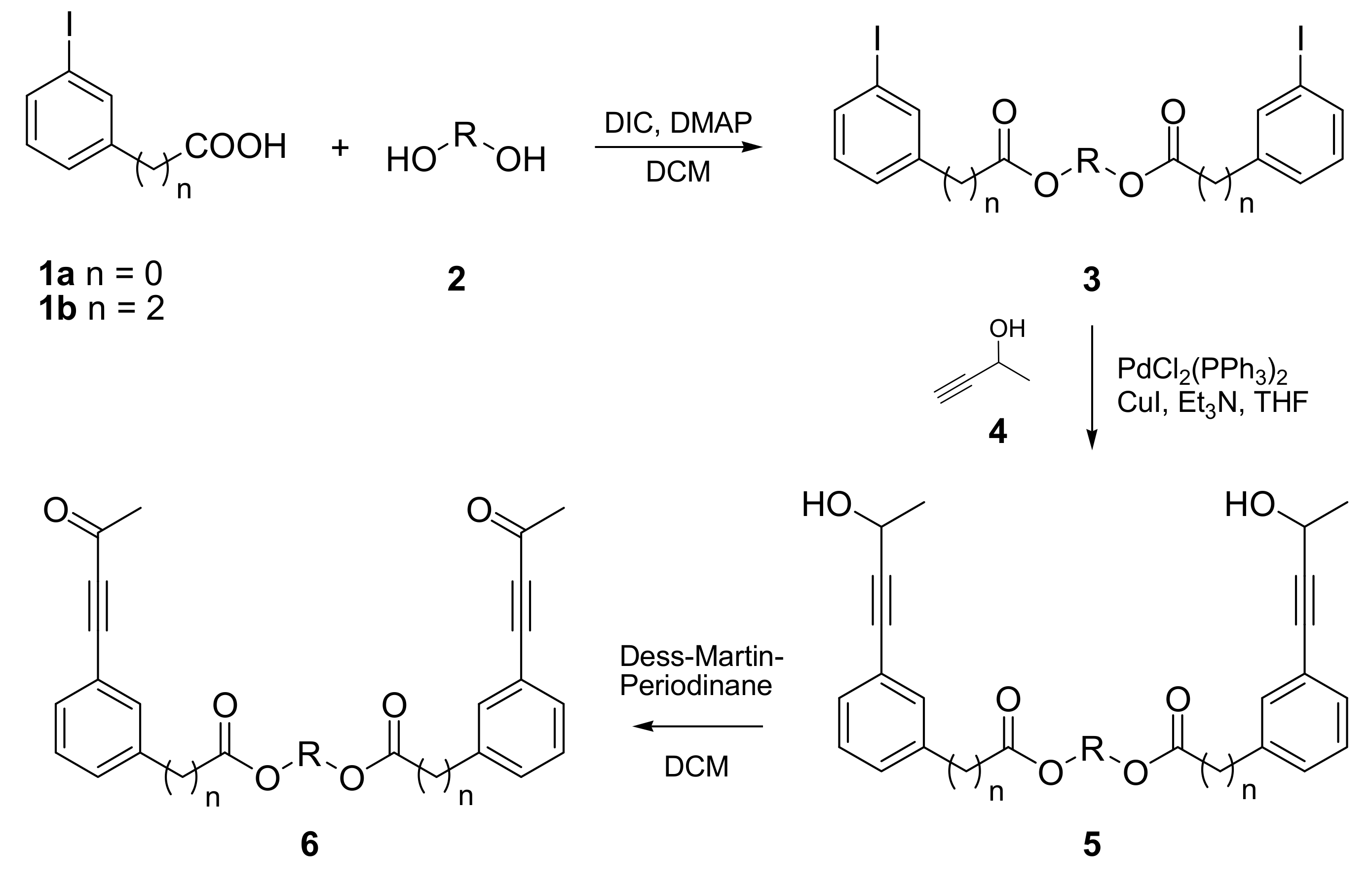
2.1. Synthesis of PDDA Reactants
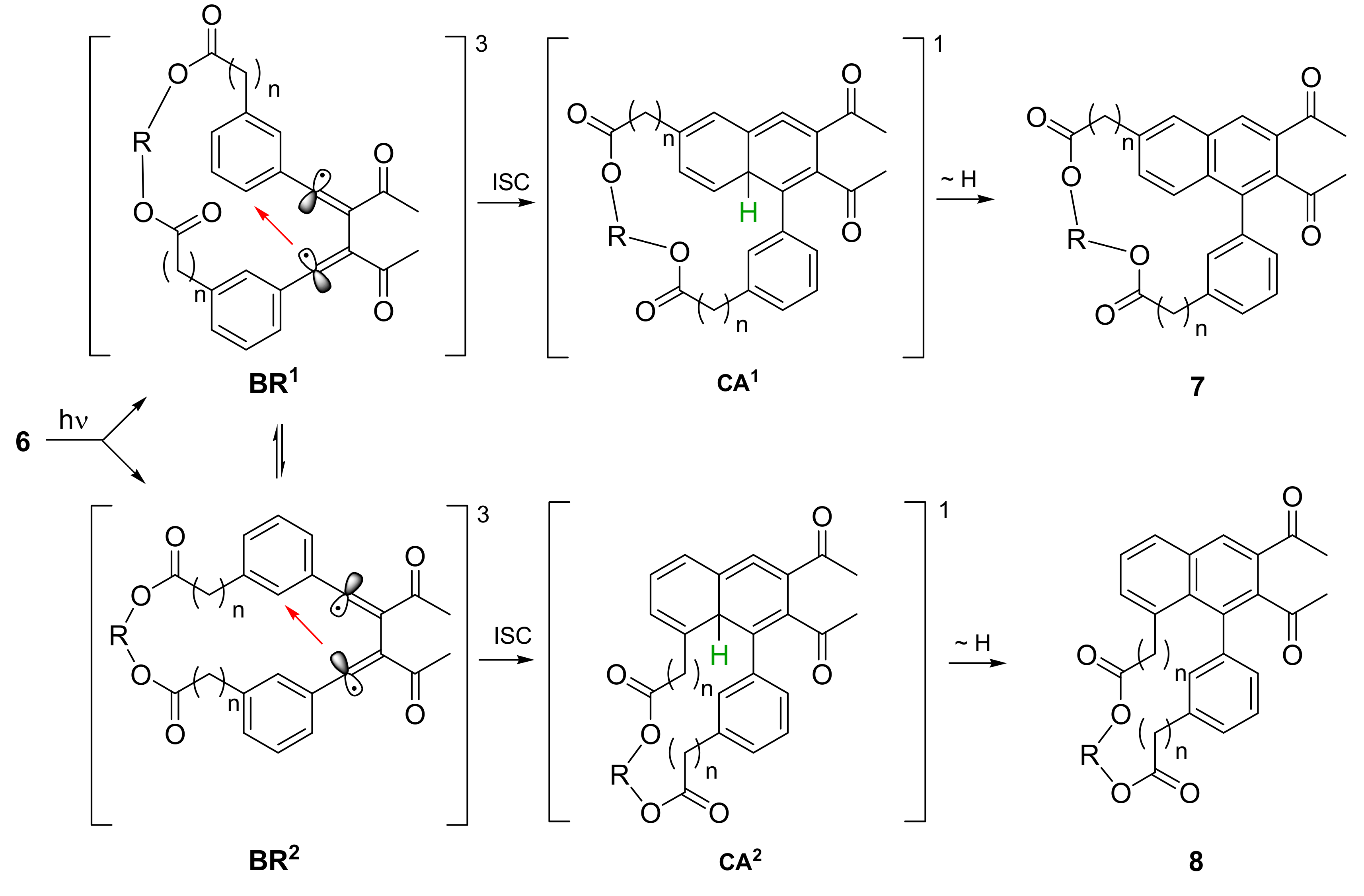
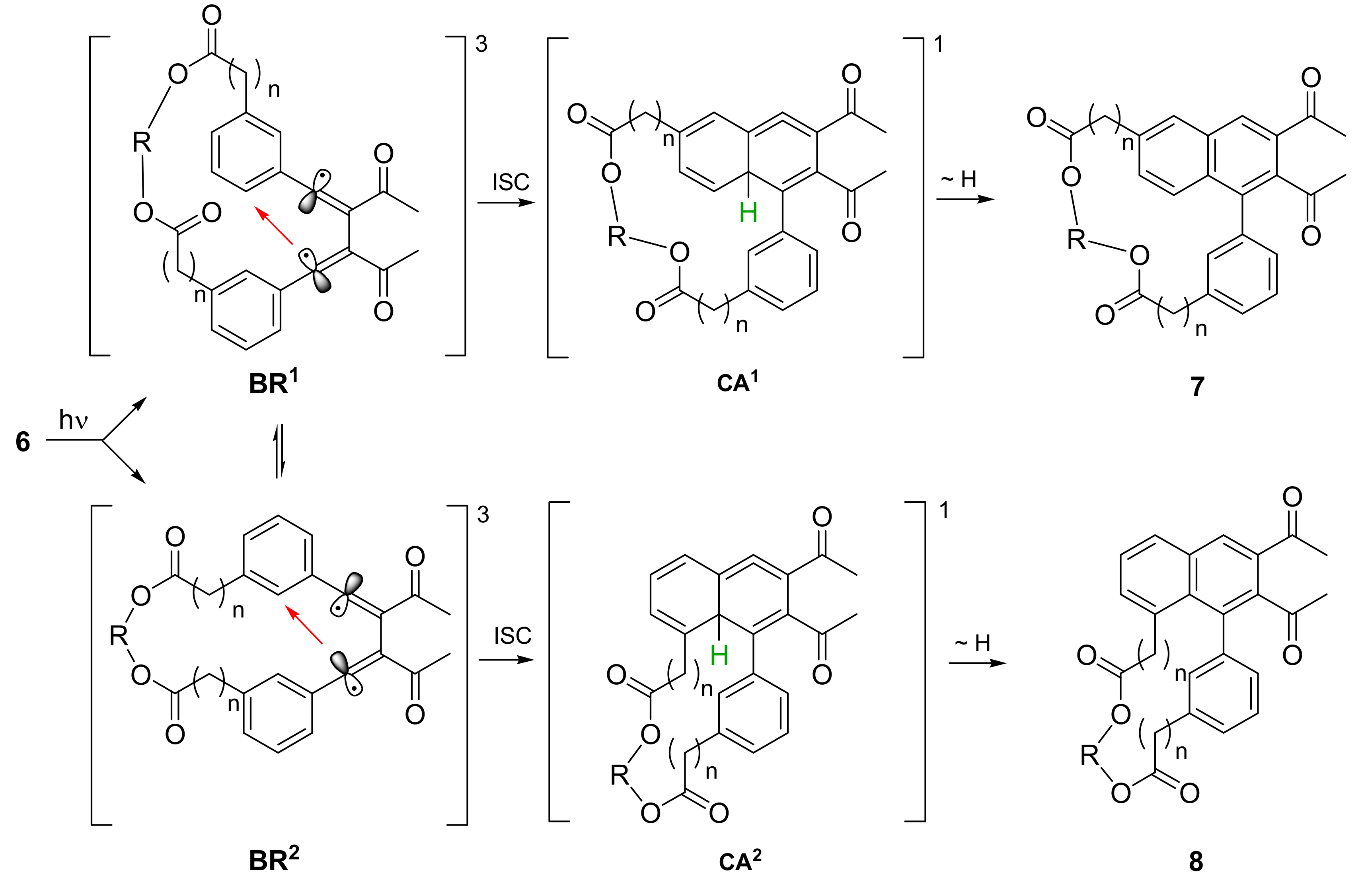
2.2. Photochemical Synthesis of Naphthalenophanes
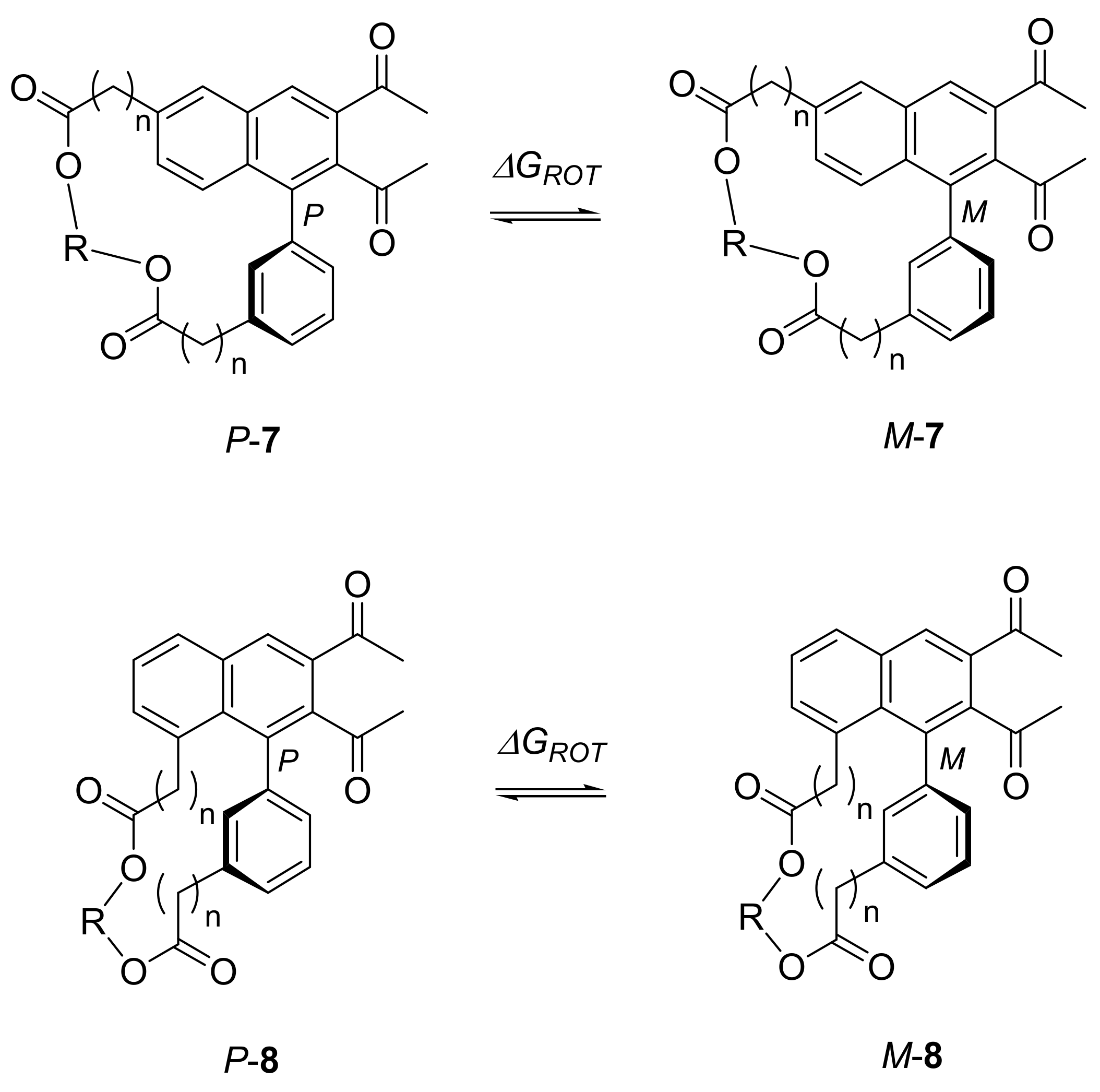
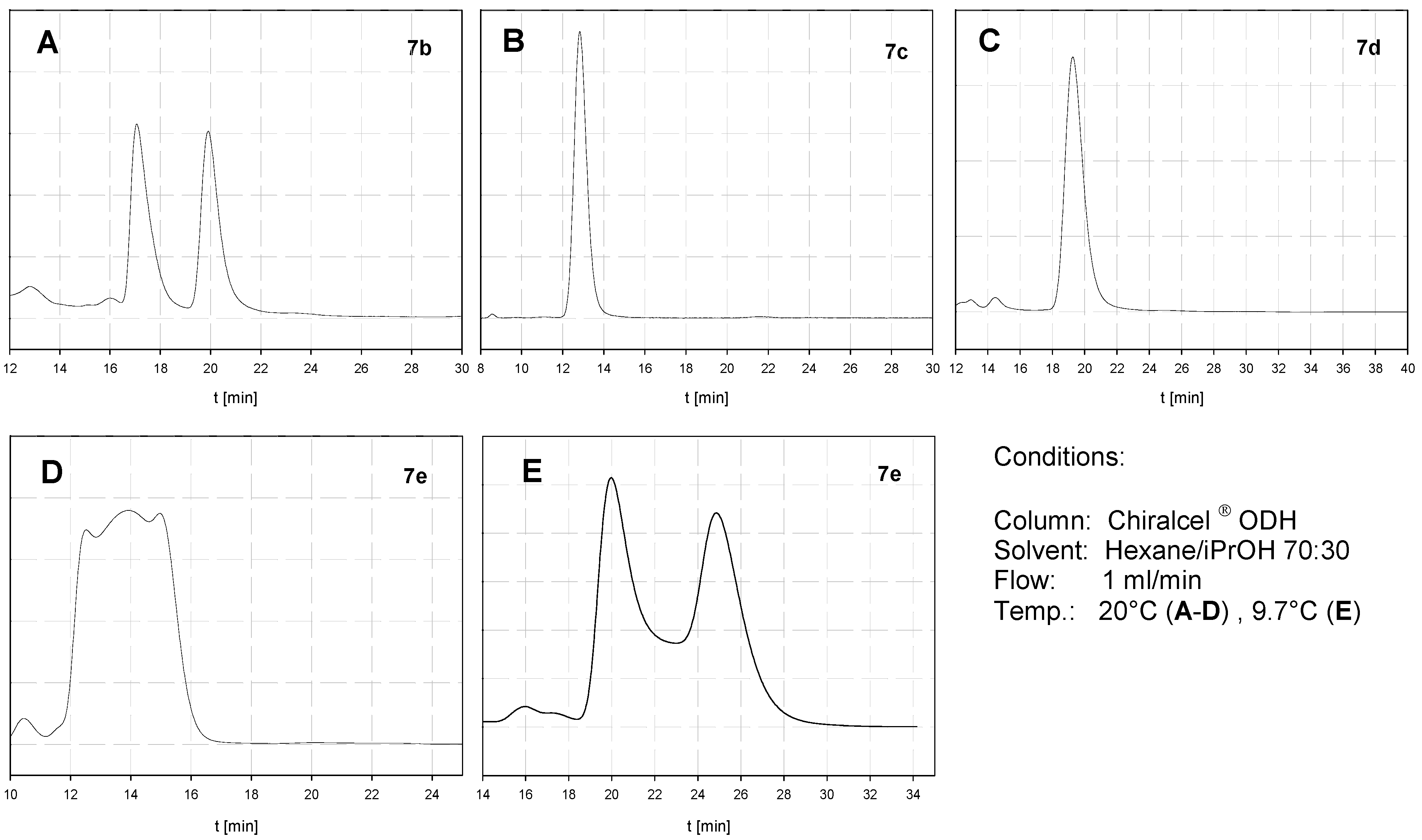
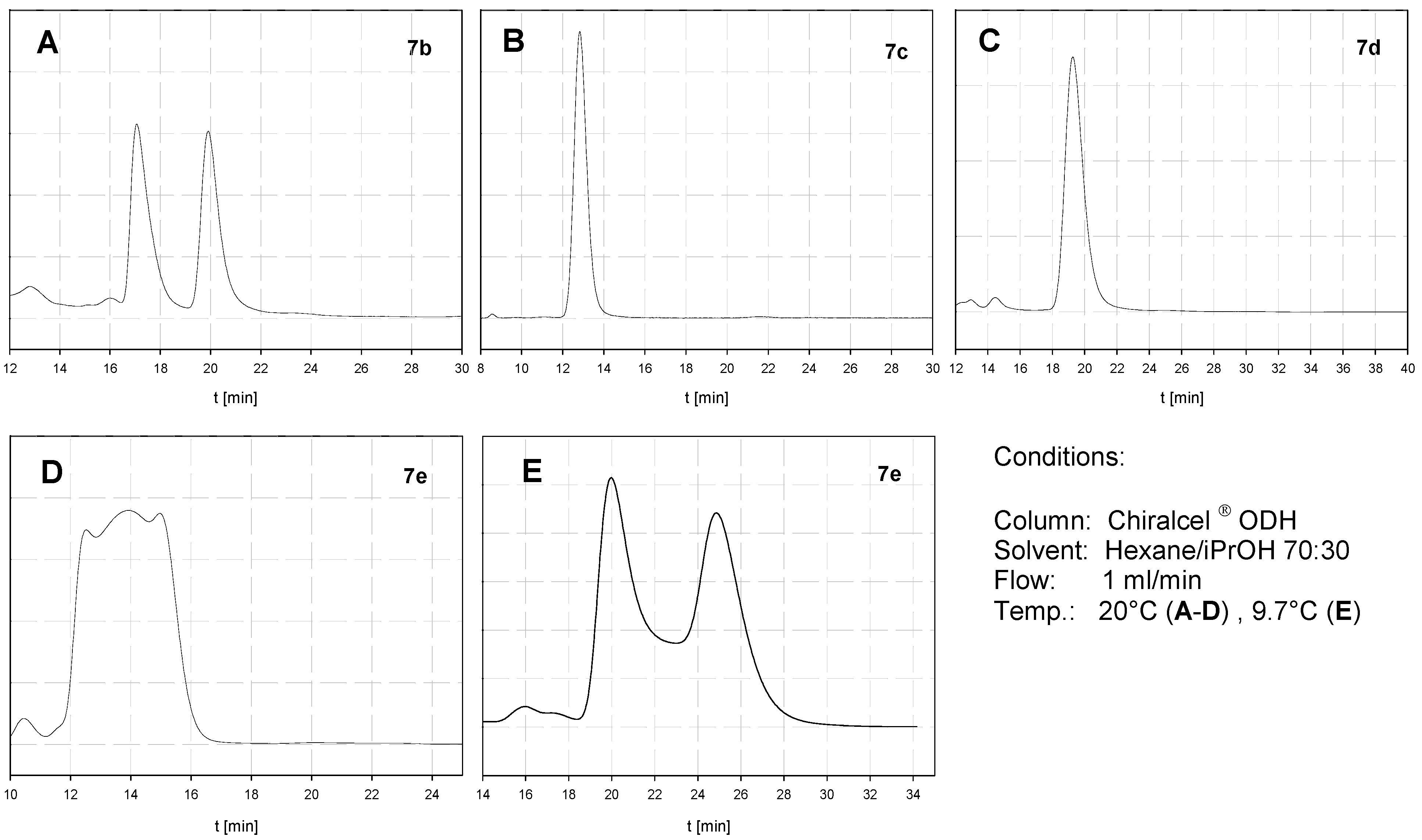
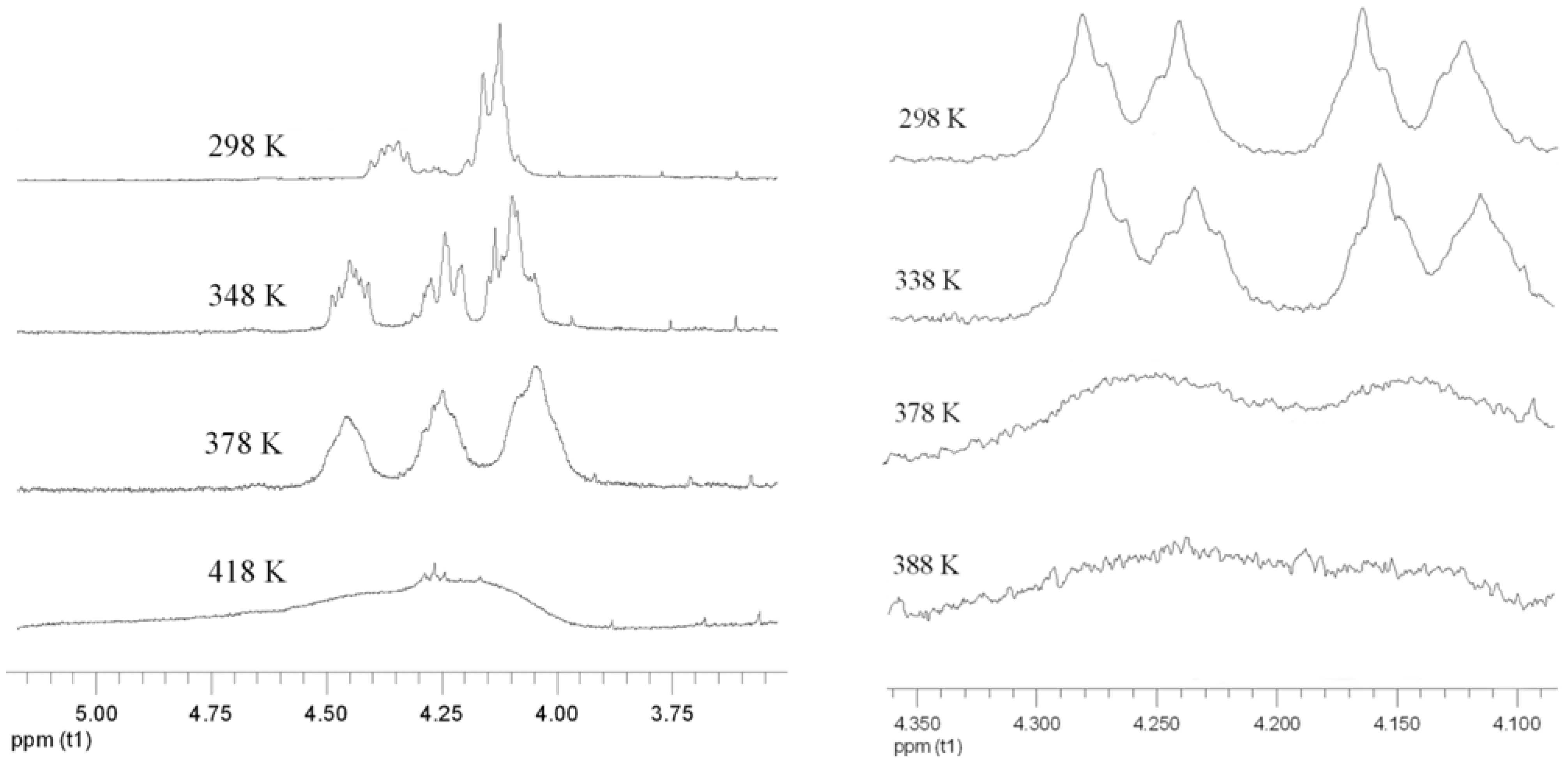
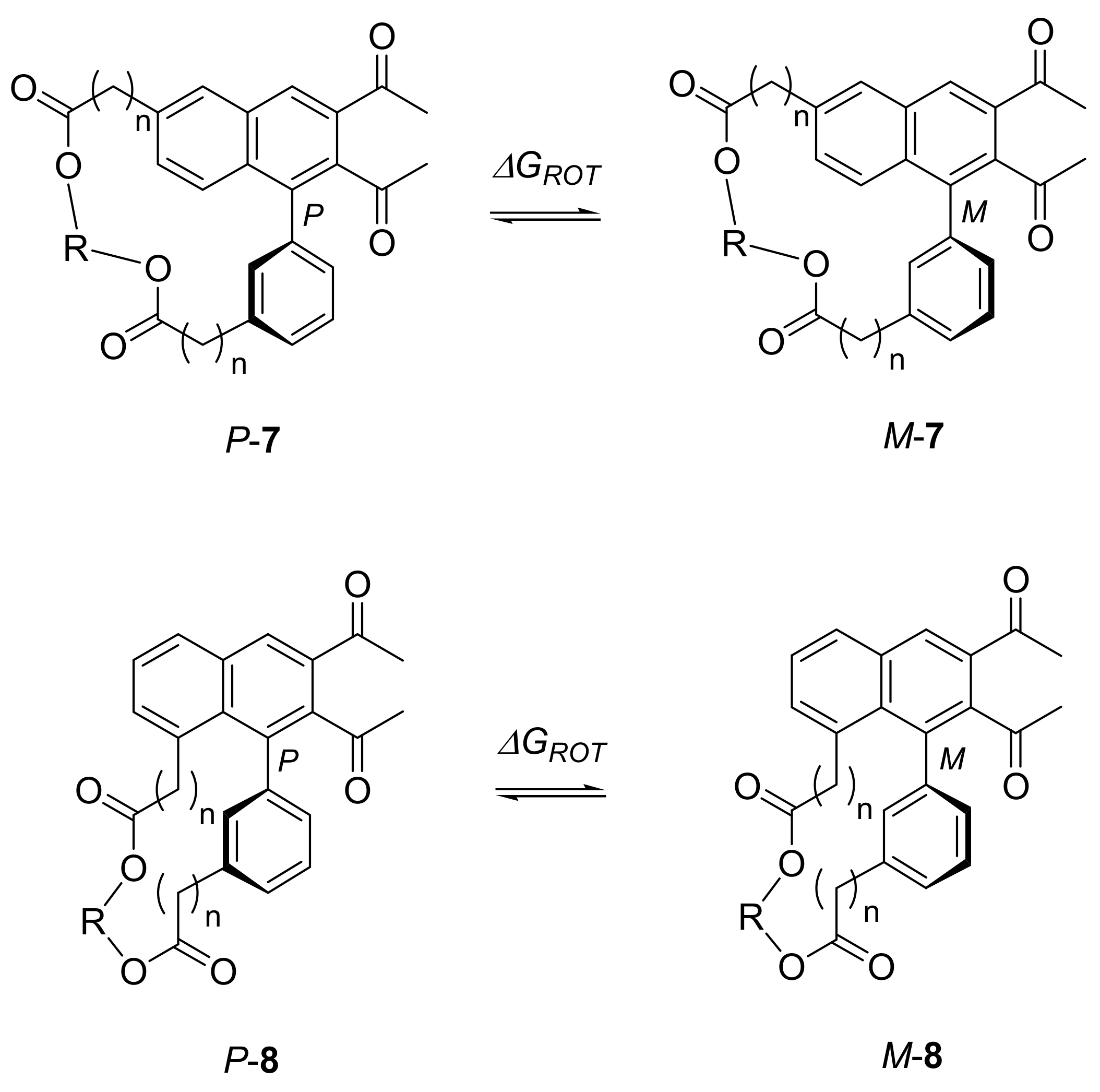
2.3. Atropisomerization of Naphthalenophanes 7 and 8
3. Experimental
3.1. General
3.2. General Procedure—Irradiation of Compounds 6
4. Conclusions
Acknowledgments
References
- Gleiter, R.; Hopf, H. (Eds.) Modern Cyclophane Chemistry; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Bogdan, N.D.; Grosu, I. [4.n]cyclophanes. Curr. Org. Chem. 2009, 13, 502–531. [Google Scholar] [CrossRef]
