Sulfur-Functionalized N-Heterocyclic Carbene Complexes of Pd(II): Syntheses, Structures and Catalytic Activities

Abstract
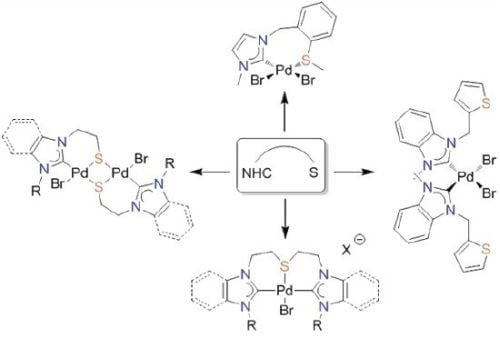
:
1. Introduction
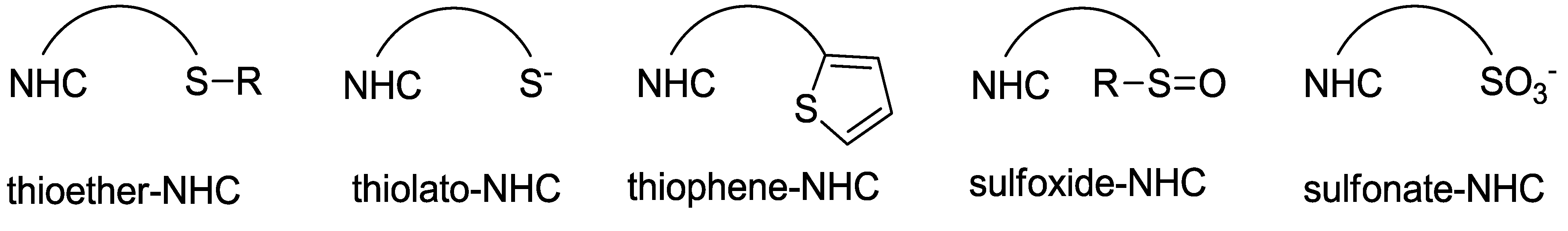
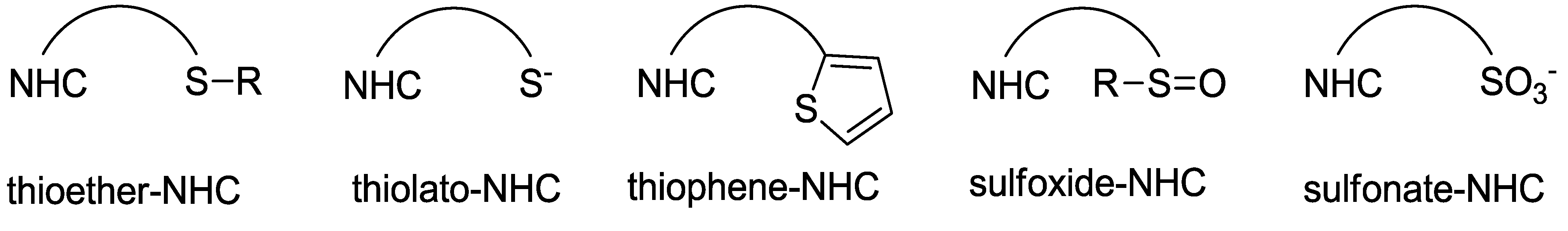
2. Sulfur-Functionalized Azolium Salts
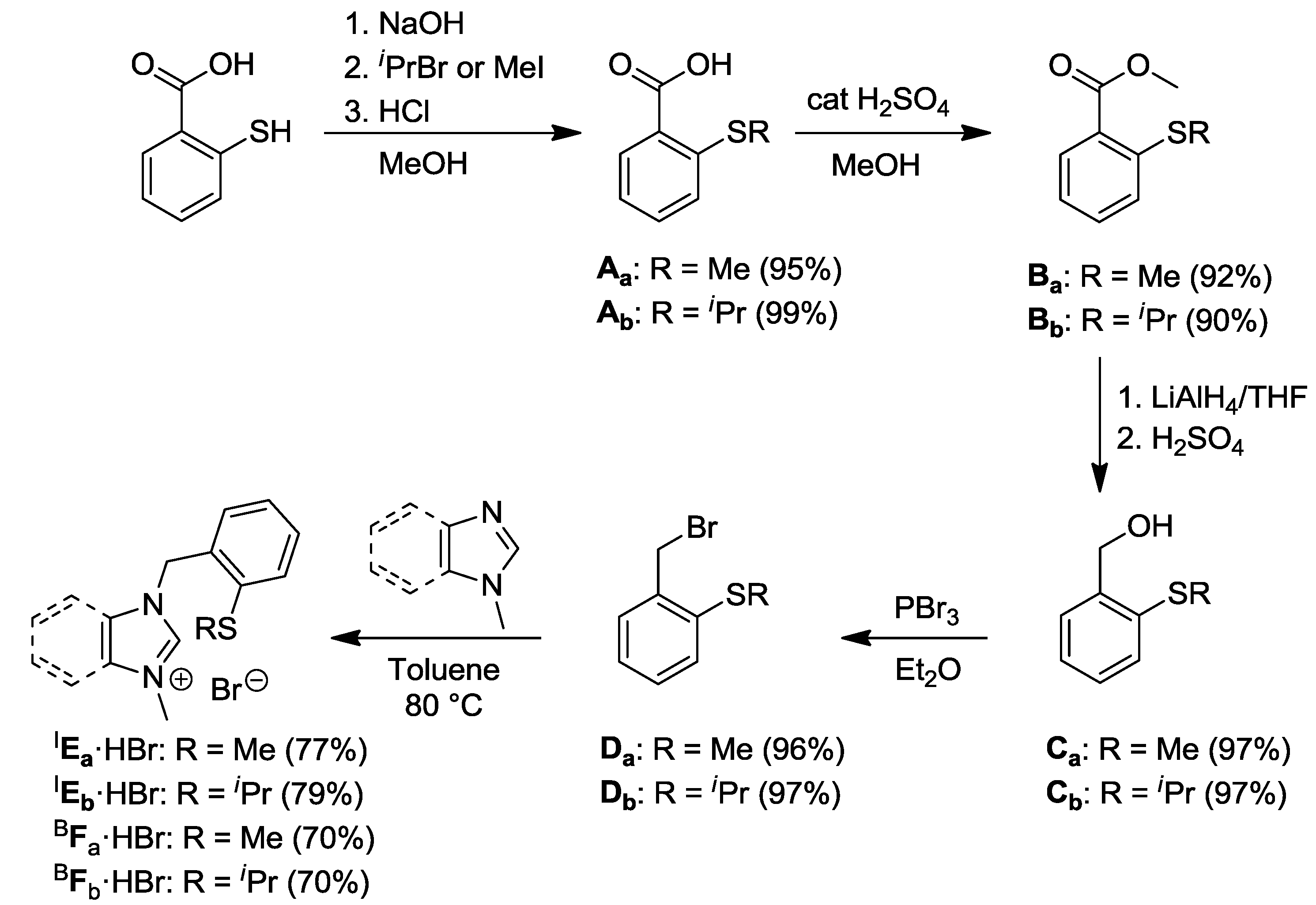
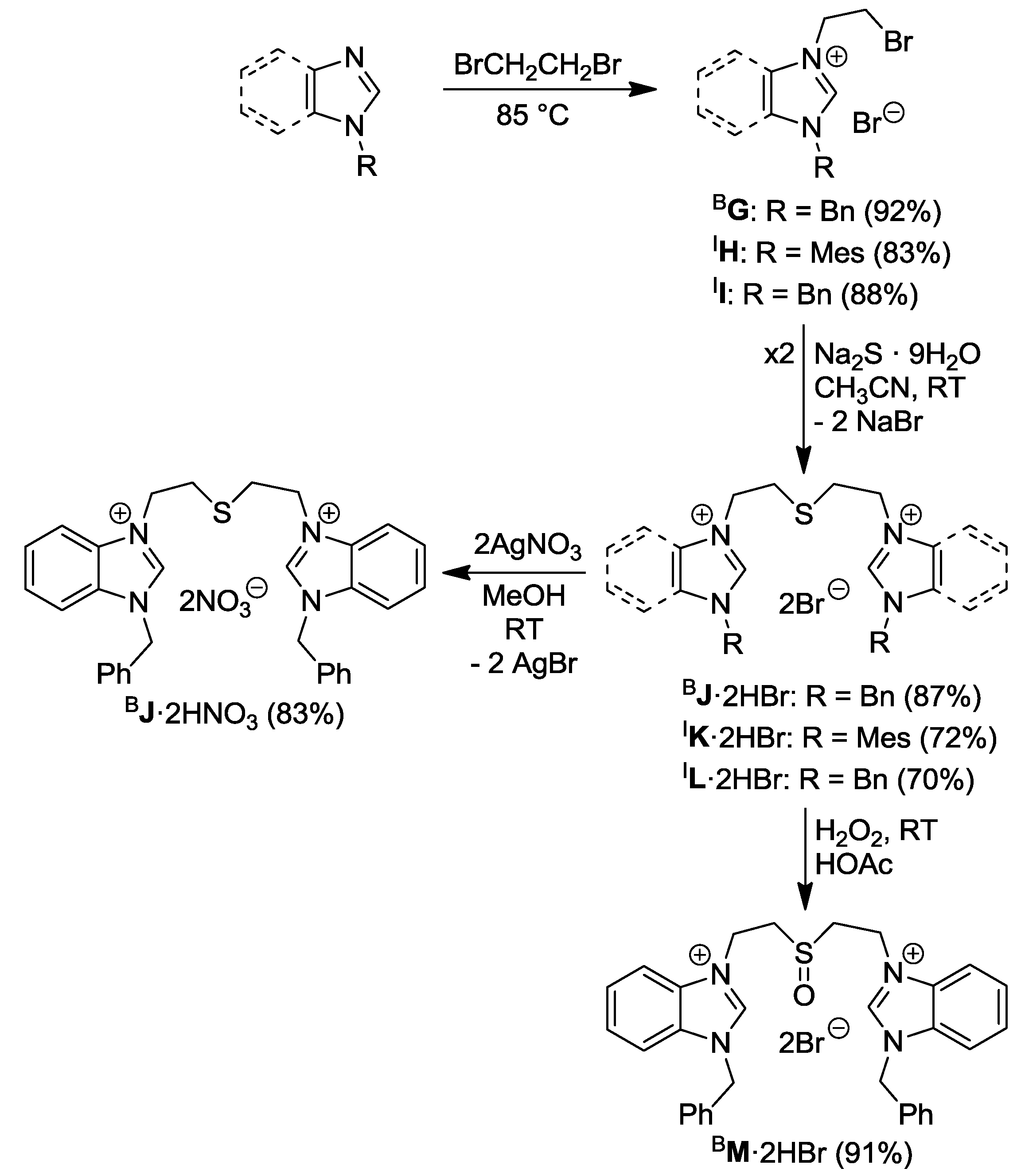
2.1. Thioether-Functionalized Azolium Salts

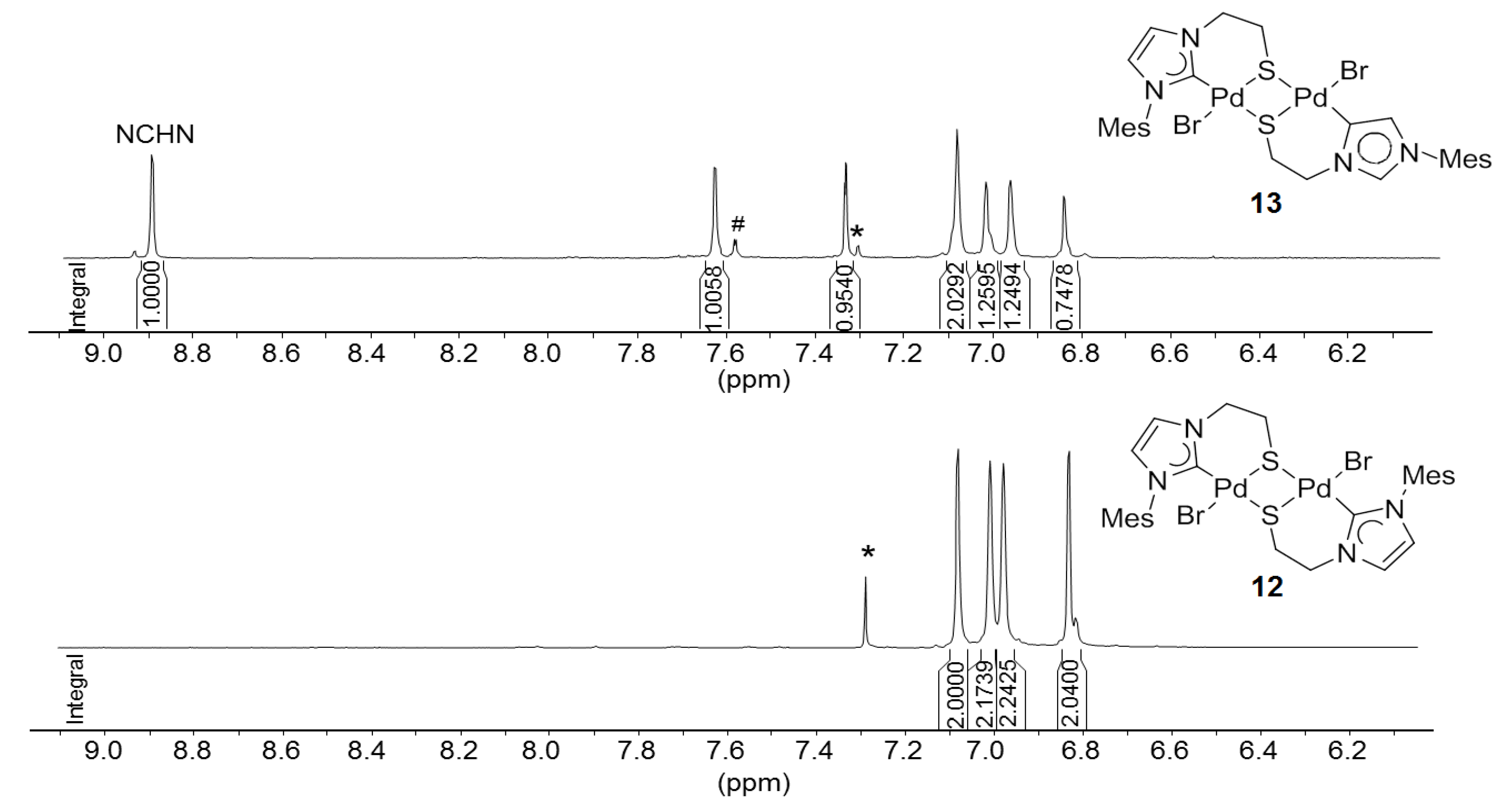
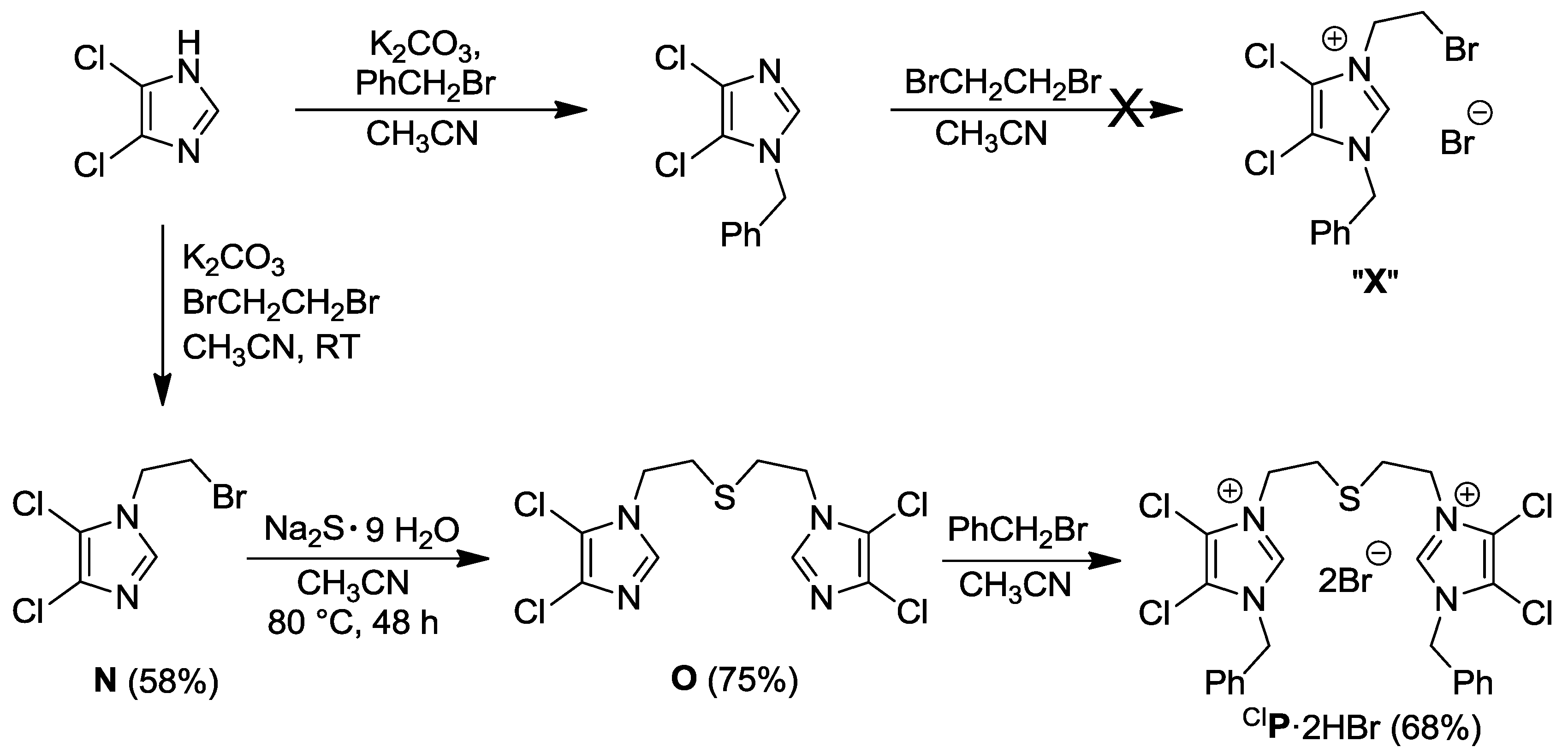
2.2. Thioether-Bridged Diazolium Salts
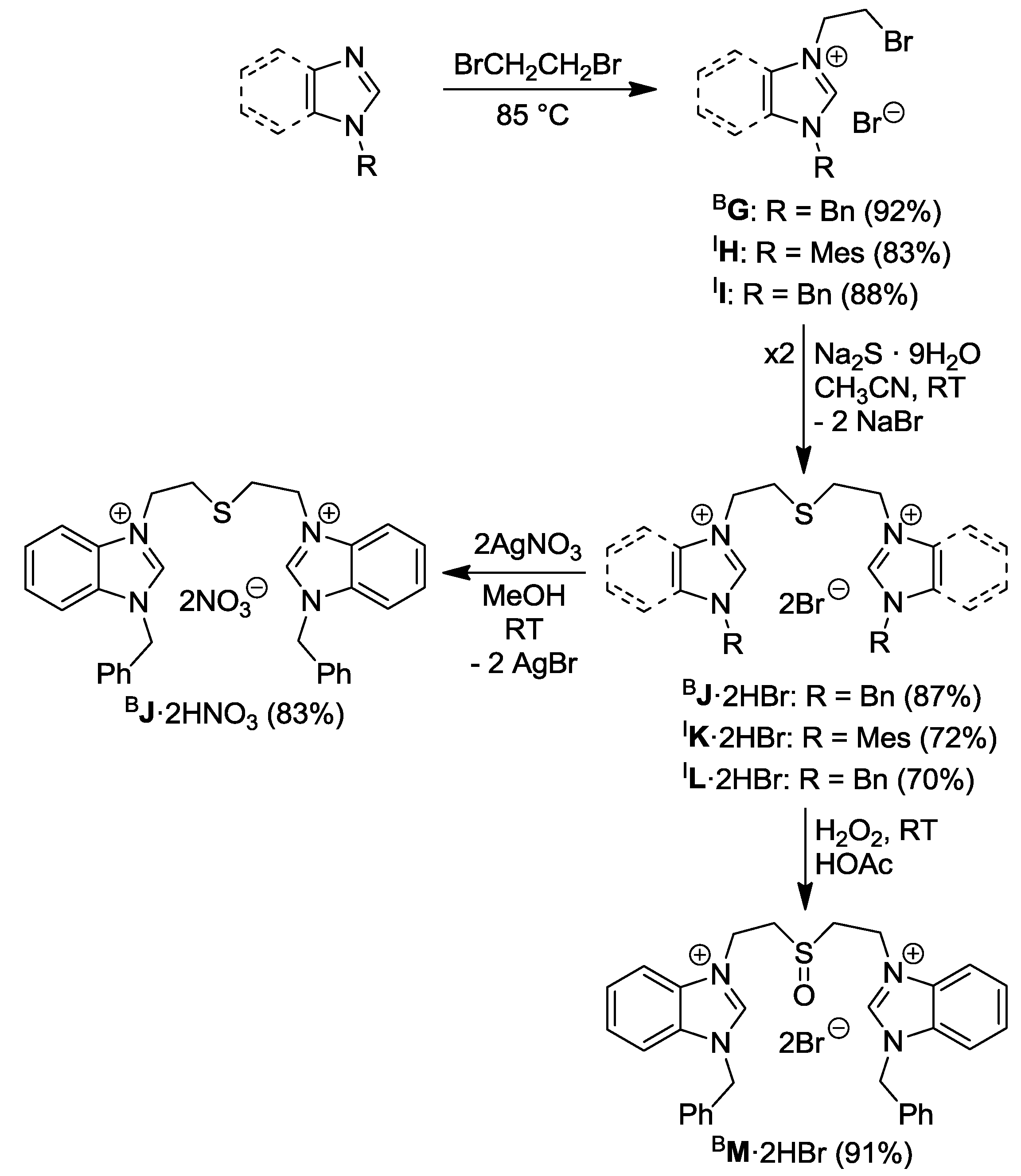
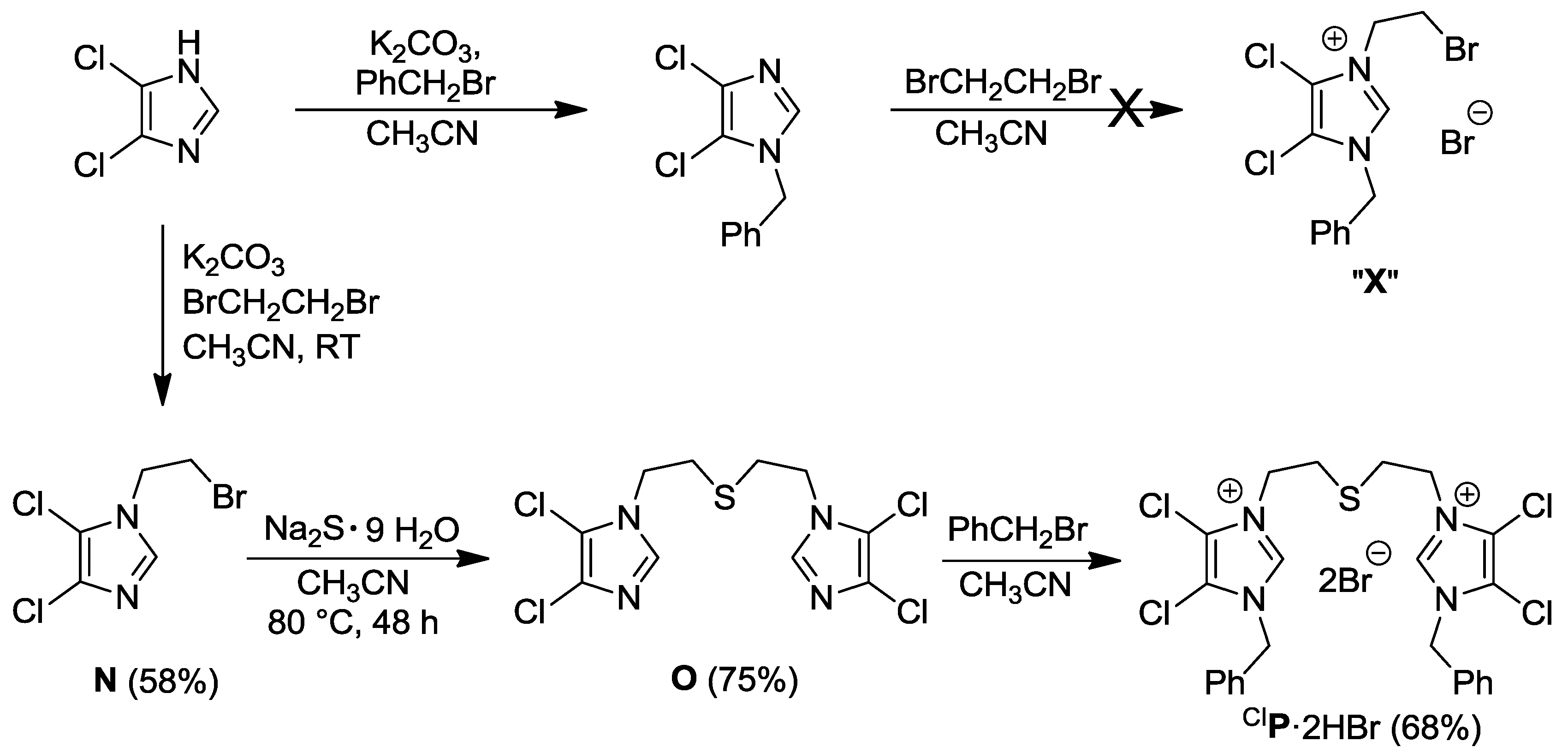
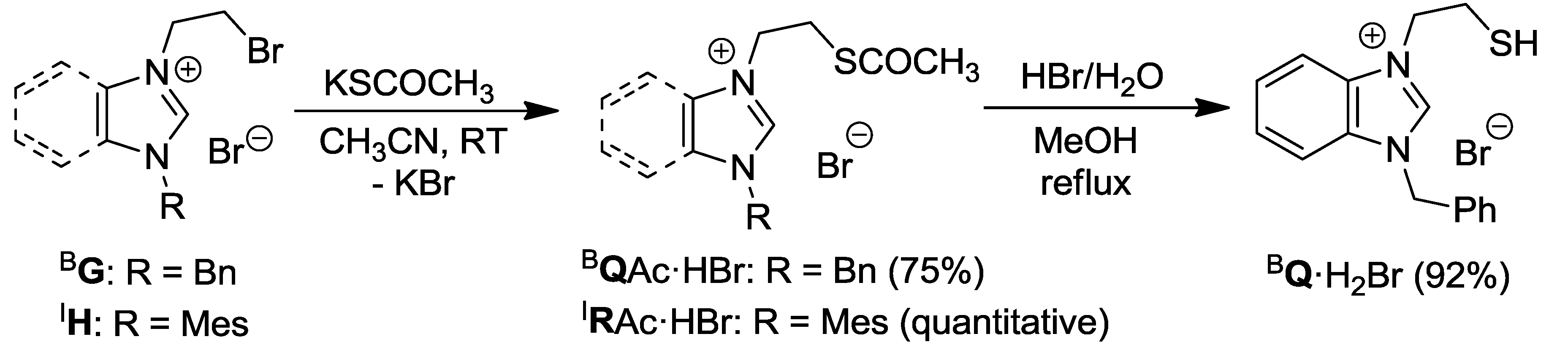
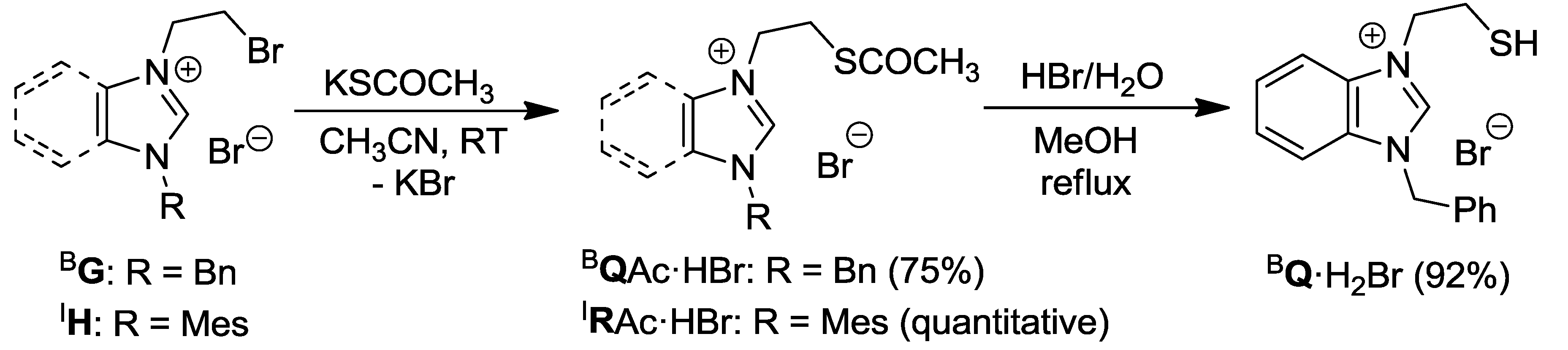
2.3. Thioester- and Thiol-Functionalized Azolium Salts
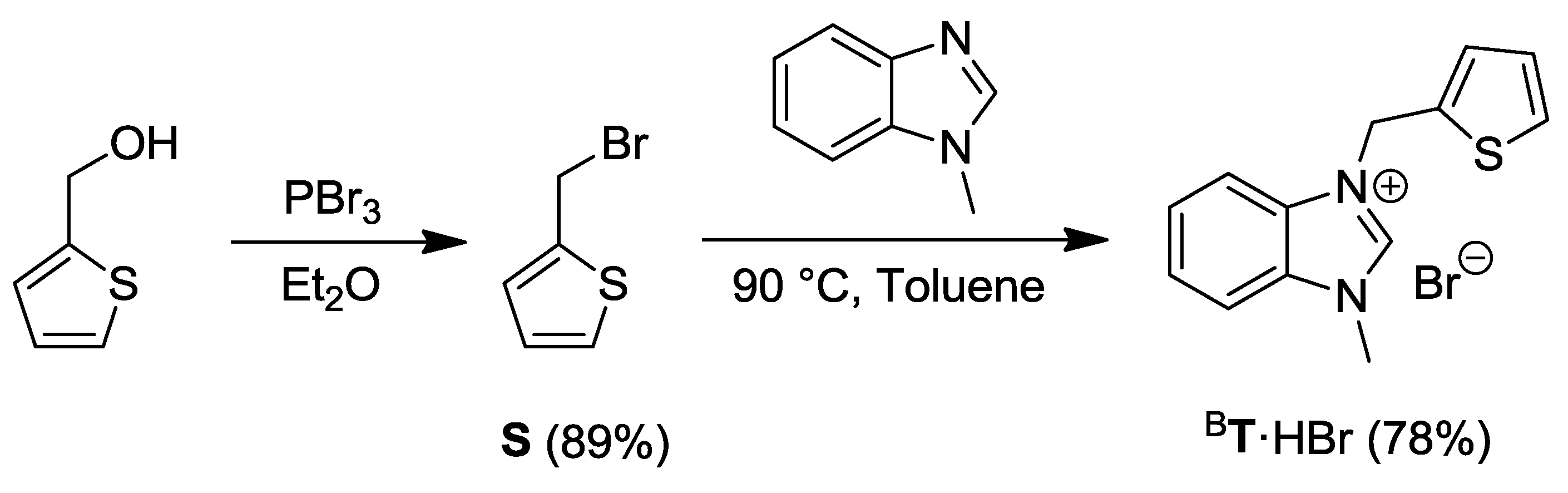
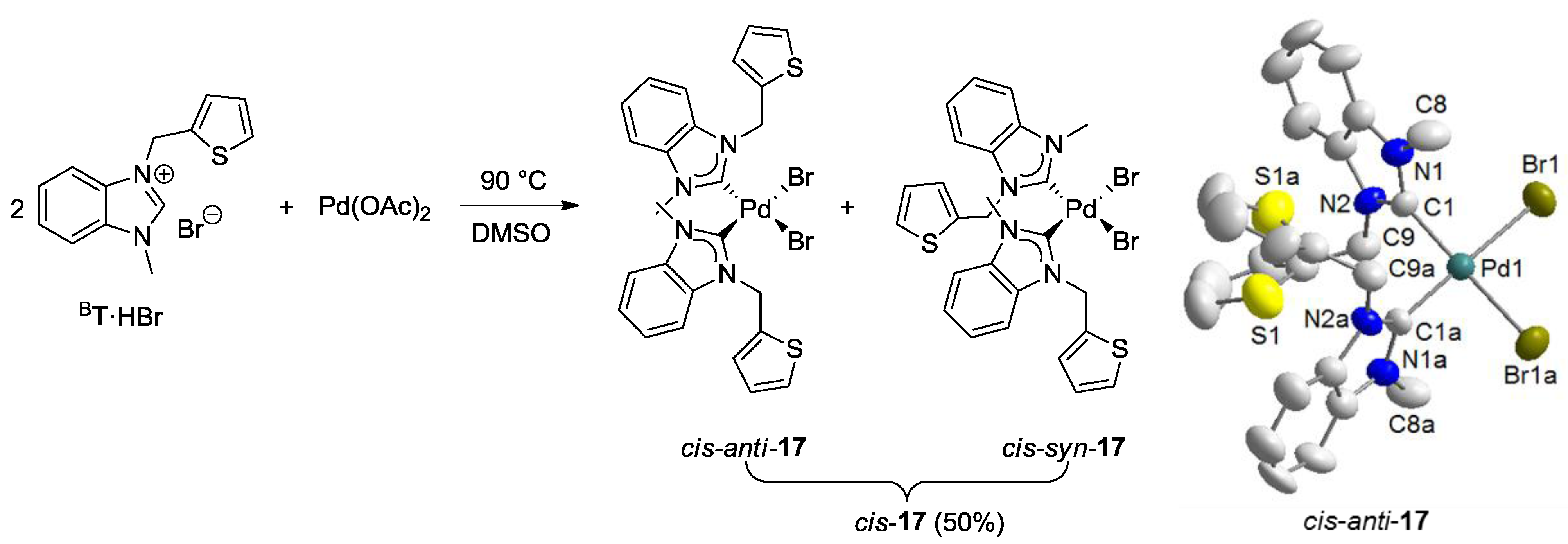
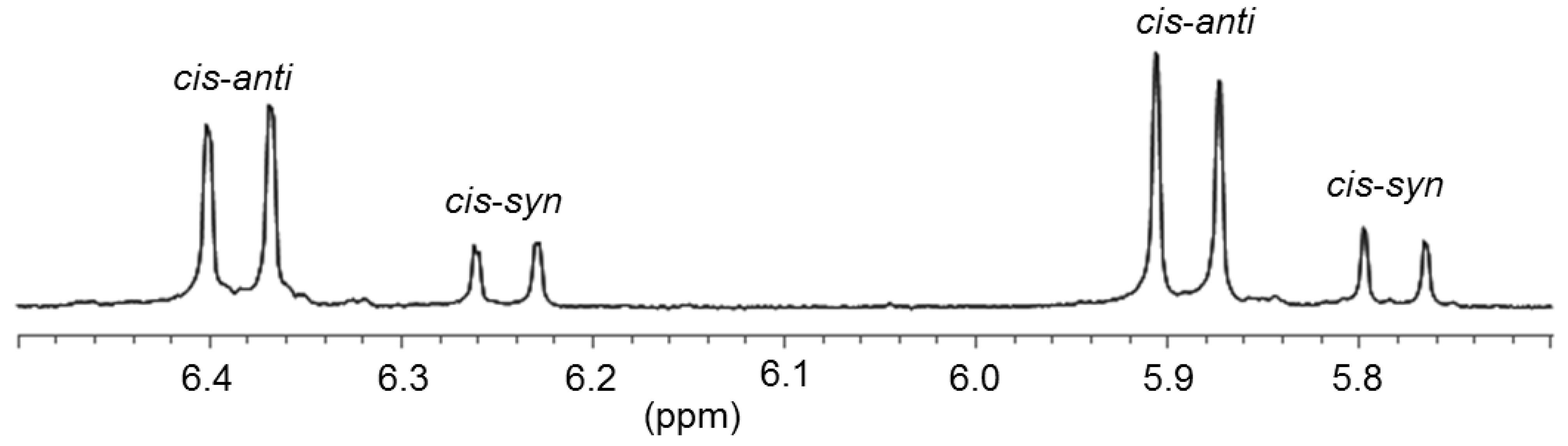
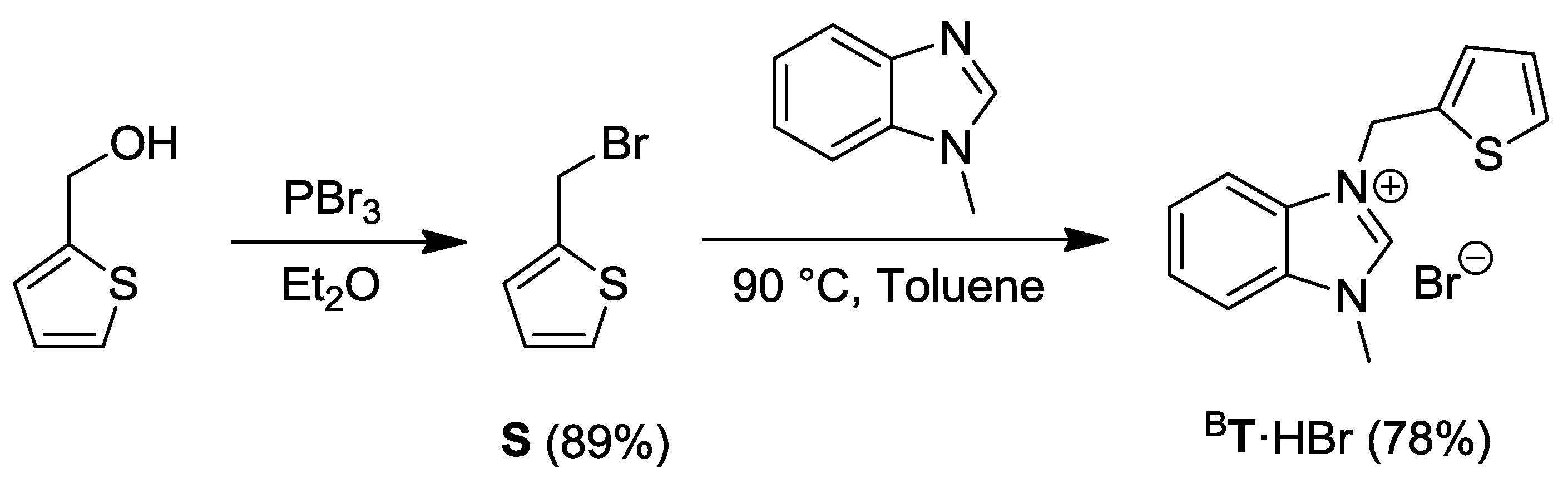
2.4. Thiophene-Functionalized Azolium Salt
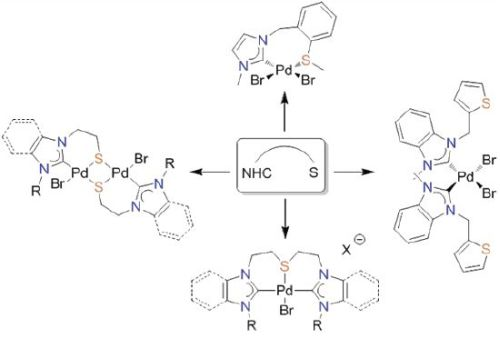
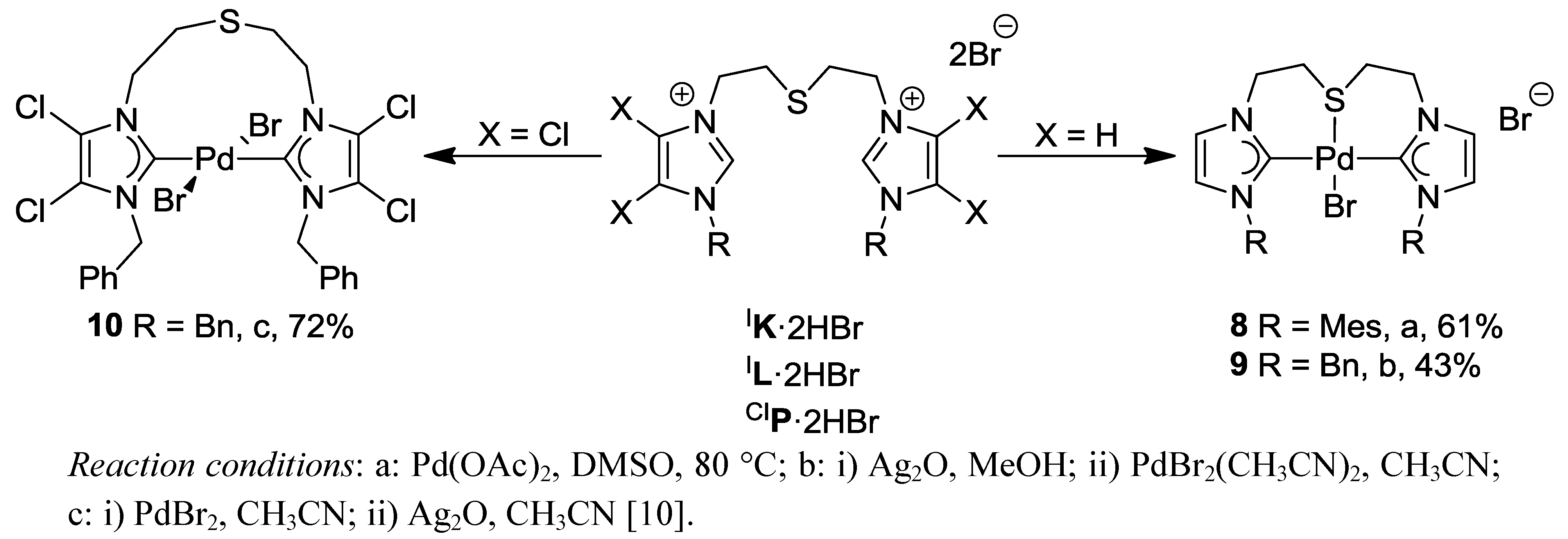
3. Thioether-NHC Complexes
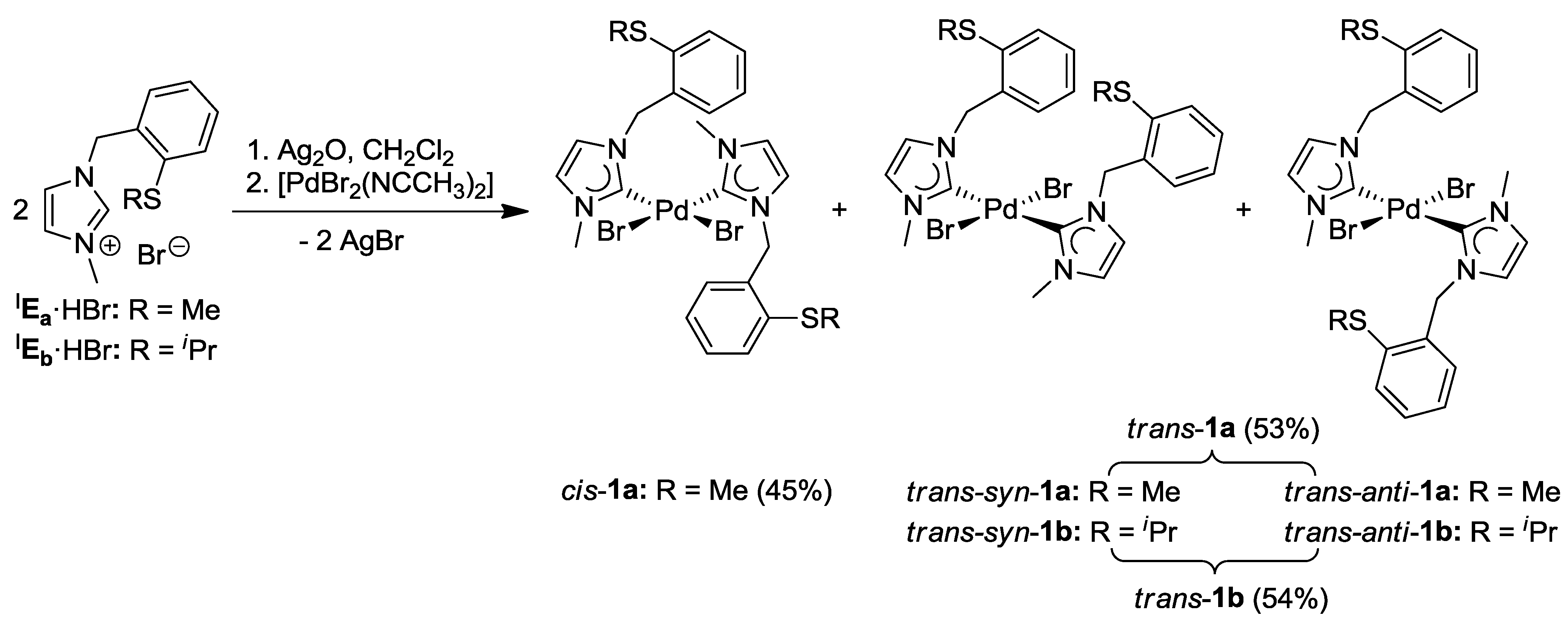
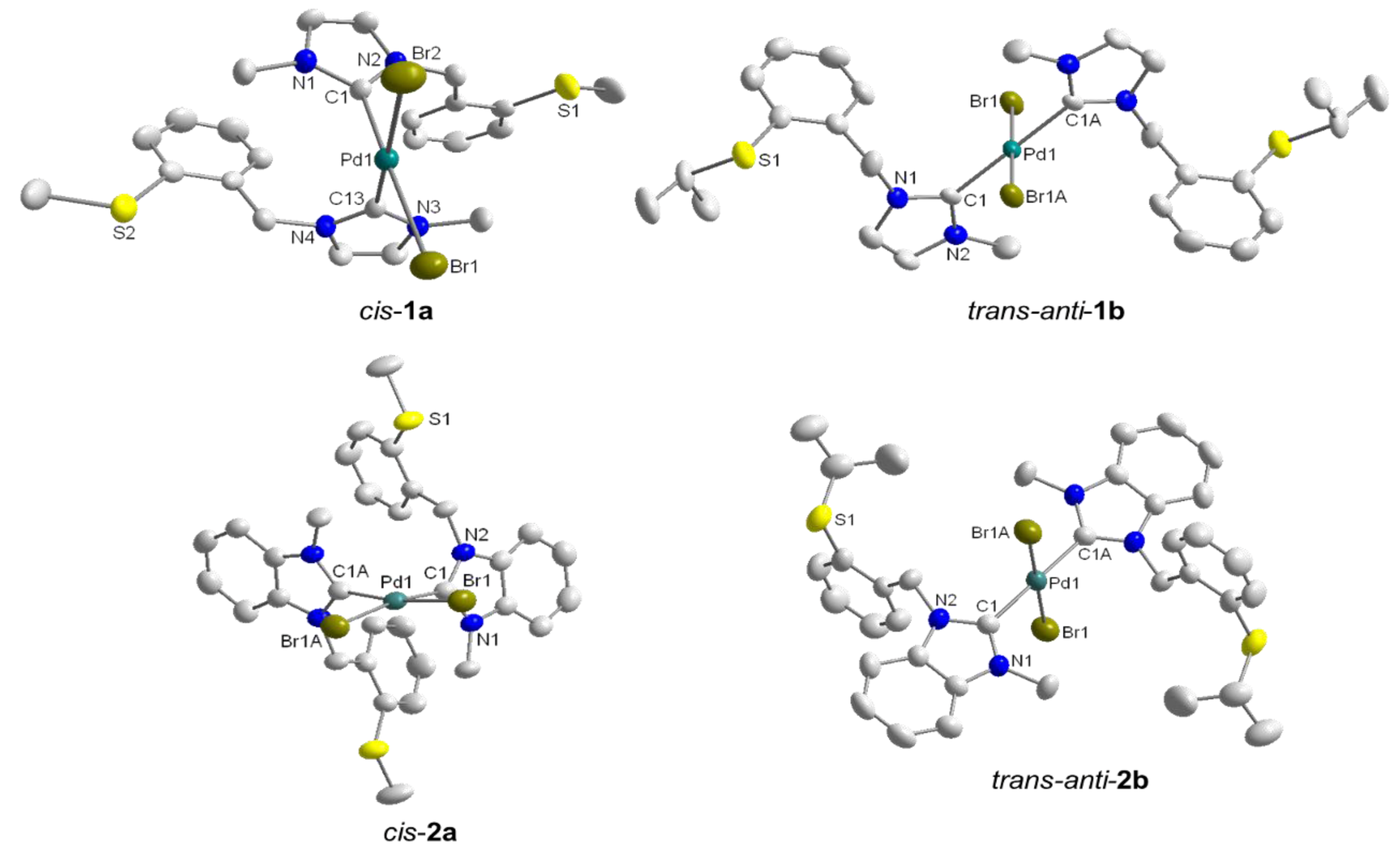
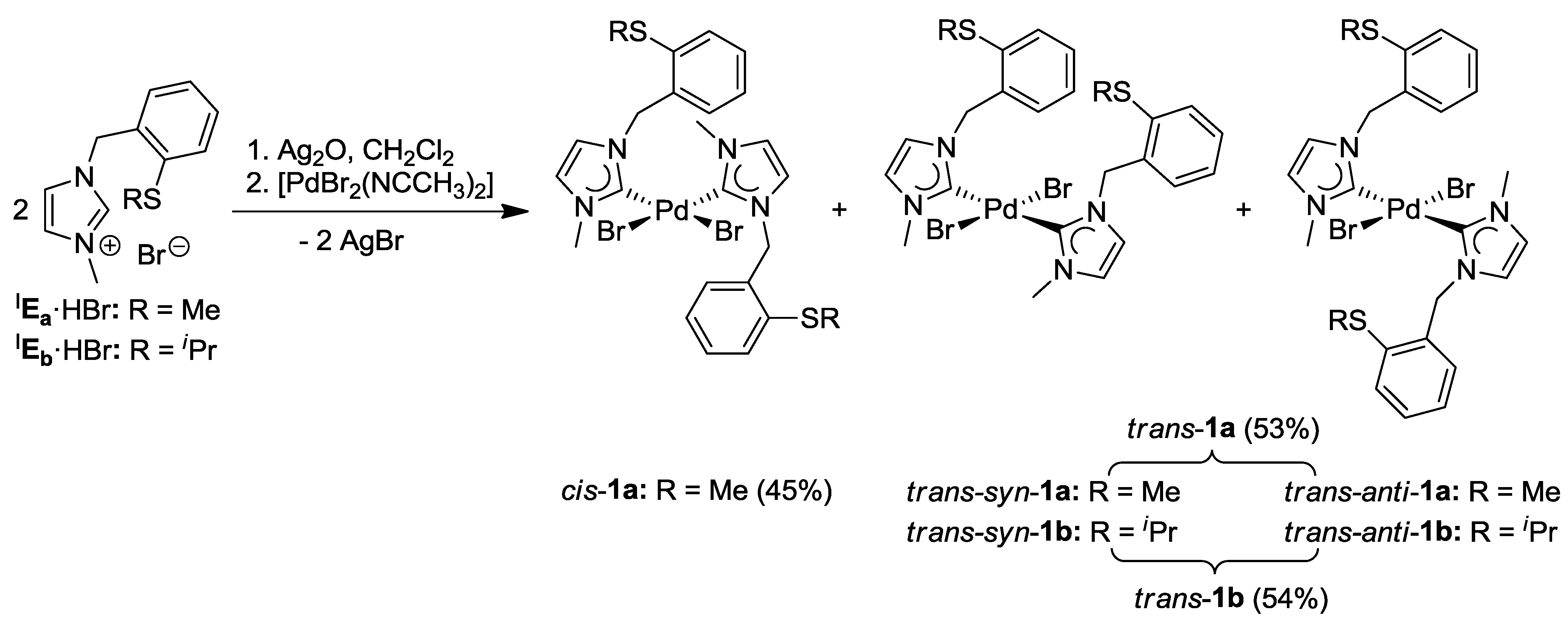
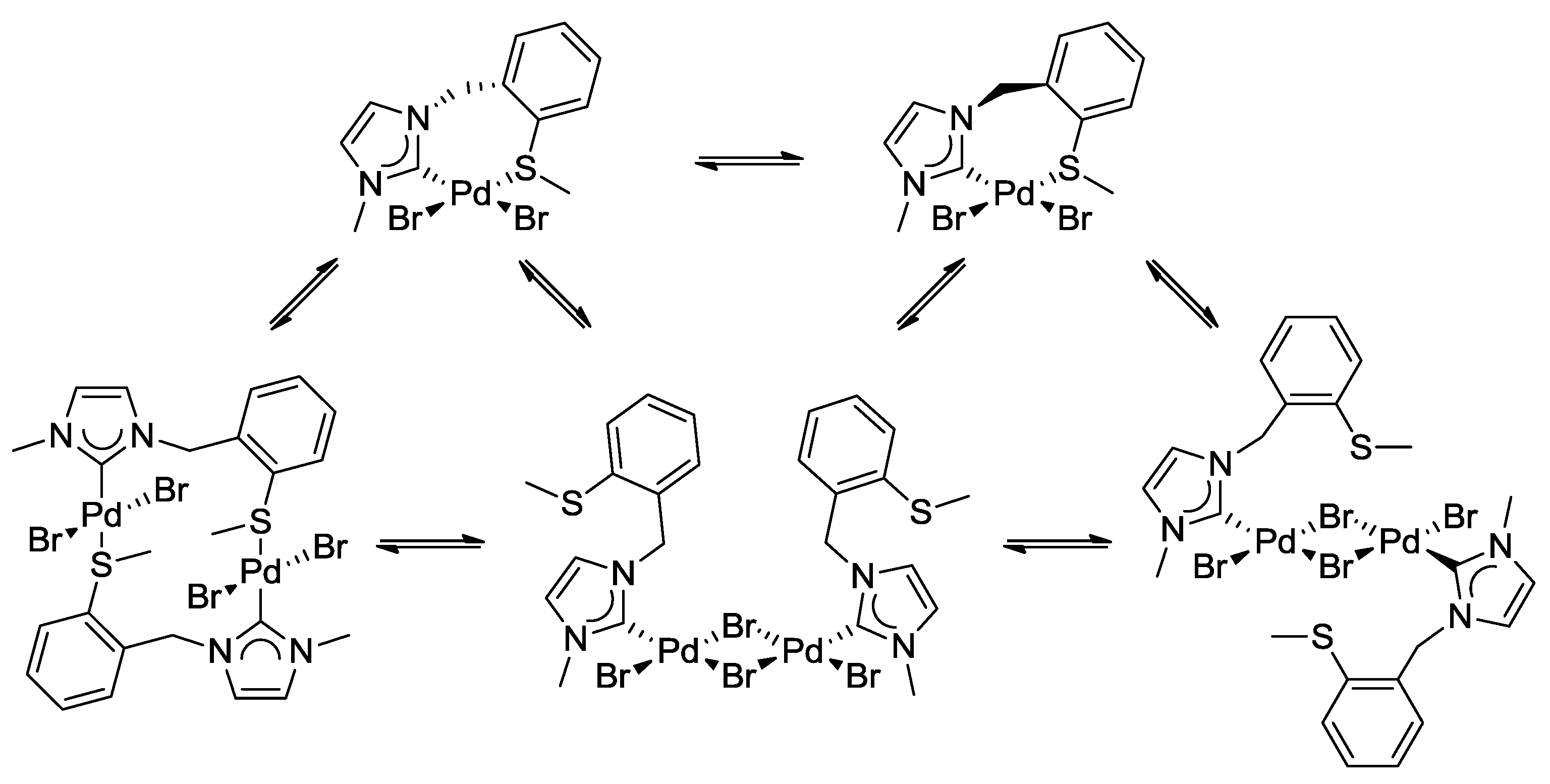
3.1. Thioether-Functionalized Pd(II) Bis(carbene) Complexes
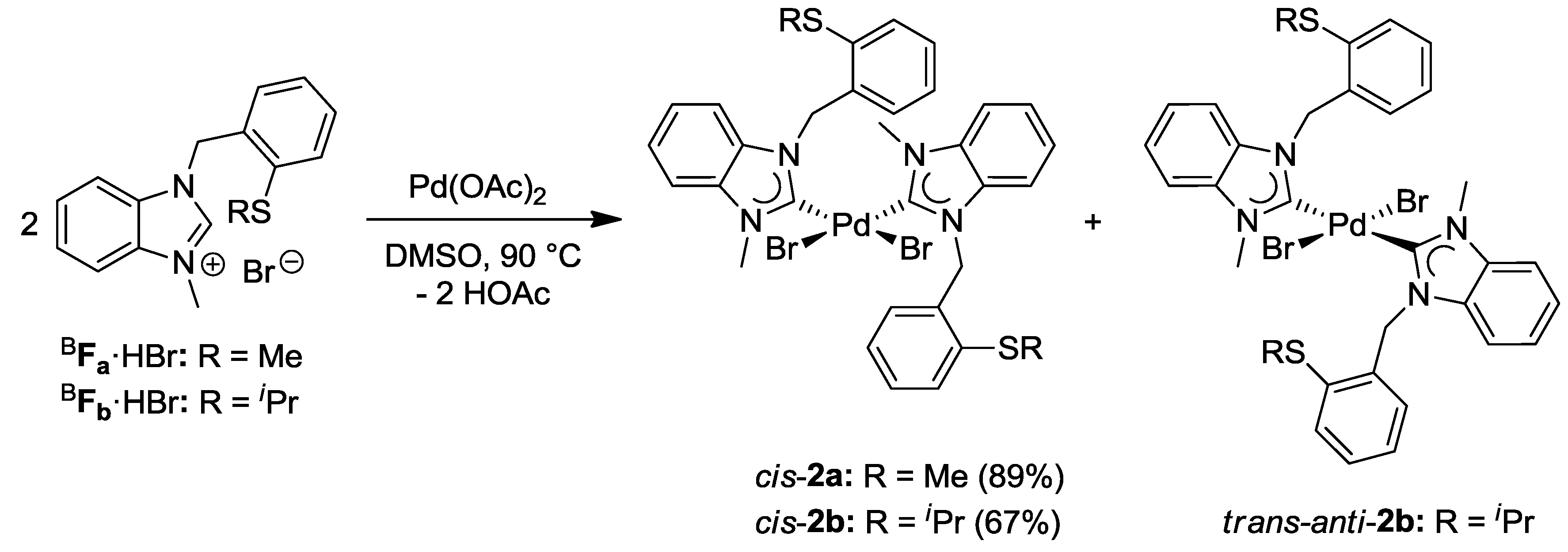
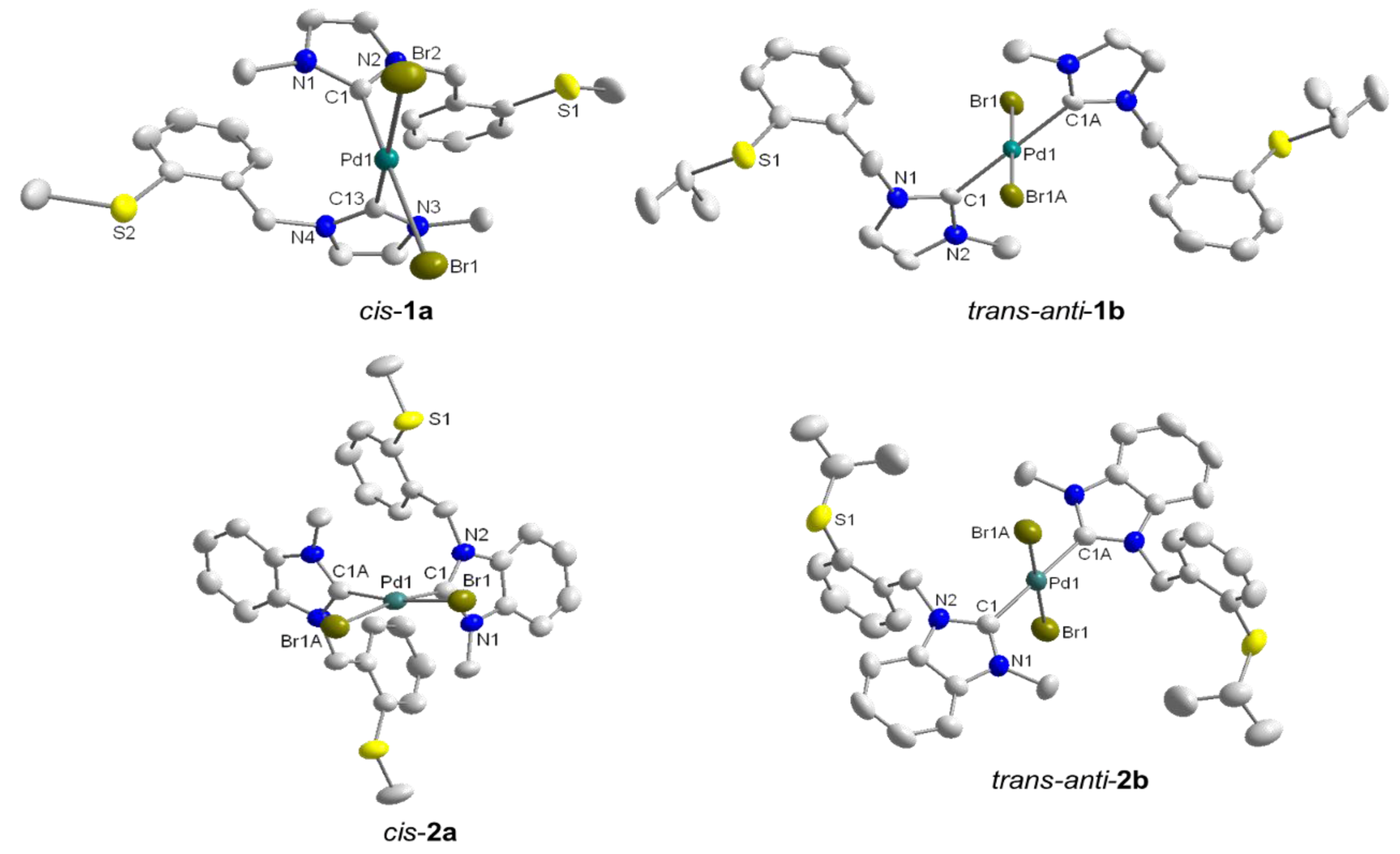
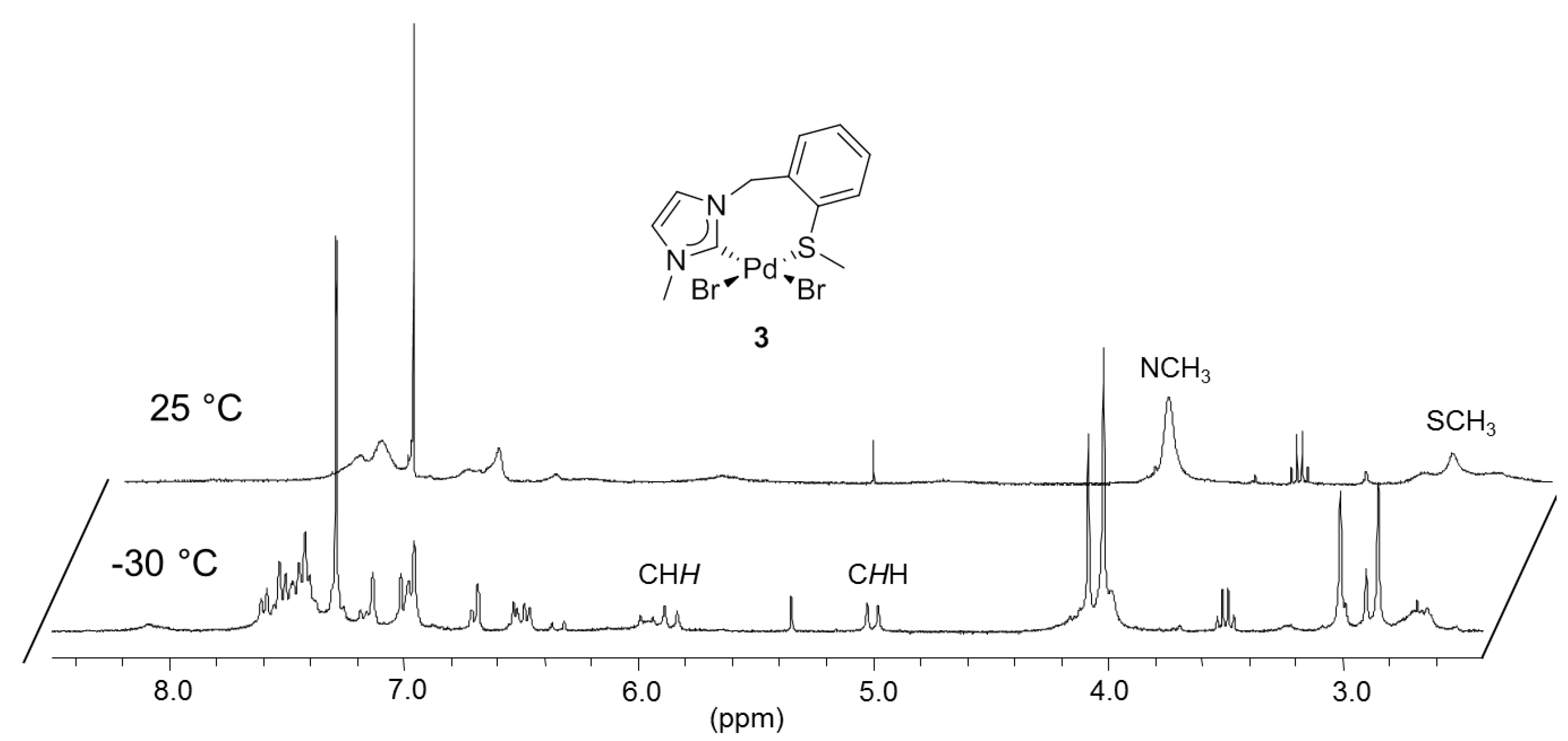