- Vögtle, F.; Schäfer, R.; Schunder, L.; Neumann, P. Steric interactions of inner atoms in cyclic compounds. XIII. Naphthalenophanes. Liebigs Ann. Chem. 1970, 734, 102–105. [Google Scholar] [CrossRef]
- Vögtle, F.; Neumann, P. Nomenclature of phanes [cyclic bridged aromatic compounds]. Tetrahedron Lett. 1969, 60, 5329–5334. [Google Scholar] [CrossRef]
- Wessig, P.; Matthes, A. Preparation of Strained Axially Chiral (1,5)Naphthalenophanes by Photo-dehydro-Diels-Alder Reaction. J. Am. Chem. Soc. 2011, 133, 2642–2650. [Google Scholar] [CrossRef] [PubMed]
- Wessig, P.; Müller, G.; Pick, C.; Matthes, A. The Photo-Dehydro-Diels–Alder Reaction for the Preparation of Biaryls. Synthesis 2007, 464–477. [Google Scholar] [CrossRef]
- Wessig, P.; Matthes, A.; Pick, C. The Photo-dehydro-Diels-Alder (PDDA) reaction. Org. Biomol. Chem. 2011, 9, 7599–7605. [Google Scholar] [CrossRef] [PubMed]
- Wessig, P.; Müller, G. The Dehydro-Diels-Alder Reaction. Chem. Rev. 2008, 108, 2051–2063. [Google Scholar] [CrossRef] [PubMed]
- Wessig, P.; Müller, G.; Kühn, A.; Herre, R.; Blumenthal, H.; Troelenberg, S. The Photo-Dehydro-Diels-Alder Reaction: An efficient route to naphthalenes. Synthesis 2005, 1445–1454. [Google Scholar] [CrossRef]
- Wessig, P.; Müller, G. Synthesis of 1,1'-Binaphthyls by Photo-Dehydro-Diels-Alder reaction. Chem. Commun. 2006, 4524–4526. [Google Scholar] [CrossRef]