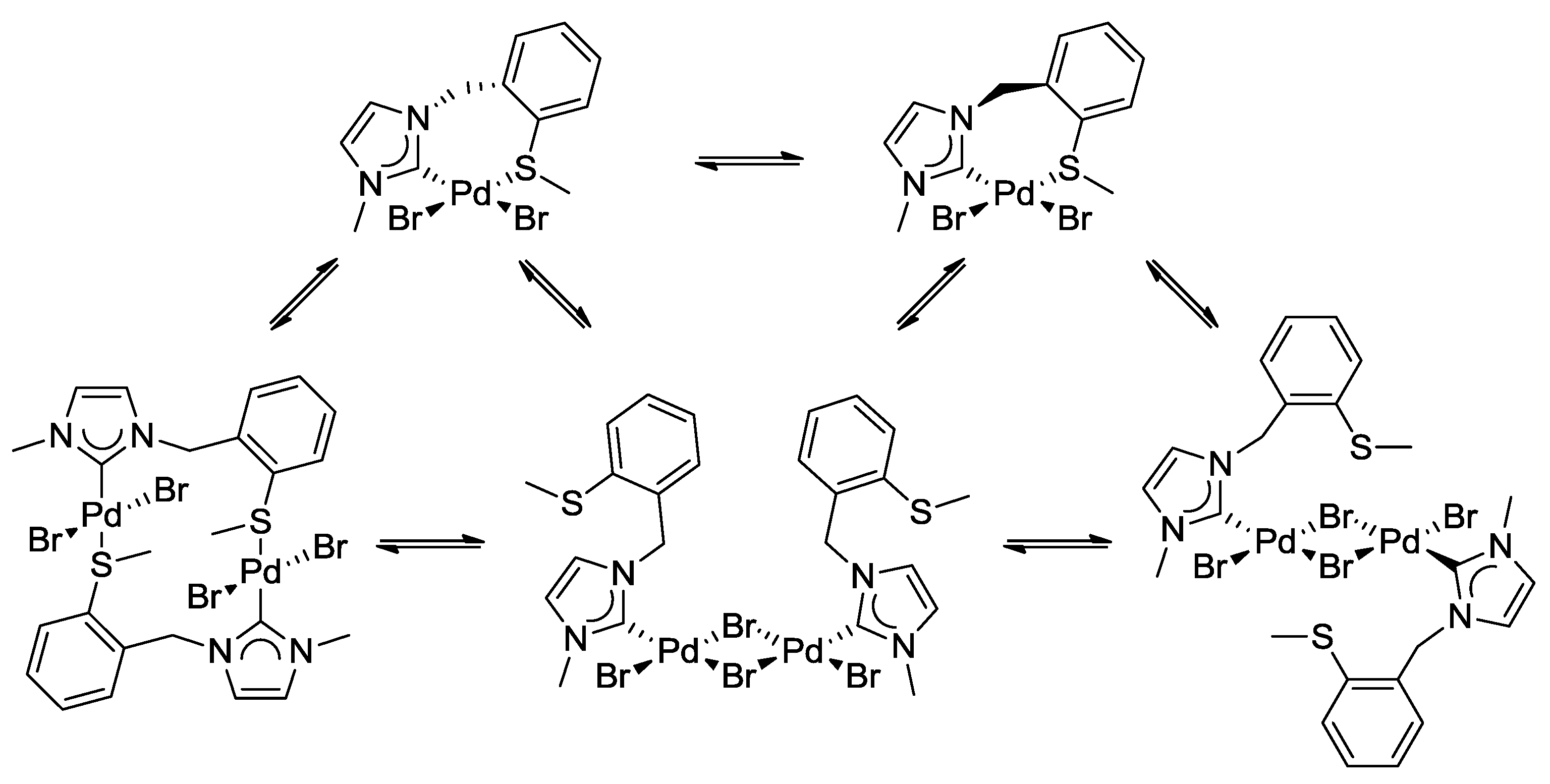
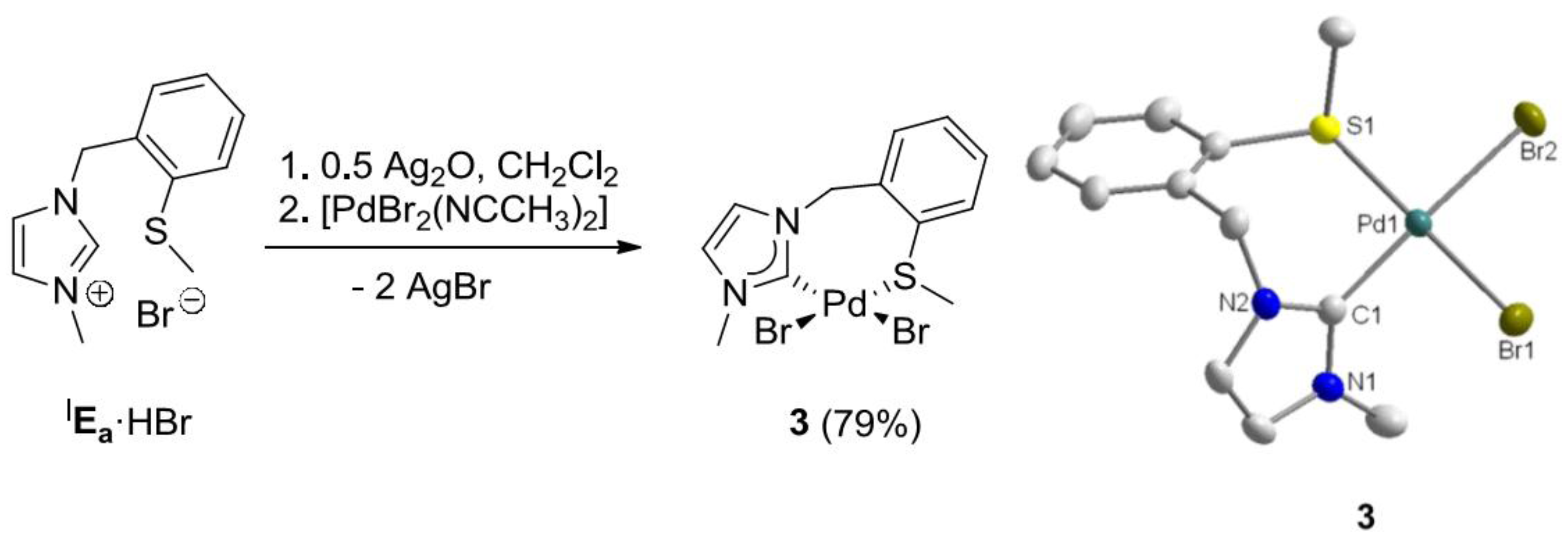
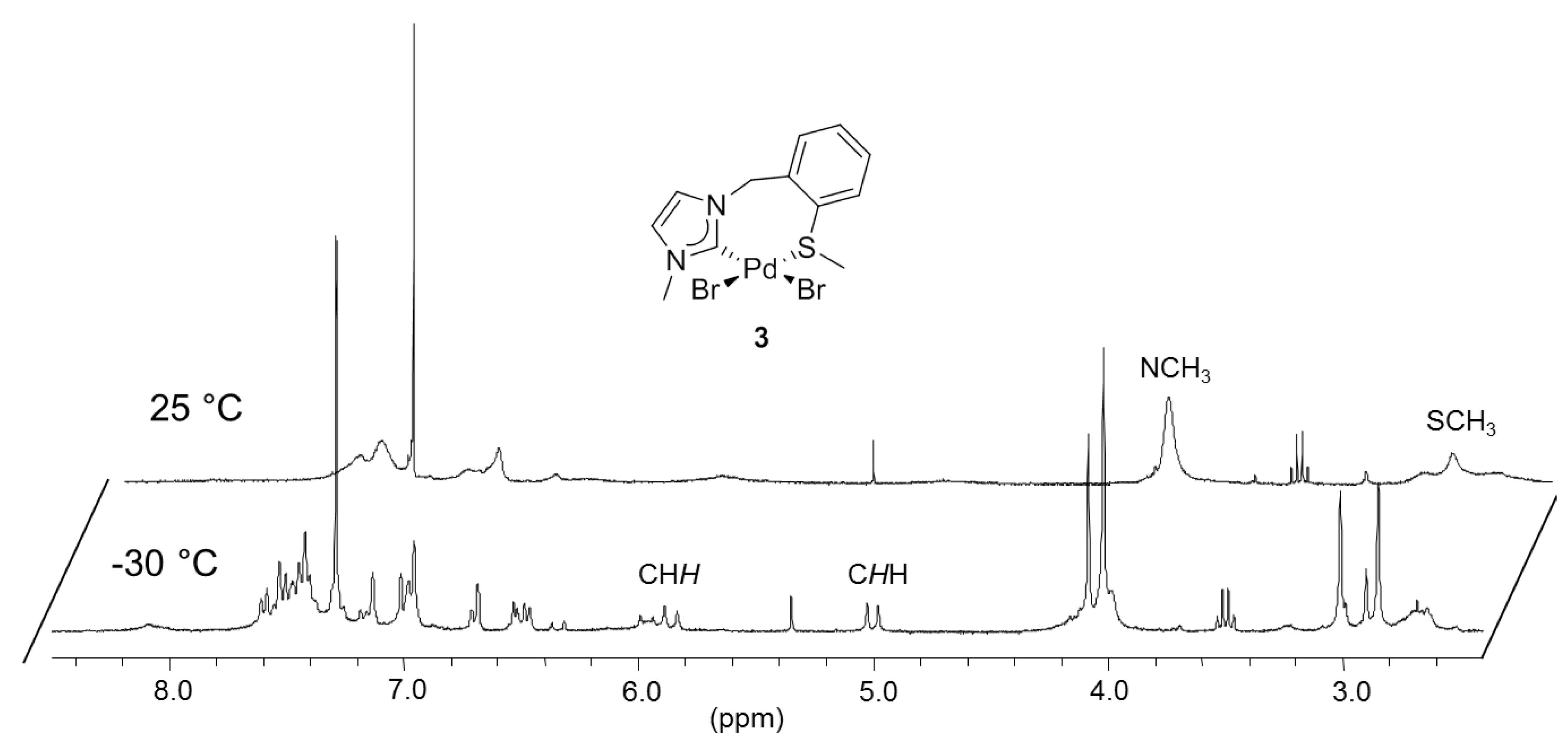
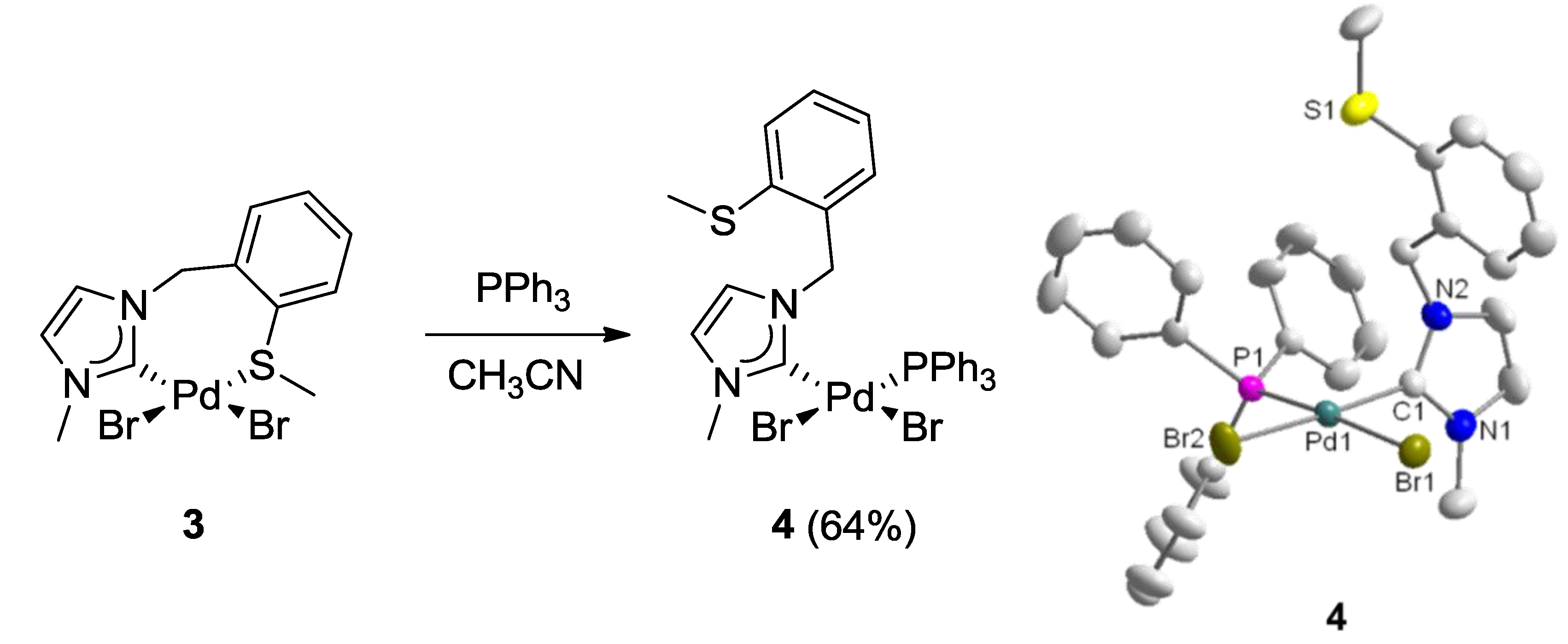
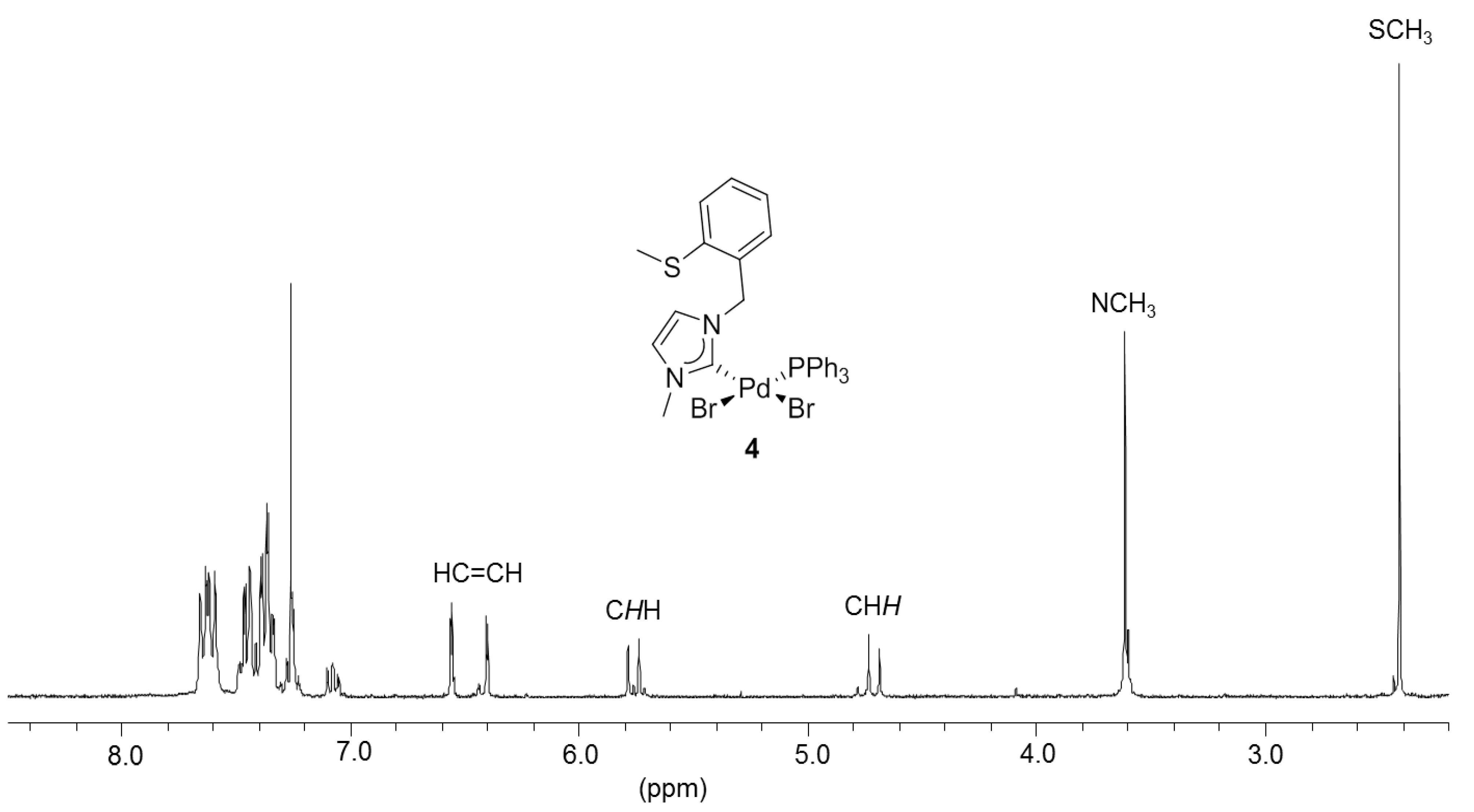
3.2. Hemilabile Thioether-Functionalized Pd(II) Monocarbene Complexes

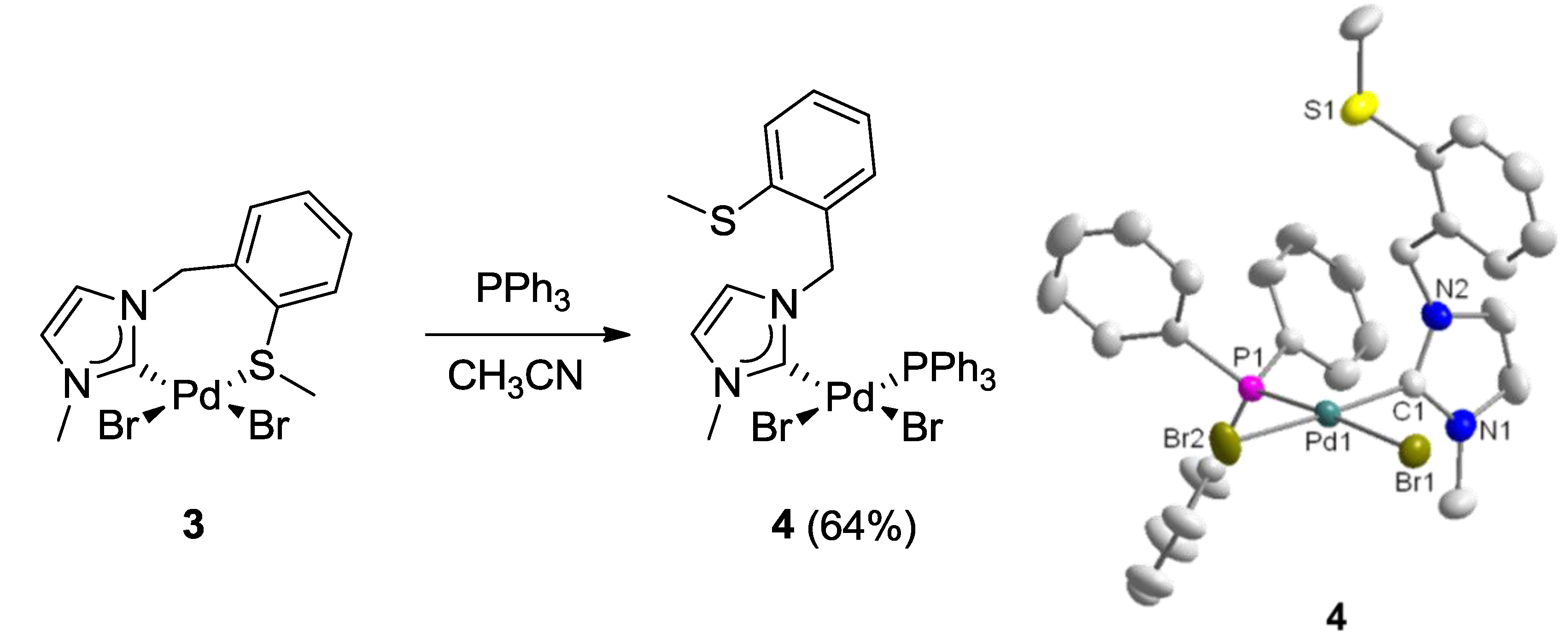

4. CSC-Pincer Type Pd(II) Complexes
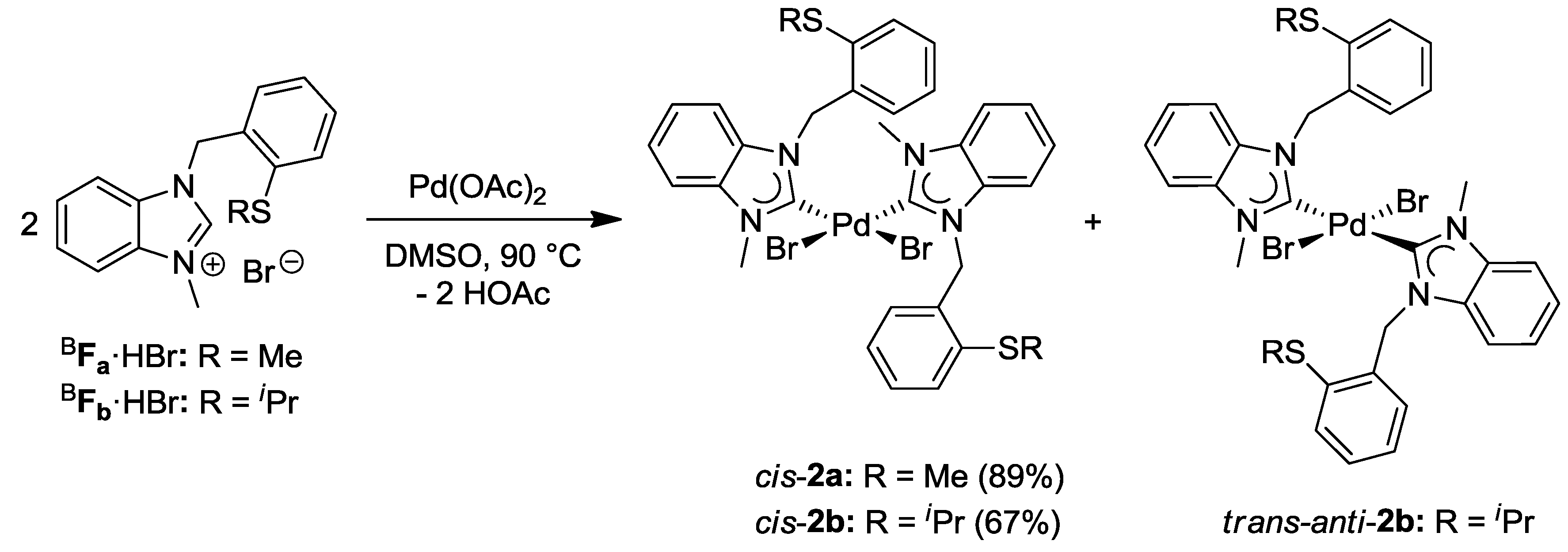
4.1. Pd(II) Benzimidazolin-2-ylidene Complexes
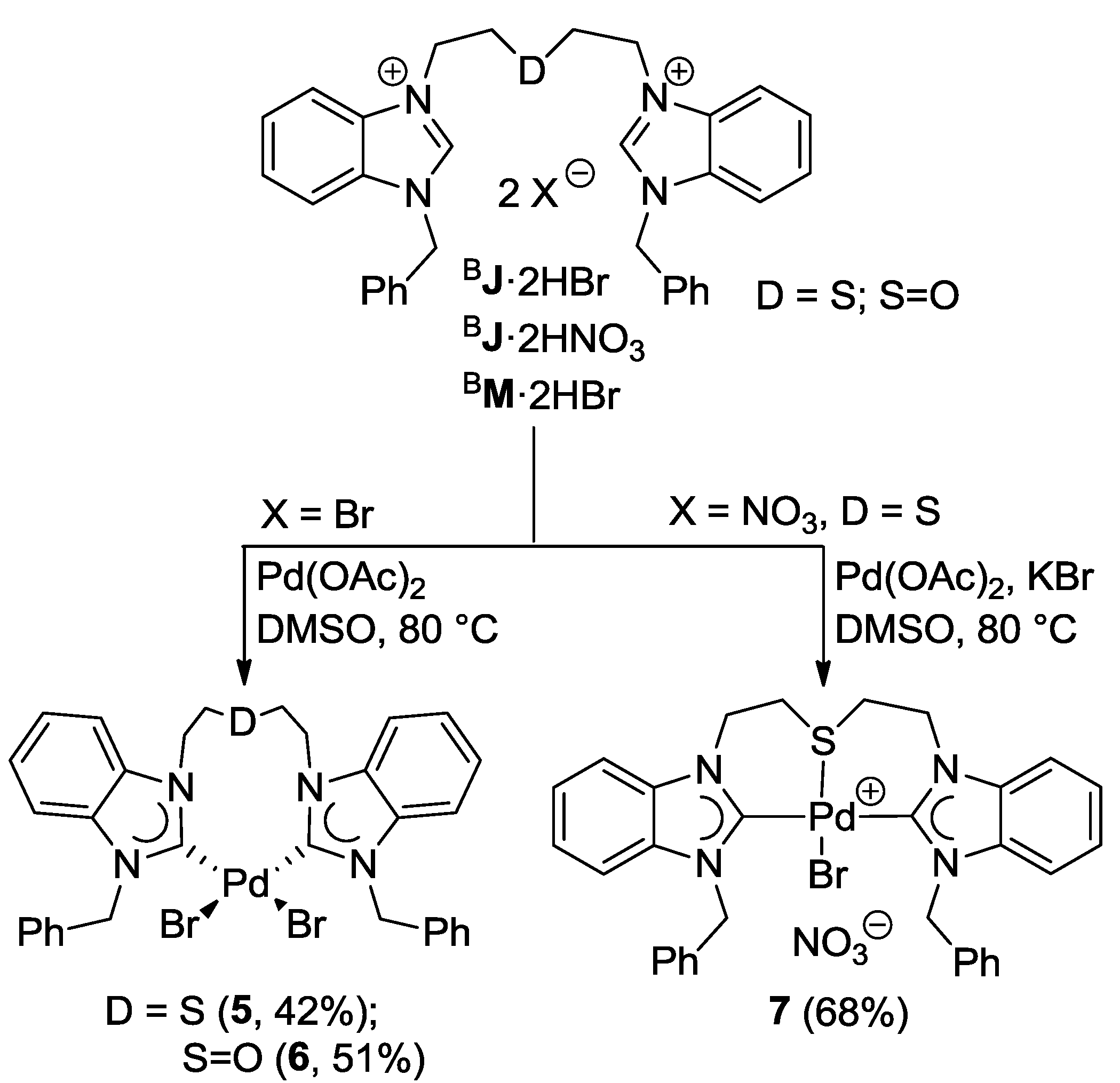
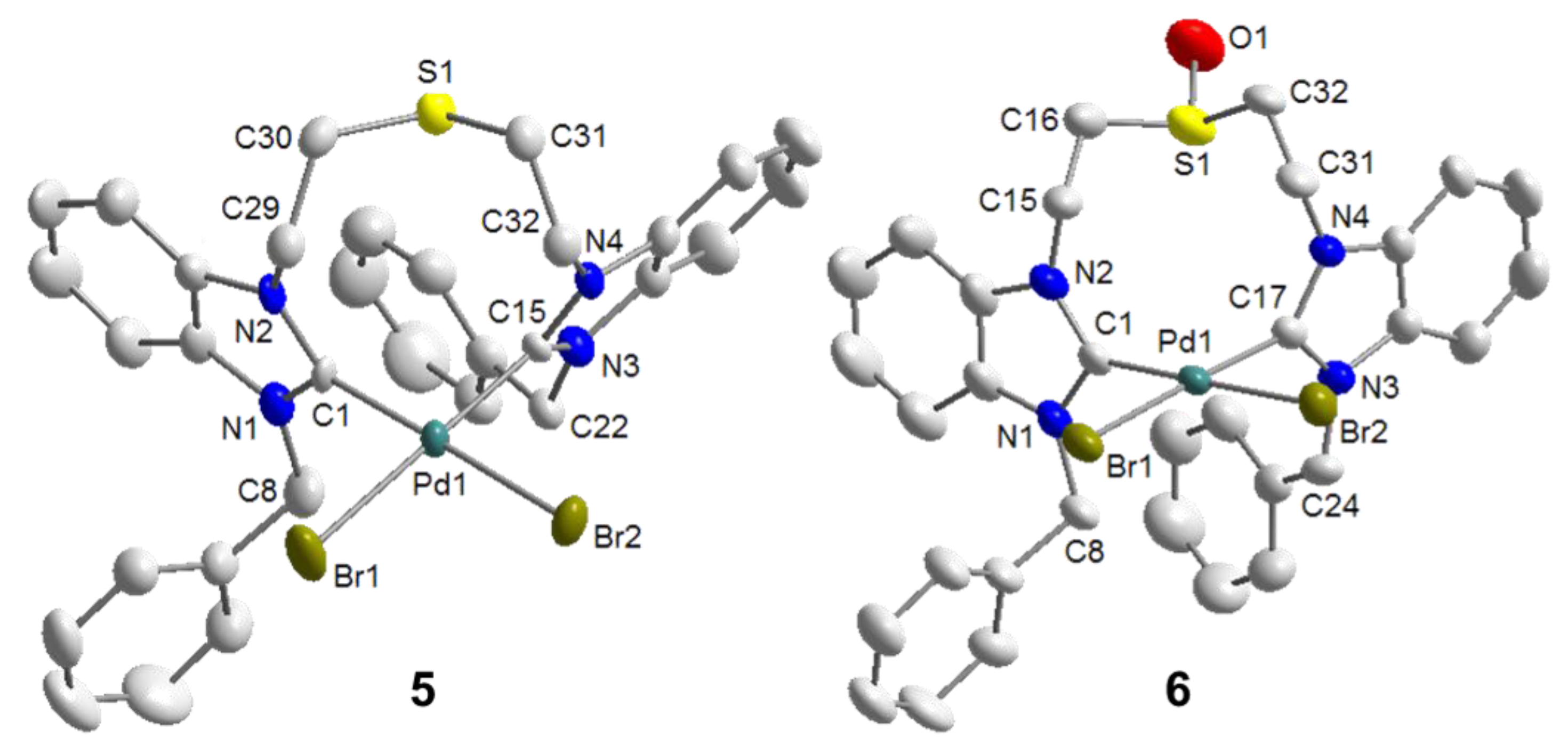
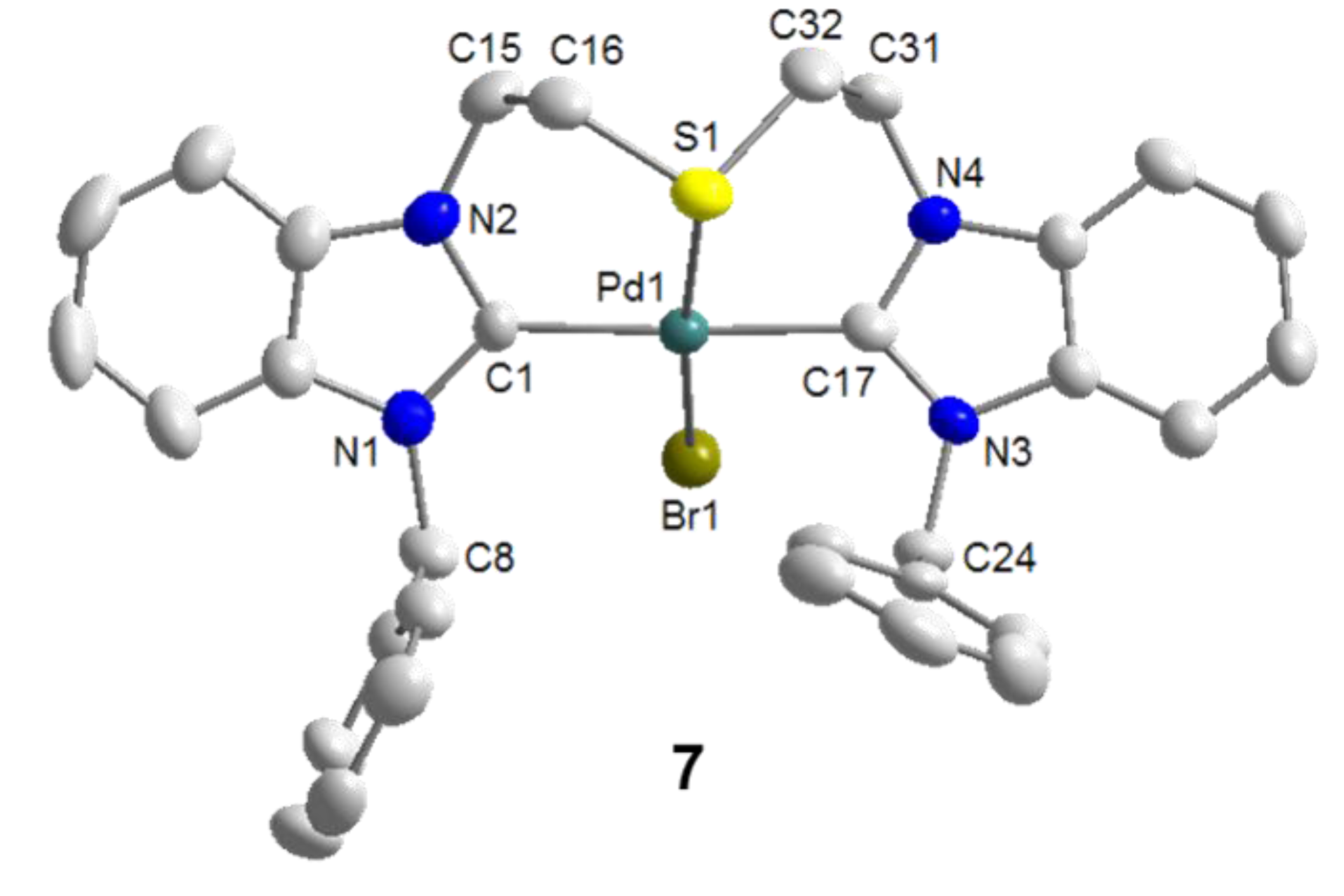
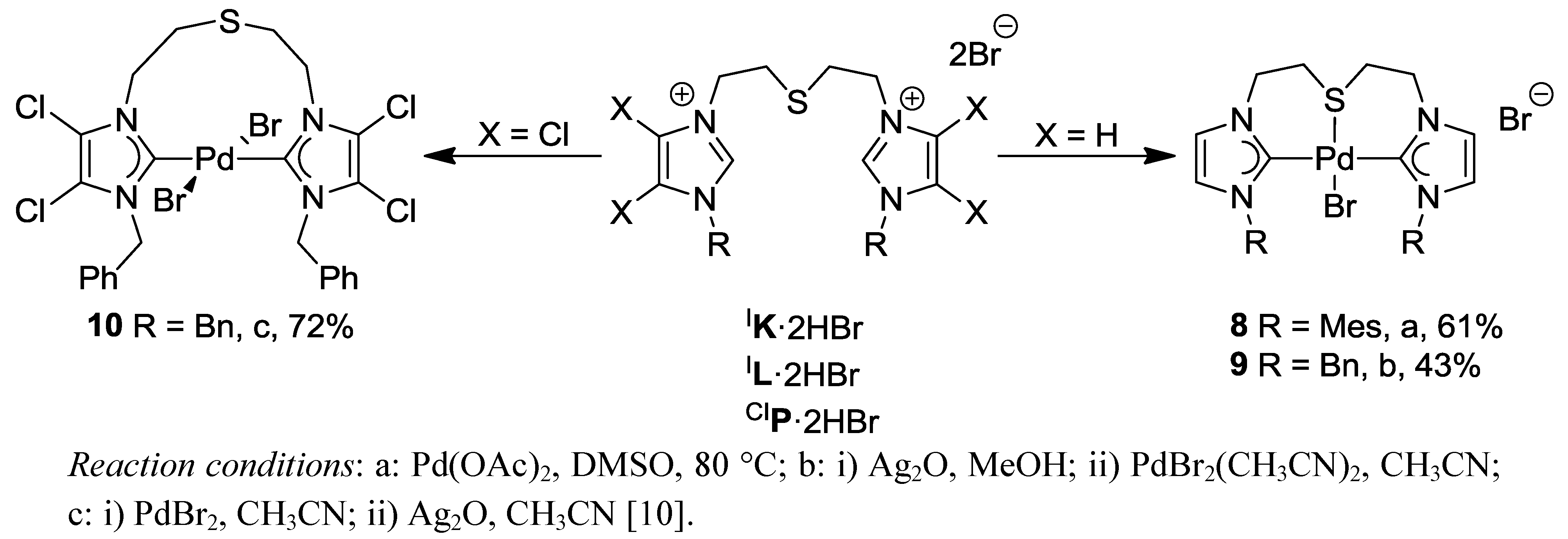
4.2. Pd(II) Imidazolin-2-ylidene Complexes
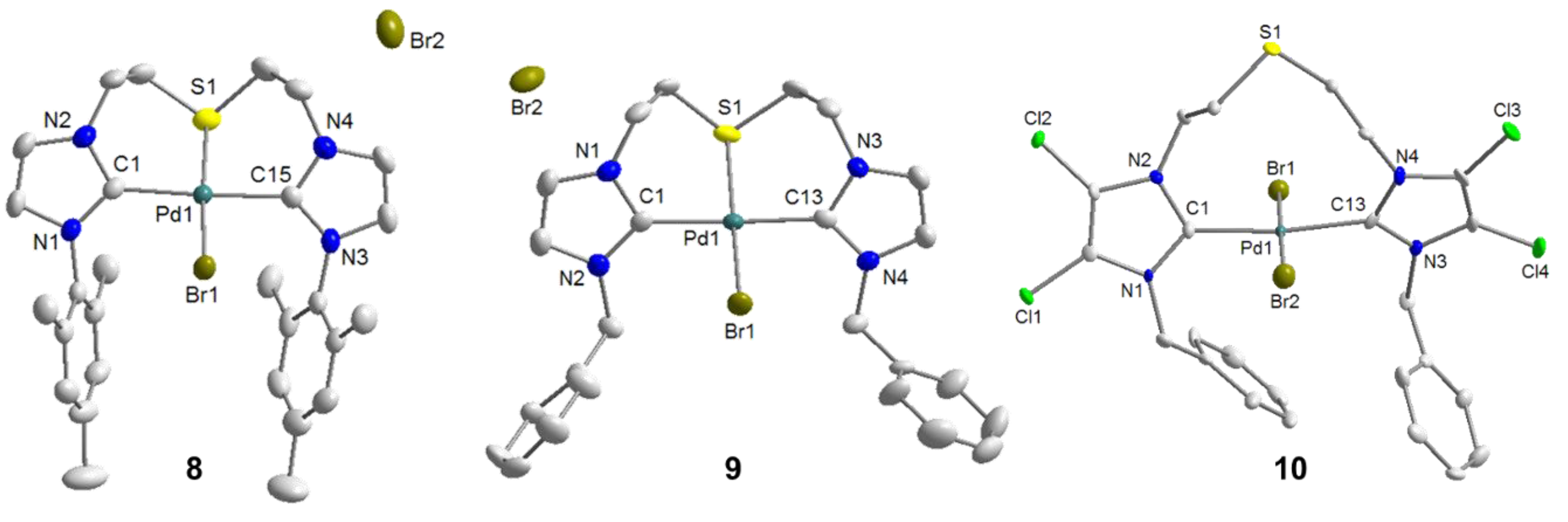
5. Thiophene-NHC Complexes
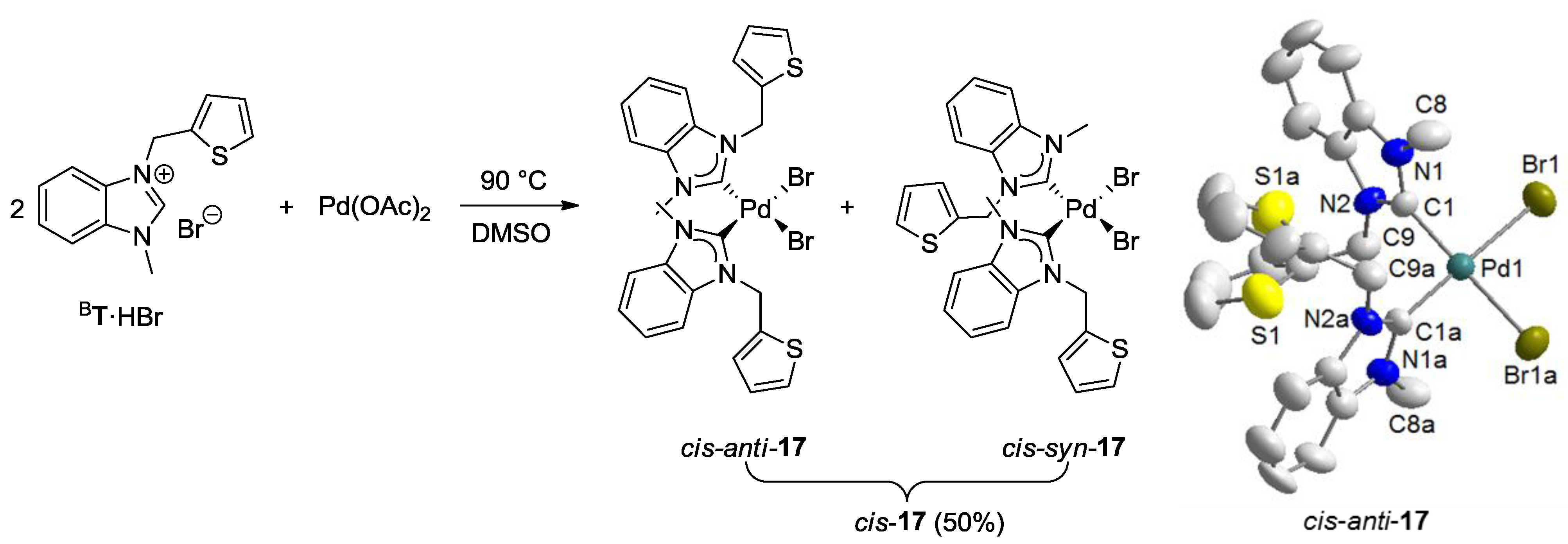
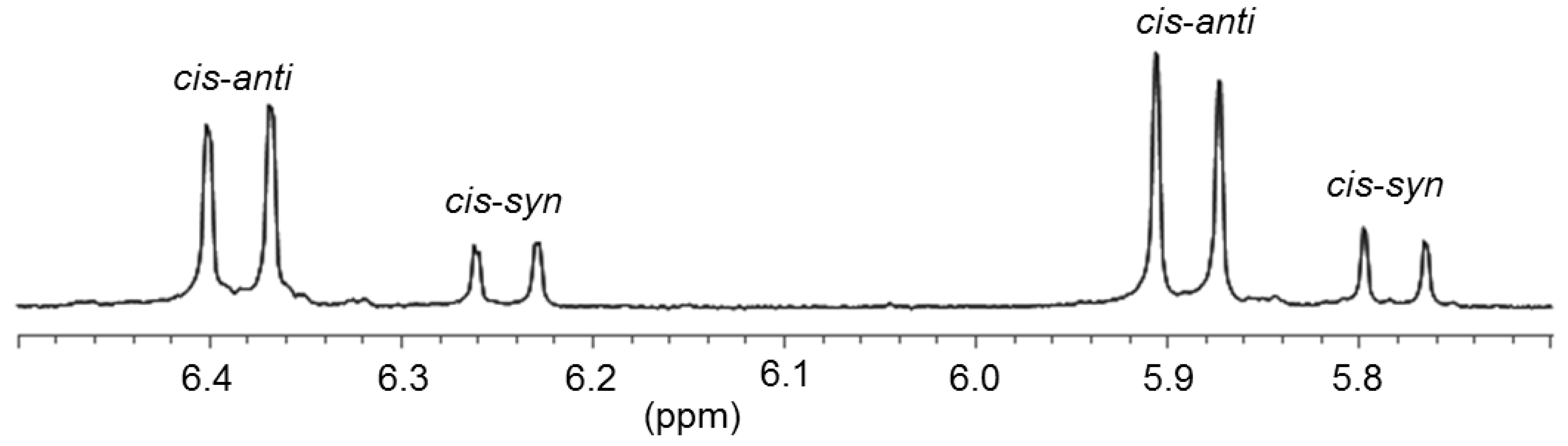
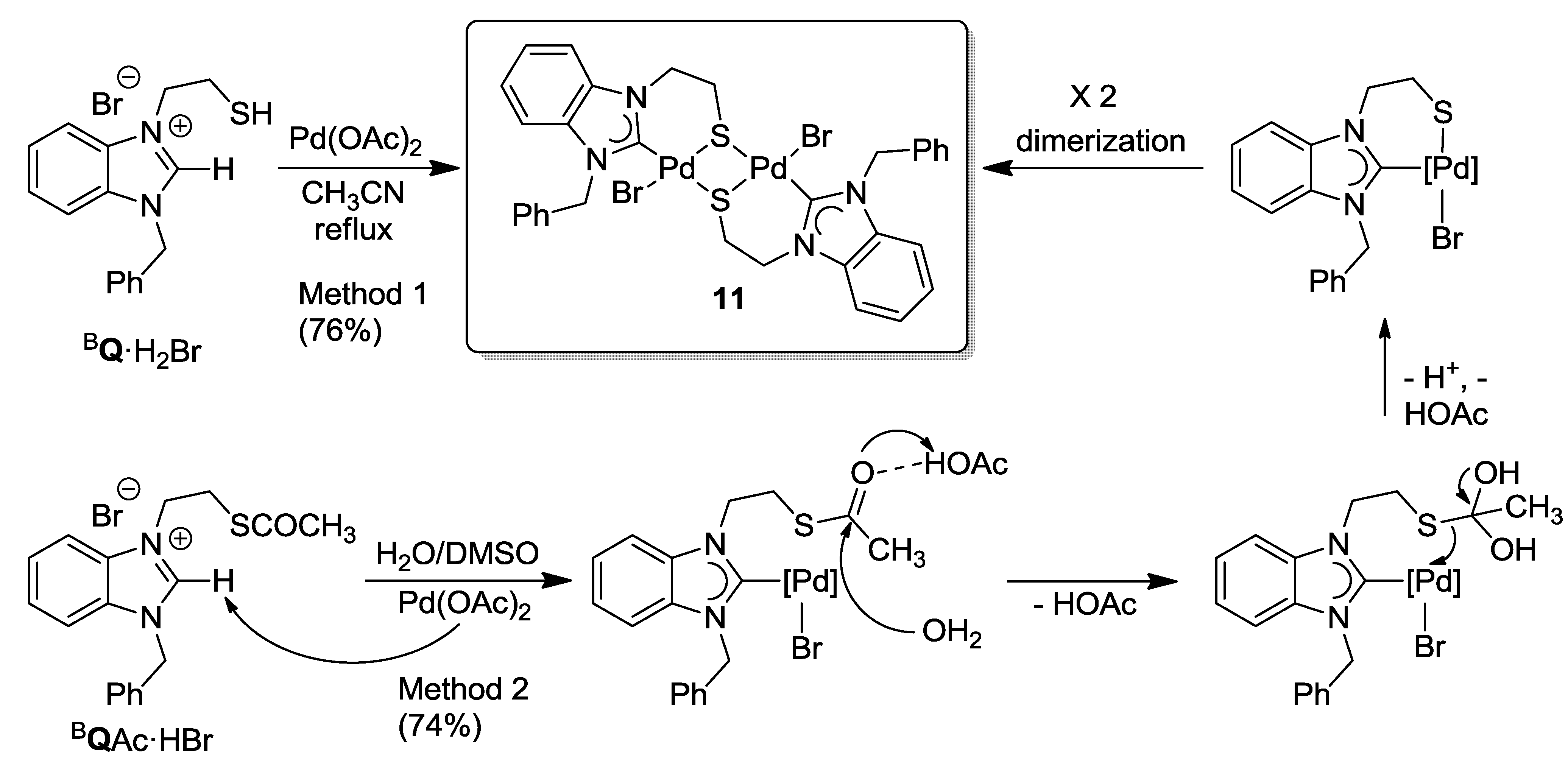
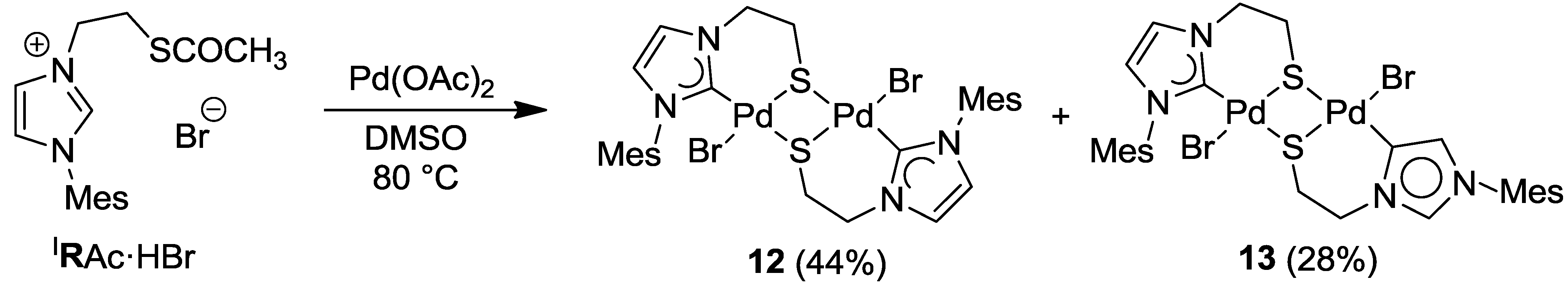
6. Thiolato-NHC Complexes
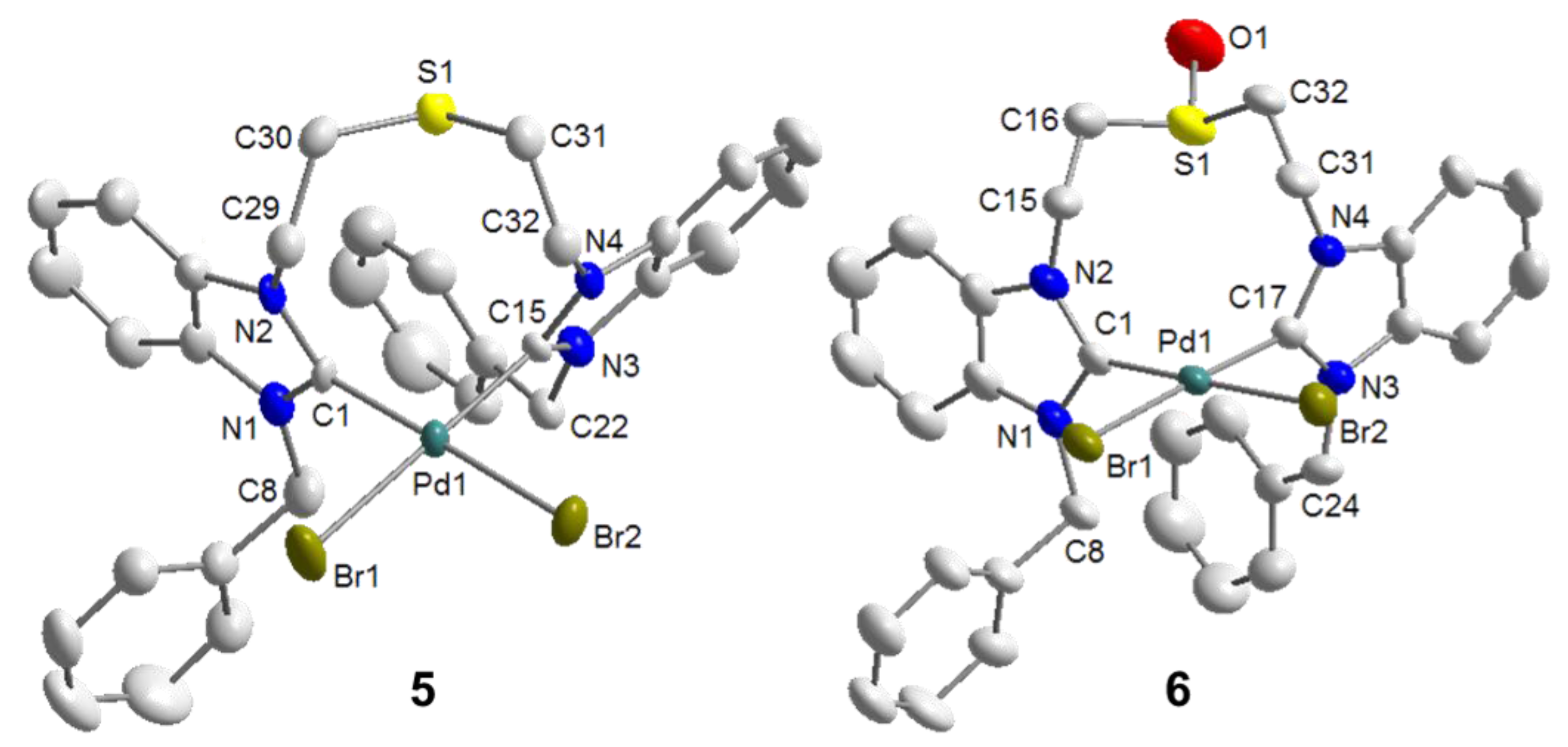
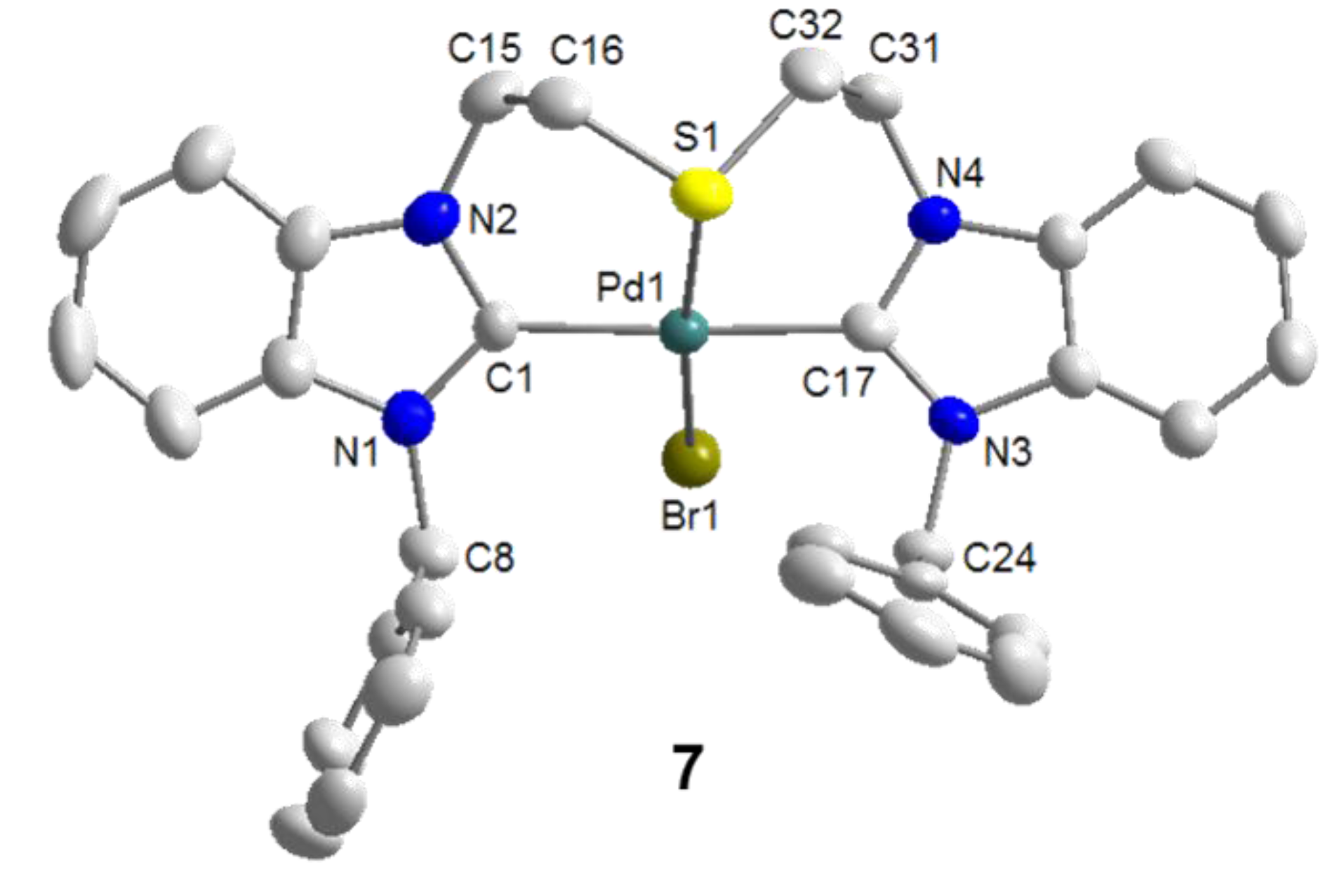
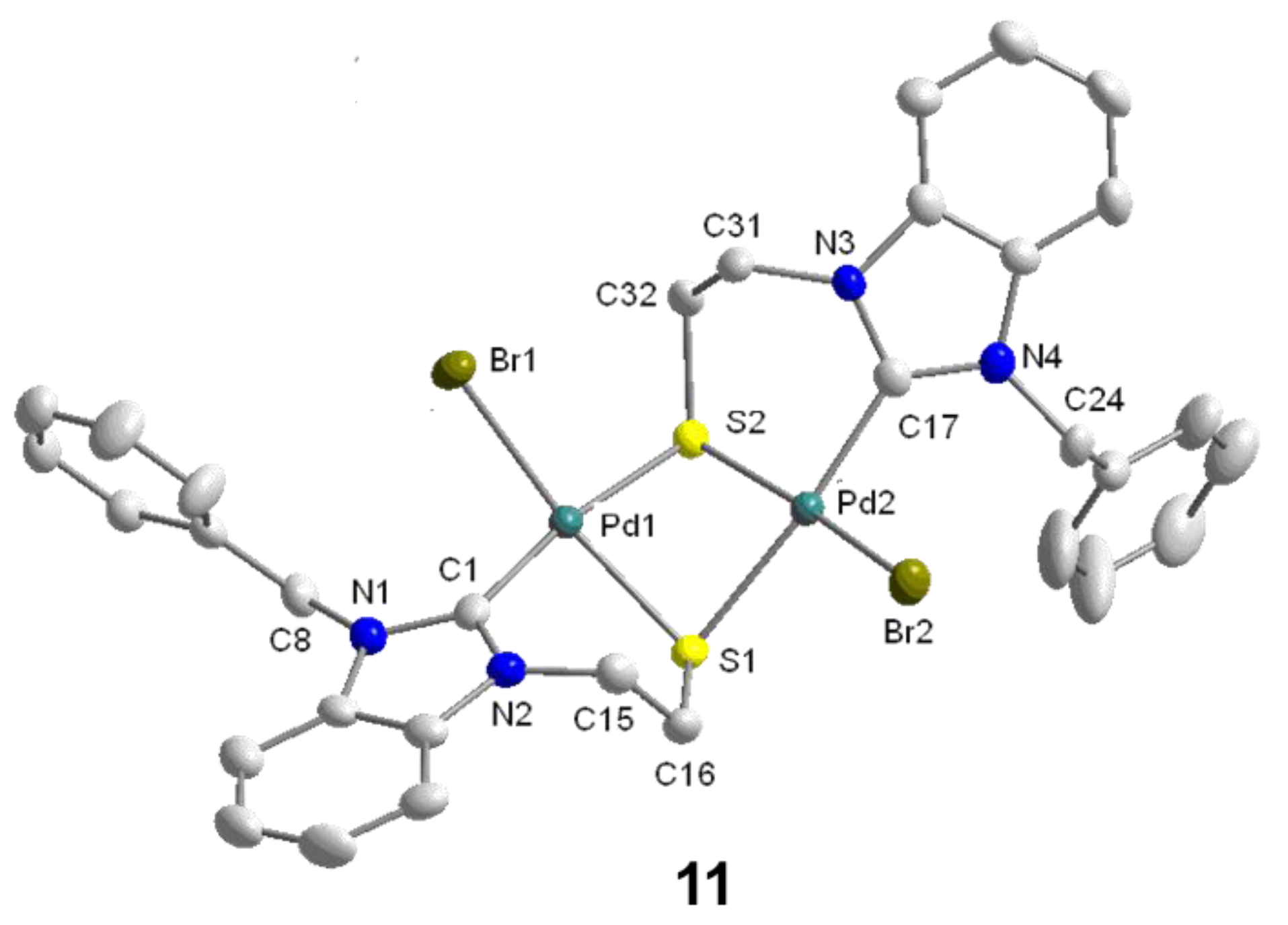
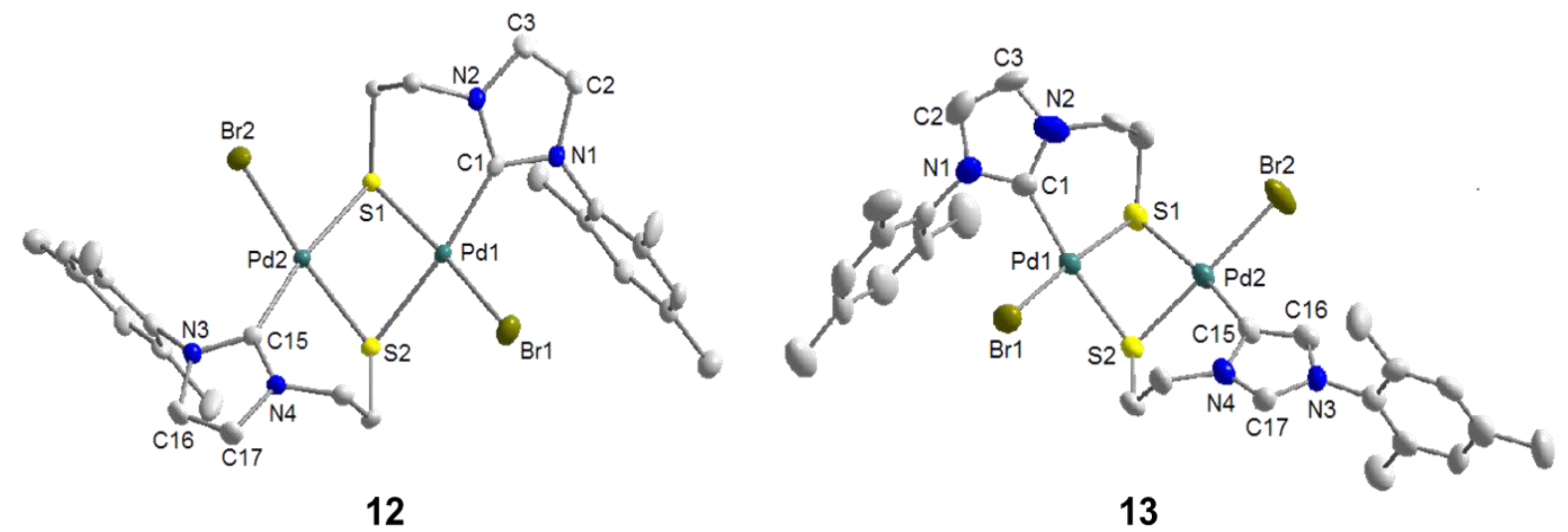
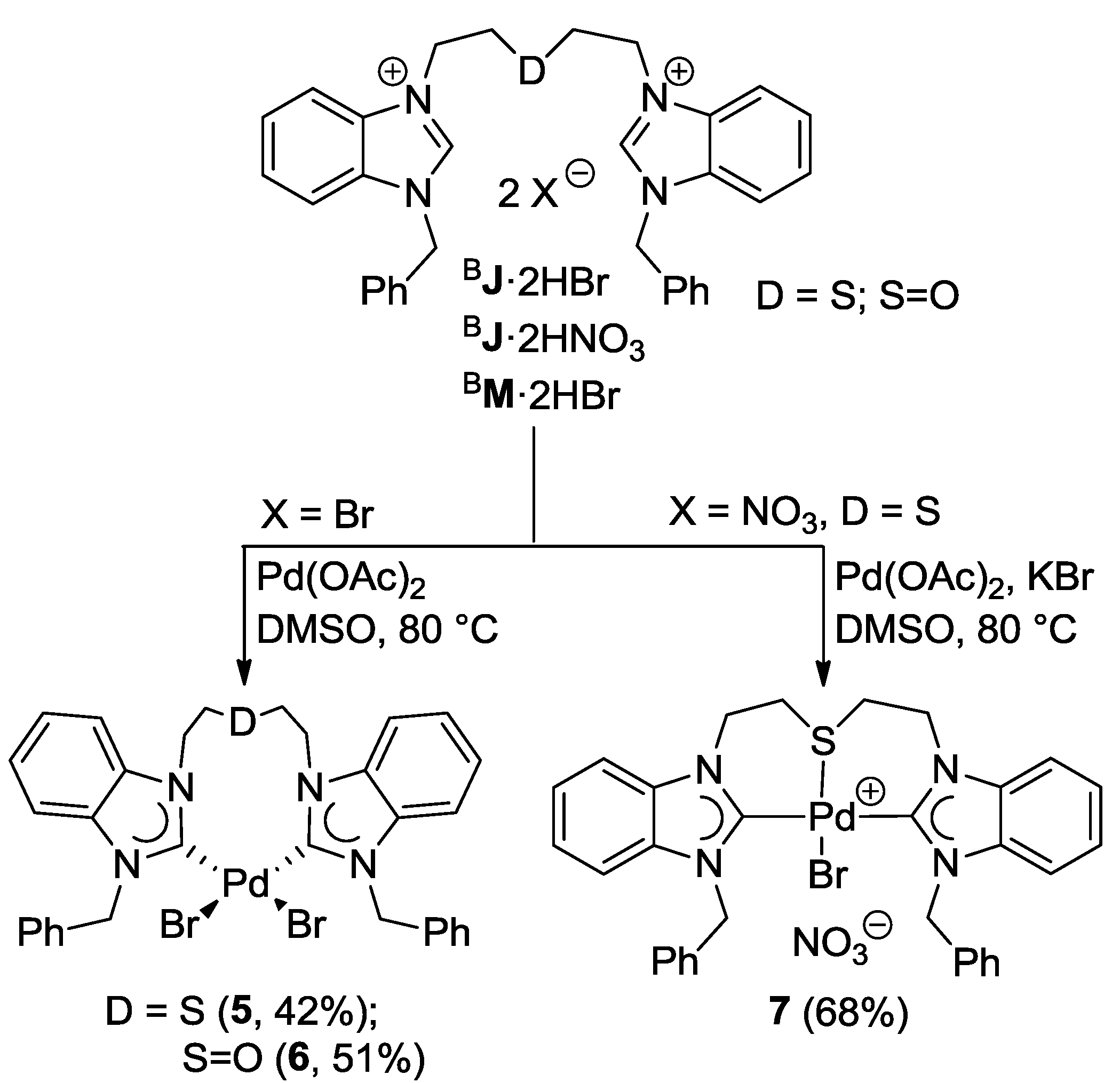
6.1. Thiolato-Bridged Pd(II) Complexes
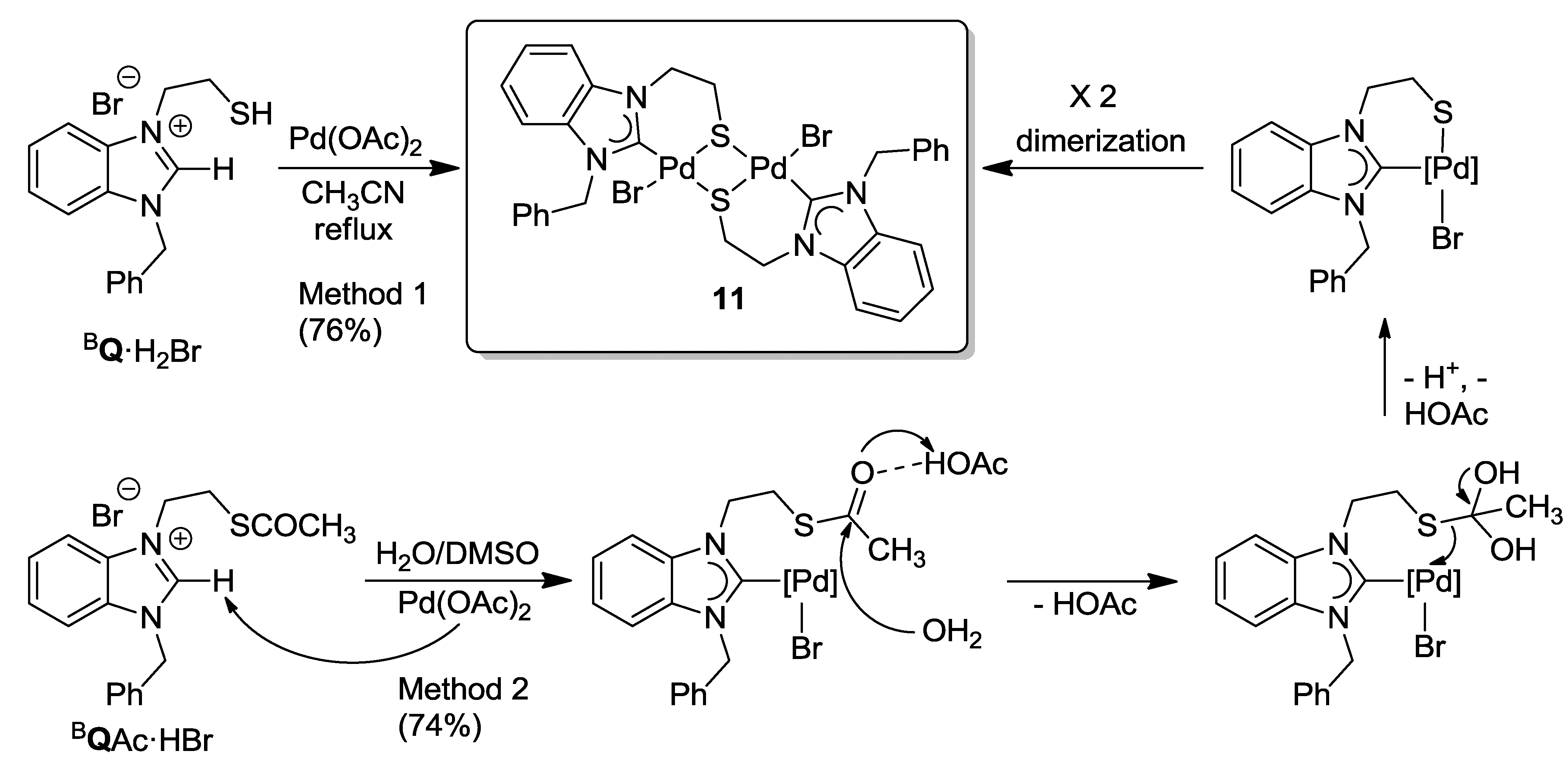

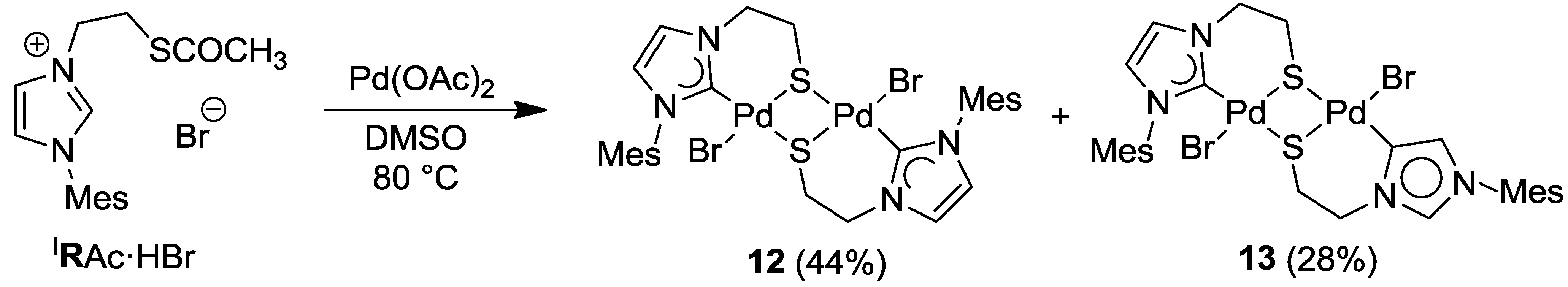
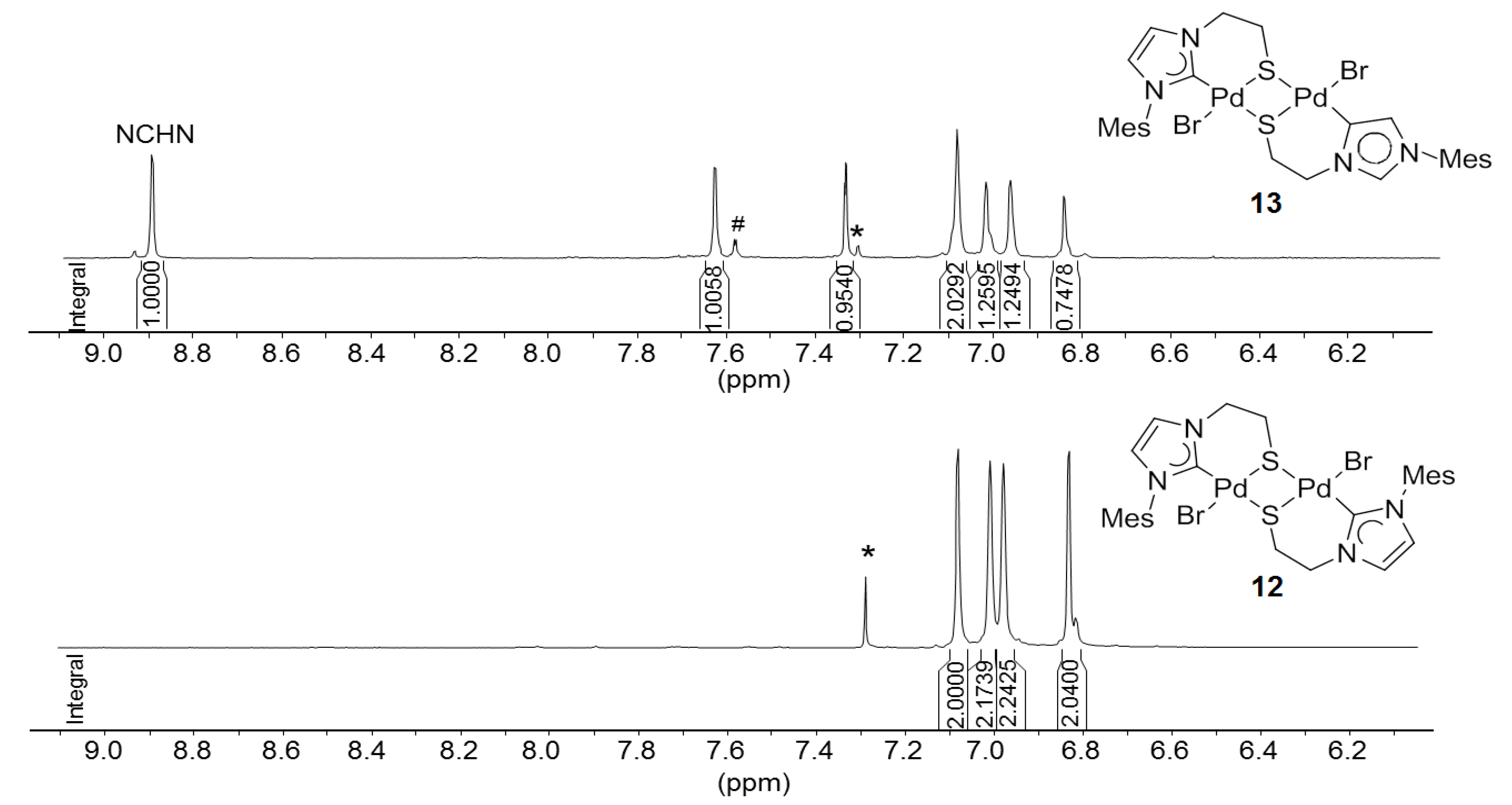
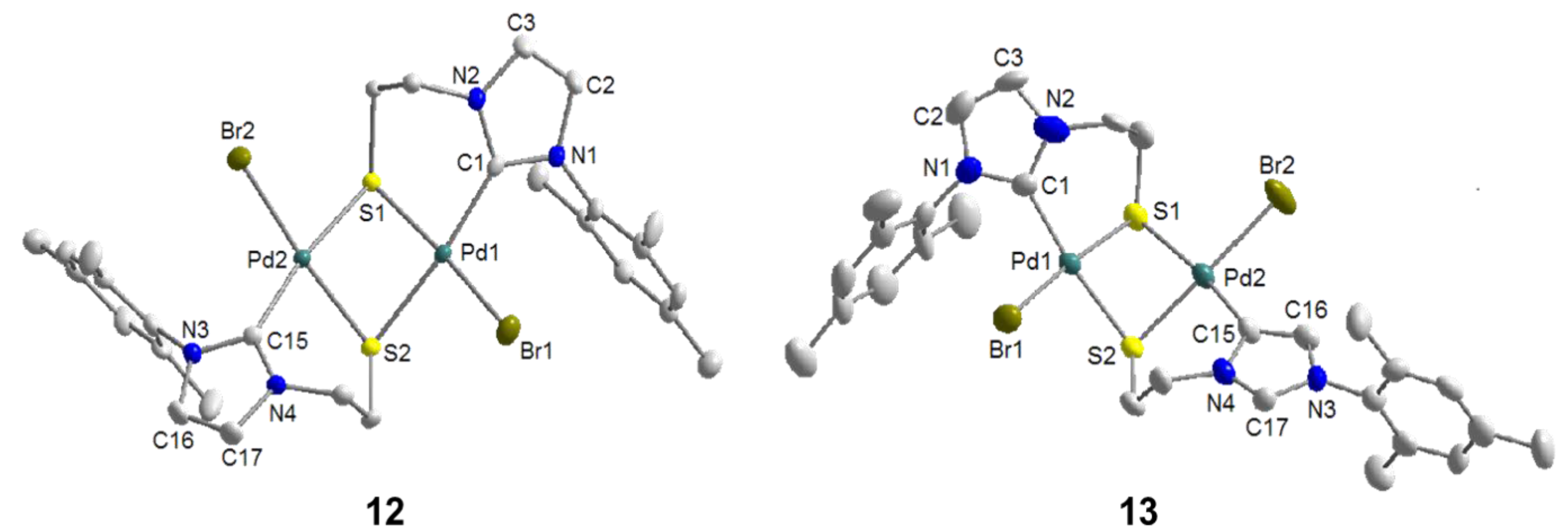
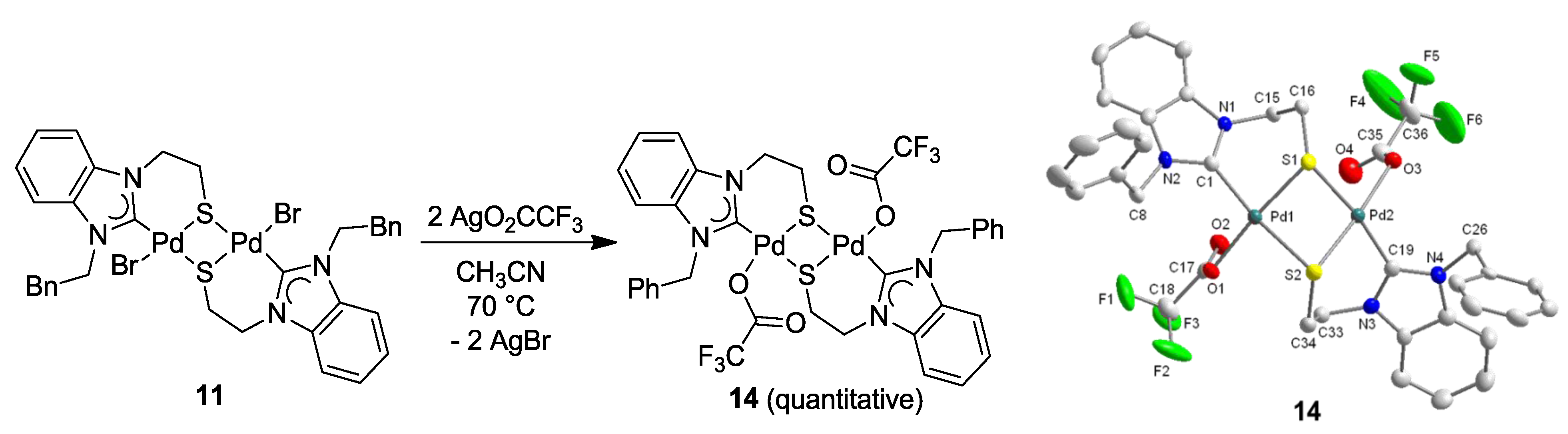
6.2. Reactivity Studies with Ag-carboxylates
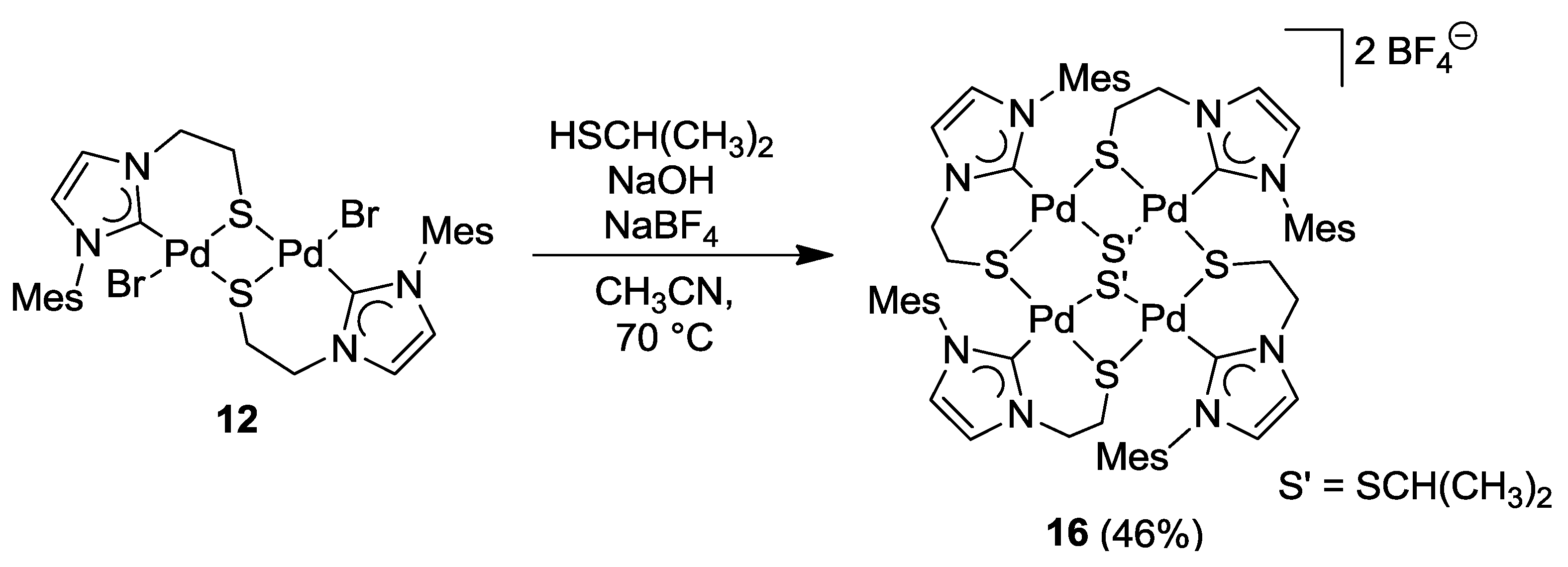
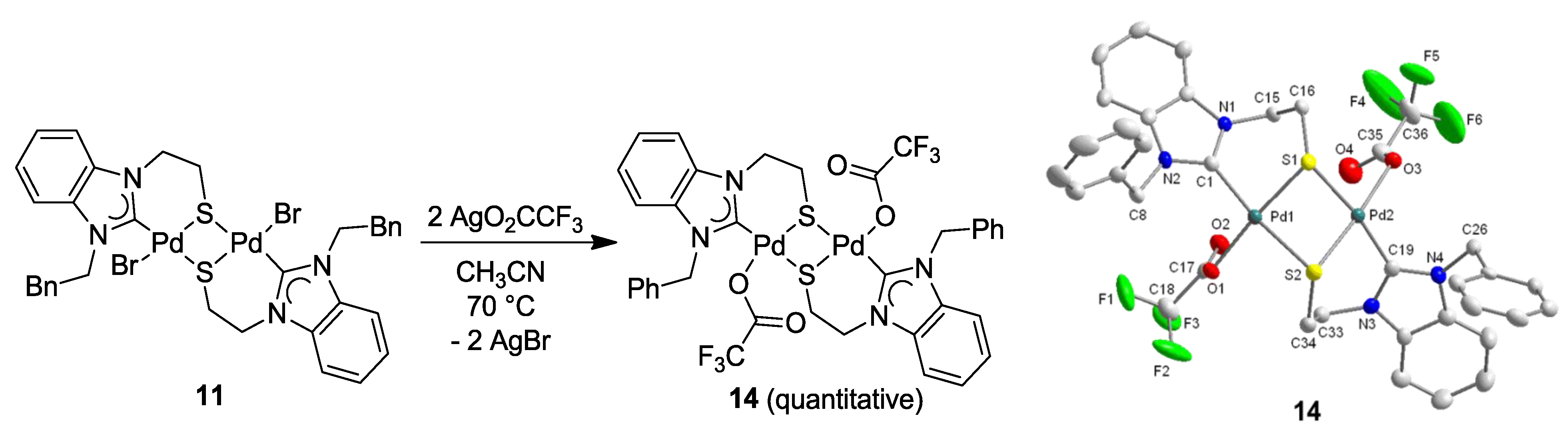
6.3. Reactivity Studies with Alkylating Reagents
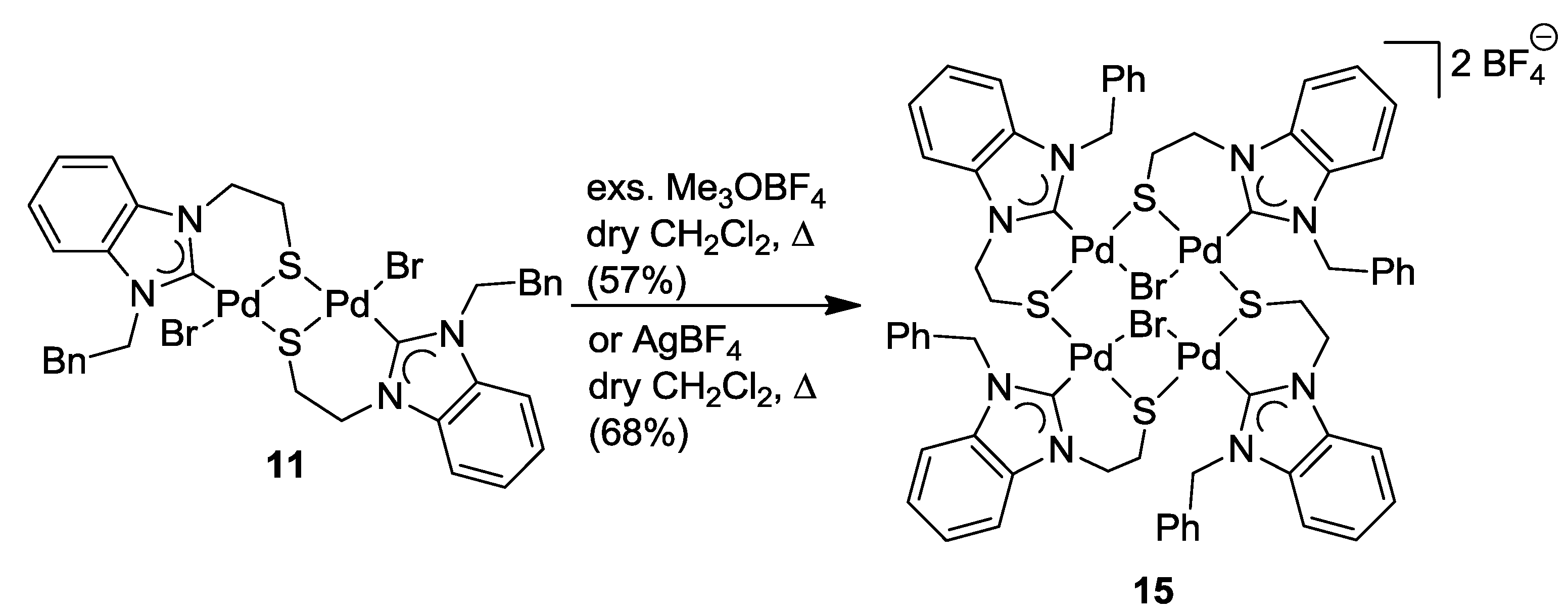
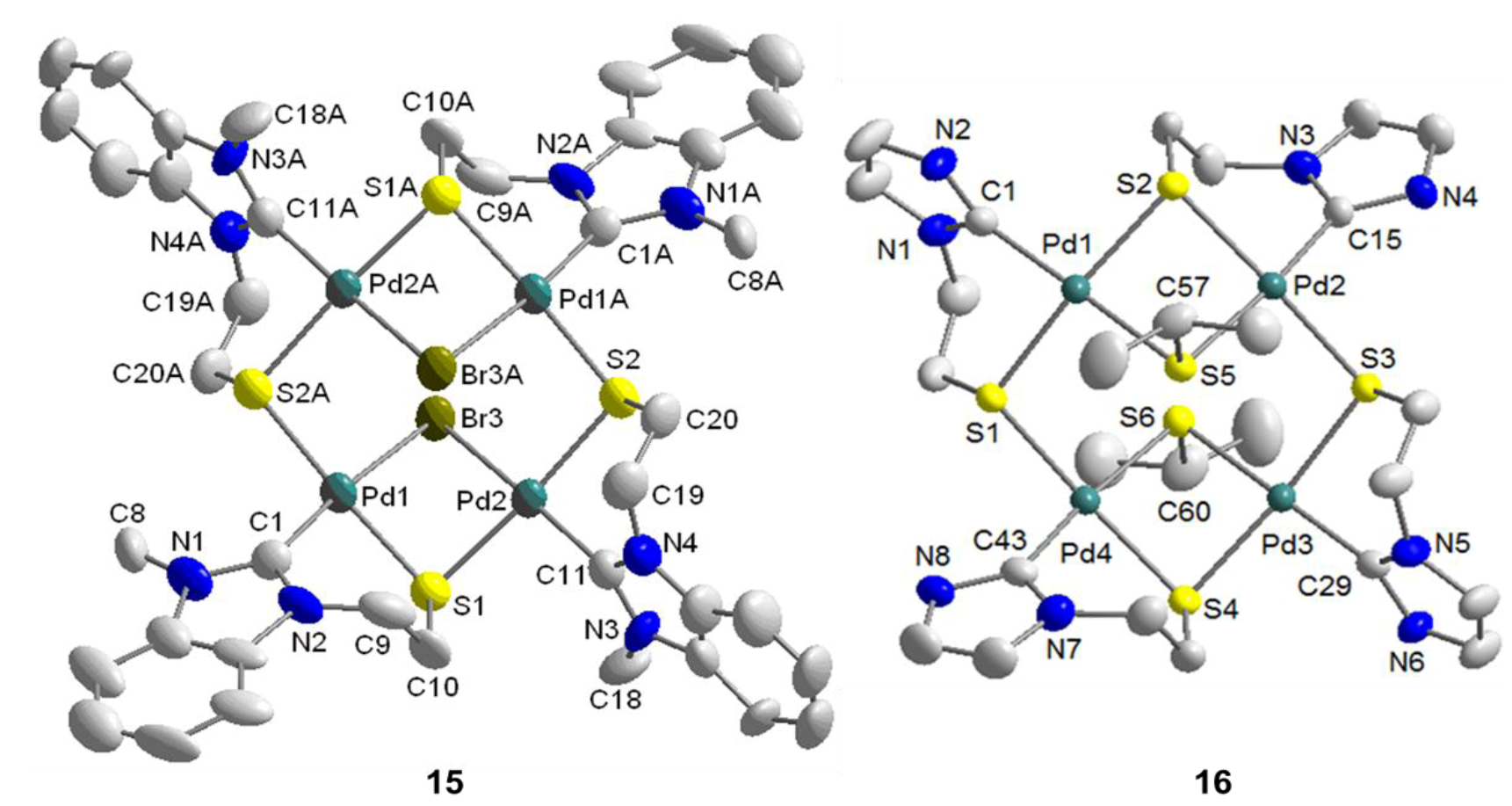
6.4. Reactivity Studies with External Thiolato Ligands
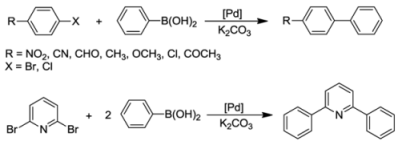
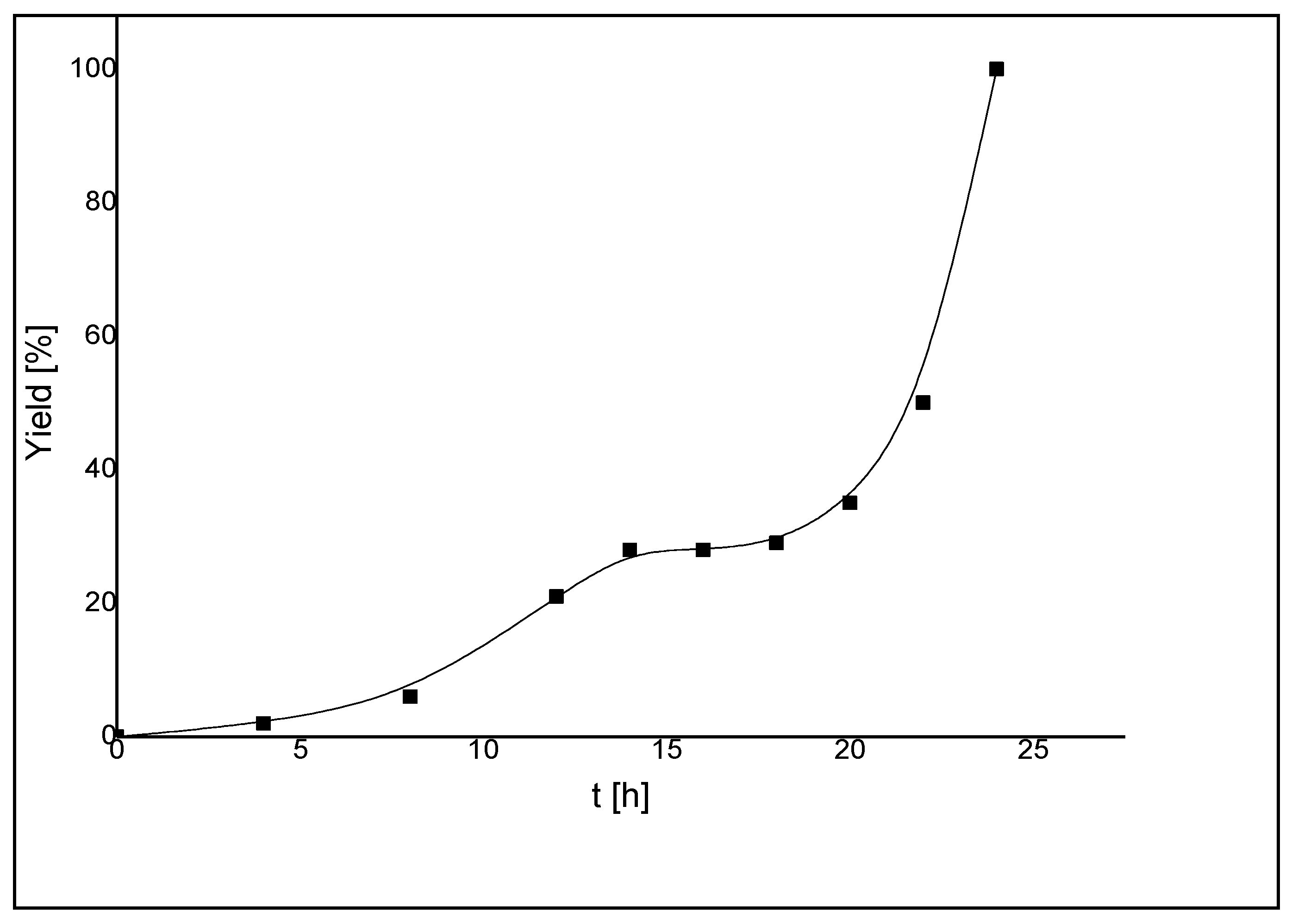
7. Catalytic Applications

| Entry | Catalyst | temp. [°C] | Aryl halide | catalyst loading [mol%] | t [h] | yield [%] b | TON | TOF [h−1] |
|---|---|---|---|---|---|---|---|---|
| 1 c | 11 | 60 | 4-bromobenzophenone | 0.001 | 24 | 0 | 0 | 0 |
| 2 | 11 | 60 | 4-bromobenzophenone | 0.001 | 24 | 84 | 84,000 | 3500 |
| 3 | 14 | 60 | 4-bromobenzophenone | 0.001 | 24 | >99 | 100,000 | 4167 |
| 4 | 15 d | 60 | 4-bromobenzophenone | 0.001 | 24 | 48 | 96,000 | 4000 |
| 5 | 14 | 60 | 4-bromobenzonitrile | 0.001 | 24 | >99 | 100,000 | 4167 |
| 6 | 14 | 60 | 1-bromo-4-nitrobenzene | 0.001 | 42 | 90 | 90,000 | 2143 |
| 7 | 14 | 80 | 4-bromobenzaldehyde | 0.001 | 23 | >99 | 100,000 | 4167 |
| 8 | 14 | 100 | 2,6-dibromopyridine e | 0.0025 | 48 | >99 | 20,000 | 417 |
| 9 | 14 | 100 | 4-bromotoluene | 0.0025 | 20 | 97 | 38,800 | 1940 |
| 10 | 14 | 100 | 4-bromoanisole | 0.0025 | 24 | 86 | 34,400 | 1433 |
| 11 | 14 | 100 | 1-bromo-4-chlorobenzene | 0.0025 | 24 | 72 f | 28,800 | 1200 |
| 12 g | 14 | 100 | 4-chlorobenzaldehyde | 0.1 | 72 | 41 | 410 | 5.7 |