- Wessig, P.; Müller, G.; Herre, R.; Kühn, A. Synthesis of Benzo[g]isochromenes through Photo-Dehydro-Diels-Alder Reaction. Helv. Chim. Acta 2006, 89, 2694–2719. [Google Scholar] [CrossRef]
- Wessig, P.; Müller, G. Facile Photochemical Synthesis of 1,1'-Binaphthyls. Aust. J. Chem. 2008, 61, 569–572. [Google Scholar] [CrossRef]
- Wessig, P.; Pick, C. Photochemical synthesis and properties of axially chiral naphthylpyridines. J. Photochem. Photobiol. A 2011, 222, 263–265. [Google Scholar] [CrossRef]
- Kawasaki, N.; Goto, M.; Kawabata, S.; Kometani, T. The effect of vinyl esters on the enantioselectivity of the lipase-catalysed transesterification of alcohols. Tetrahedron: Asymmetry 2001, 12, 585–596. [Google Scholar] [CrossRef]
- Neises, B.; Steglich, W. Simple Method for the Esterification of Carboxylic Acids. Angew. Chem. Int. Ed. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975, 50, 4467–4470. [Google Scholar] [CrossRef]
- Dess, D.; Martin, B. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J. Org. Chem. 1983, 48, 4155–4156. [Google Scholar] [CrossRef]
- Dess, D.; Martin, B. A useful 12-I-5 triacetoxyperiodinane (the Dess-Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species. J. Am. Chem. Soc. 1991, 113, 7277–7287. [Google Scholar] [CrossRef]
- Cope, A.C.; Martin, M.M.; McKervey, M.A. Transannular reactions in medium-sized rings. Q. Rev. Chem. Soc. 1966, 20, 119–152. [Google Scholar] [CrossRef]
- Bringmann, G.; Gulder, T.; Gulder, T.A.M.; Breuning, M. Atroposelective total synthesis of axially chiral biaryl natural products. Chem. Rev. 2011, 111, 563–639. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, G.; Mortimer, A.J.P.; Keller, P.A.; Gresser, M.J.; Garner, J.; Breuning, M. Atroposelective synthesis of axially chiral biaryl compounds. Angew. Chem. Int. Edit. 2005, 44, 5384–5427. [Google Scholar] [CrossRef] [PubMed]
- Sandström, J. Dynamic NMR Spectroscopy; Academic Press: New York, NY, USA, 1982. [Google Scholar]
- Trapp, O. Fast and precise access to enantiomerization rate constants in dynamic chromatography Chirality. Chirality 2006, 18, 489–497. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | n | HO-R-OH | Yield 3 [%] | Yield 5 [%] | Yield 6 [%] |
|---|---|---|---|---|---|
| 6a | 0 | HO-(CH2)2-OH | 97 | 97 | 90 |
| 6b | 0 | HO-(CH2)4-OH | 69 | 95 | 86 |
| 6c | 0 | HO-(CH2)6-OH | 93 | 92 | 85 |
| 6d | 0 | HO-(CH2CH2O)3-H | 99 | 93 | 62 |
| 6e | 2 | HO-(CH2)5-OH | 100 | 100 | 100 |
| 6f | 2 | HO-(CH2CH2O)3-H | 90 | 93 | 67 |
| Reactant | n | k (a) | Yield 7 [%] | Yield 8 [%] | Yield 7 + 8 [%] |
|---|---|---|---|---|---|
| 6a | 0 | 9 | 0 | 0 | 0 |
| 6b | 0 | 11 | 3 | 12 | 15 |
| 6c | 0 | 13 | 15 | 26 | 41 |
| 6d | 0 | 15 | 33 | 33 | 66 |
| 6e | 2 | 16 | (b) | (b) | 21 |
| 6f | 2 | 19 | (b) | (b) | 61 |
| Compound | DHPLC ΔGROT [kcal/mol] | DNMR ΔGROT [kcal/mol] |
|---|---|---|
| 7b | - | - |
| 7c | - | 20.6 |
| 7d | - | 19.6 |
| 7e | 20.4 | - |
| 8b | - | - |
| 8c | <20.0 | ≈20.0 |
| 8d | 21.5 | - |
| 8e | 22.5 | - |
© 2013 by the authors. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wessig, P.; Matthes, A. Photochemical Synthesis and Properties of 1,6- and 1,8-Naphthalenophanes. Molecules 2013, 18, 1314-1324. https://doi.org/10.3390/molecules18011314
Wessig P, Matthes A. Photochemical Synthesis and Properties of 1,6- and 1,8-Naphthalenophanes. Molecules. 2013; 18(1):1314-1324. https://doi.org/10.3390/molecules18011314
Chicago/Turabian StyleWessig, Pablo, and Annika Matthes. 2013. "Photochemical Synthesis and Properties of 1,6- and 1,8-Naphthalenophanes" Molecules 18, no. 1: 1314-1324. https://doi.org/10.3390/molecules18011314
APA StyleWessig, P., & Matthes, A. (2013). Photochemical Synthesis and Properties of 1,6- and 1,8-Naphthalenophanes. Molecules, 18(1), 1314-1324. https://doi.org/10.3390/molecules18011314