7.1. Suzuki-Miyaura Coupling Reactions
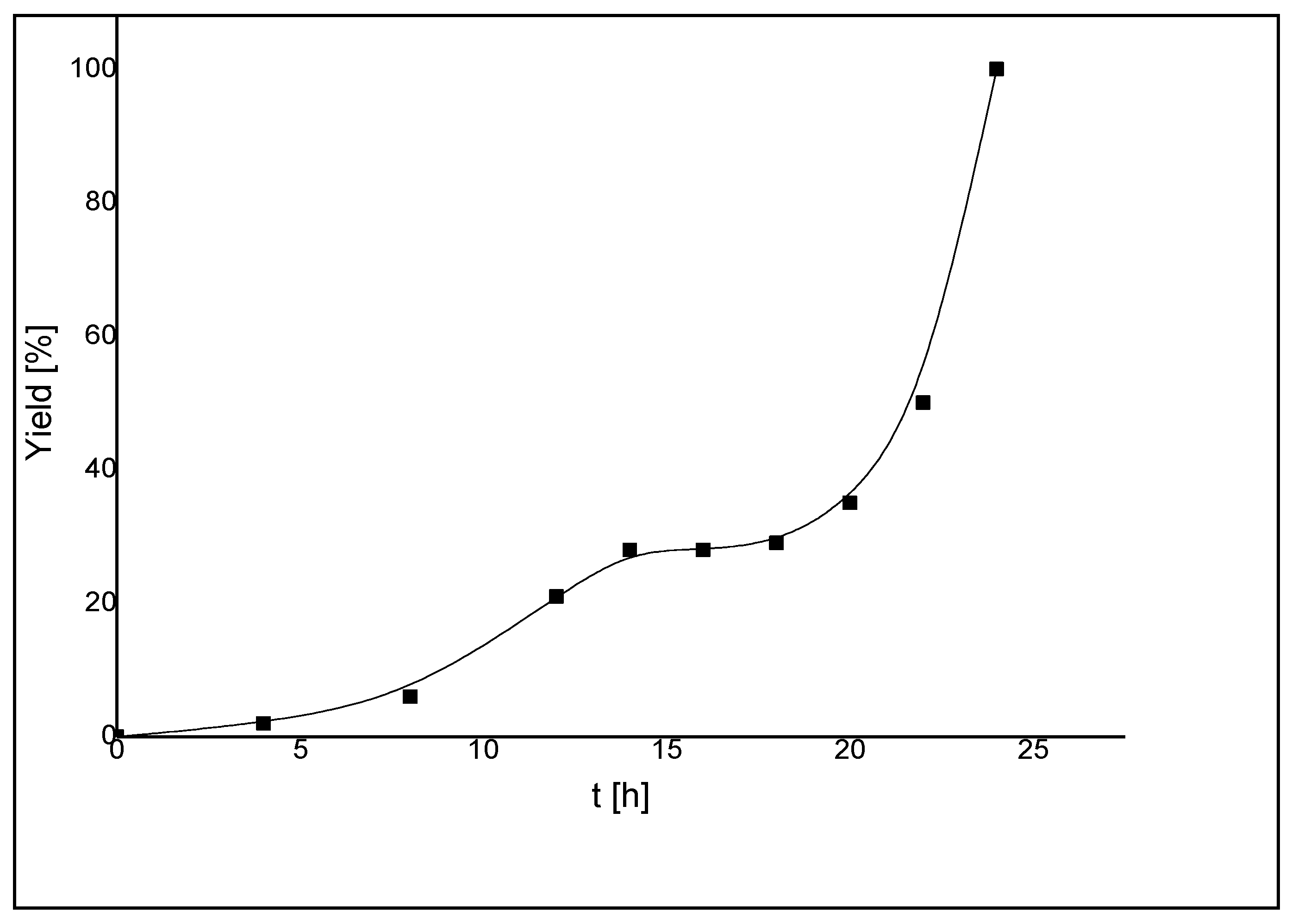
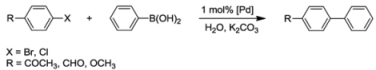
| Entry a | Catalyst | Aryl halide | t [h] | temp. [°C] | yield [%] b |
|---|---|---|---|---|---|
| 1 | cis-1a | 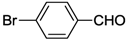 | 8 | RT | >99 |
| 2 | trans-1a | >99 | |||
| 3 | cis-2a | >99 | |||
| 4 | 3 | >99 | |||
| 5 | 4 | >99 | |||
| 6 | cis-17 | >99 | |||
| 7 | cis-1a | 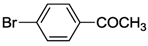 | 8 | RT | 98 |
| 8 | trans-1a | 84 | |||
| 9 | cis-2a | 90 | |||
| 10 | 3 | 85 | |||
| 11 | 4 | >99 | |||
| 12 | cis-17 | 85 | |||
| 13 c | cis-1a | 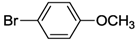 | 21 | 85 | >99 |
| 14 c | trans-1a | >99 | |||
| 15 c | cis-2a | >99 | |||
| 16 c | 3 | >99 | |||
| 17 c | 4 | >99 | |||
| 18 c | cis-17 | >99 | |||
| 19 c | cis-1a | 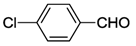 | 21 | 85 | 7 |
| 20 c | trans-1a | 11 | |||
| 21 c | cis-2a | 3 | |||
| 22 c | 3 | 8 | |||
| 23 c | 4 | 52 | |||
| 24 c | cis-17 | 9 | |||
| 25 c | cis-1a |  | 21 | 85 | 3 |
| 26 c | trans-1a | 9 | |||
| 27 c | cis-2a | 23 | |||
| 28 c | 3 | 25 | |||
| 29 c | 4 | 44 | |||
| 30 c | cis-17 | 14 |
7.2. Mizoroki-Heck Coupling Reactions
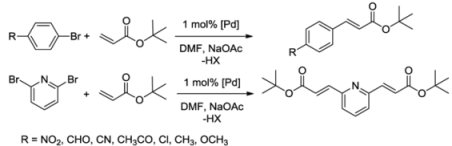
| Entry | Catalyst | Aryl halide | t [h] | temp [°C] | yield [%] b |
|---|---|---|---|---|---|
| 1 | 5 | 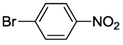 | 24 | 120 | >99 |
| 2 | 6 | >99 | |||
| 3 | 7 | >99 | |||
| 4 | 5 | 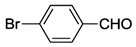 | 24 | 120 | >99 |
| 5 | 6 | >99 | |||
| 6 | 7 | >99 | |||
| 7 | 5 | 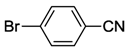 | 24 | 120 | >99 |
| 8 | 6 | >99 | |||
| 9 | 7 | >99 | |||
| 10 | 5 | 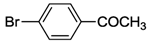 | 24 | 120 | >99 |
| 11 | 6 | >99 | |||
| 12 | 7 | >99 | |||
| 13 | 5 |  | 24 | 120 | >99 |
| 14 | 6 | >99 | |||
| 15 | 7 | >99 | |||
| 16 | 5 | 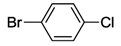 | 24 | 120 | 50 |
| 17 | 6 | 58 | |||
| 18 | 7 | 45 | |||
| 19 c | 5 | 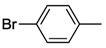 | 24 | 140 | 53 |
| 20 c | 6 | 52 | |||
| 21 c | 7 | 47 | |||
| 22 c | 5 | 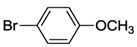 | 24 | 140 | 41 |
| 23 c | 6 | 35 | |||
| 24 c | 7 | 51 |
7.3. Hydroamination of Alkynes
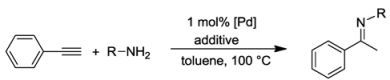
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Aniline | complex 8 yield b | complex 9 yield b | complex 10 yield b |
|---|---|---|---|---|
| 1 |  | - | 87 c | - |
| 2 | 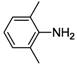 | 55 | 94 | 53 |
| 3 | 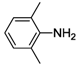 | - | 0 d | - |
| 4 | 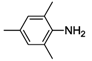 | 43 | 75 | 53 |
| 5 | 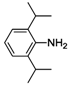 | 39 | 58 | 42 |
| 6 |  | 26 | 48 | 35 |
| 7 | 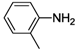 | 22 | 51 | 28 |
| 8 | 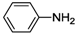 | 23 | 30 | 20 |
8. Conclusions and Outlook
Acknowledgments
References
- Hahn, F.E.; Jahnke, M.C. Heterocyclic carbenes: Synthesis and coordination chemistry. Angew. Chem. Int. Ed. 2008, 47, 3122–3172. [Google Scholar] [CrossRef]
- Kühl, O. The Chemistry of functionalised N-heterocyclic carbenes. Chem. Soc. Rev. 2007, 36, 592–607. [Google Scholar] [CrossRef]
- Normand, A.T.; Cavell, K.J. Donor-functionalised N-heterocyclic carbene complexes of group 9 and 10 metals in catalysis: Trends and directions. Eur.J.Inorg. Chem. 2008, 2781–2800. [Google Scholar]
- John, A.; Ghosh, P. Fascinating Frontiers of N/O-Functionalized N-Heterocyclic Carbene Chemistry: From chemical catalysis to biomedical applications. Dalton Trans. 2010, 39, 7183–7206. [Google Scholar] [CrossRef]
- Braunstein, P.; Naud, F. Hemilability of Hybrid Ligands and the Coordination Chemistry of Oxazoline-Based Systems. Angew. Chem. Int. Ed. 2001, 40, 680–699. [Google Scholar] [CrossRef]
- Bierenstiel, M.; Cross, E.D. Sulfur-Functionalized N-Heterocyclic Carbenes and Their Transition Metal Complexes. (References therein). Coord. Chem. Rev. 2011, 255, 574–590. [Google Scholar]
- Huynh, H.V.; Yeo, C.H.; Tan, G.K. Hemilabile Behavior of a Thioether-Functionalized N-Heterocyclic Carbene Ligand. Chem. Commun. 2006, 3833–3835. [Google Scholar]
- Huynh, H.V.; Yeo, C.H.; Chew, Y.X. Syntheses, Structures, and Catalytic Activities of Hemilabile Thioether-Functionalized NHC Complexes. Organometallics 2010, 29, 1479–1486. [Google Scholar]
- Huynh, H.V.; Yuan, D.; Han, Y. Syntheses and Catalytic Activities of Pseudo-Pincer and CSC Pincer-Type Pd(II) Complexes Derived from Benzannulated N-Heterocyclic Carbenes. Dalton Trans. 2009, 7262–7268. [Google Scholar] [CrossRef]
- Yuan, D.; Tang, H.; Xiao, L.; Huynh, H.V. CSC-pincer versus Pseudo-Pincer Complexes of Palladium(II): a Comparative Study on Complexation and Catalytic Activities of NHC Complexes. Dalton Trans. 2011, 40, 8788–8795. [Google Scholar] [CrossRef]
- Yuan, D.; Huynh, H.V. Dinuclear and Tetranuclear Palladium(II) Complexes of a Thiolato-Functionalized, Benzannulated N-Heterocyclic Carbene Ligand and Their Activities Toward Suzuki-Miyaura Coupling. Organometallics 2010, 29, 6020–6027. [Google Scholar] [CrossRef]
- Yuan, D.; Huynh, H.V. Syntheses and Characterizations of Thiolato-Functionalized N-Heterocyclic Carbene Pd(II) Complexes with Normal and Mesoionic Binding Modes. Dalton Trans. 2011, 40, 11698–11703. [Google Scholar] [CrossRef]
- Huynh, H.V.; Chew, Y.X. Synthesis, Structural Characterization and Catalytic Activity of a Palladium(II) Complex Bearing a New Ditopic Thiophene-N-Heterocyclic Carbene Ligand. Inorg. Chim. Acta 2010, 363, 1979–1983. [Google Scholar] [CrossRef]
- Liu, S.; Lee, C.; Fu, C.; Chen, C.; Liu, Y.; Elsevier, C.J.; Peng, S.; Chen, J. N-Heterocyclic Carbene Transfer from Gold(I) to Palladium(II). Organometallics 2009, 28, 6957–6962. [Google Scholar]
- Lin, J.C.Y.; Huang, R.T.W.; Lee, C.S.; Bhattacharyya, A.; Hwang, W.S.; Lin, I.J.B. Coinage Metal-N-Heterocyclic Carbene Complexes. Chem. Rev. 2009, 109, 3561–3598. [Google Scholar] [CrossRef]
- Slagt, M.Q.; van Zwieten, D.A.P.; Moerkerk, A.J.C.M.; Gebbink, R.J.M.K.; van Koten, G. NCN-Pincer Palladium Complexes with Multiple Anchoring Points for Functional Groups. Coord. Chem. Rev. 2004, 248, 2275–2282. [Google Scholar] [CrossRef]
- Pugh, D.; Danopoulos, A.A. Metal Complexes with ‘Pincer’-Type Ligands Incorporating N-Heterocyclic Carbene Functionalities. Coord. Chem. Rev. 2007, 251, 610–641. [Google Scholar]
- Huynh, H.V.; Han, Y.; Jothibasu, R.; Yang, J.A. 13C NMR Spectroscopic Determination of Ligand Donor Strengths Using N-Heterocyclic Carbene Complexes of Palladium(II). Organometallics 2009, 28, 5395–5404. [Google Scholar] [CrossRef]
- Sellmann, D.; Geipel, F.; Heinemann, F.W. Mononuclear Carbene Dithiolate [Ni(NHX)(′S2C′)] Complexes with HNX = HNPiPr3 or HNSPh2 Coligands (′S2C′2- = 1,3-Imidazolidinyl-N, N′-bis(2-benzenethiolate)(2-)). Z. Anorg. Allg. Chem. 2001, 627, 1034–1038. [Google Scholar] [CrossRef]
- Cabeza, J.A.; del Rio, I.; Sánchez-Vega, M.G.; Suárez, M. Methyl Levamisolium Triflate as a Precursor to a Chiral Bifunctional N-Heterocyclic Carbene-Thiolate Ligand: Palladium(II) Complexes. Organometallics 2006, 25, 1831–1834. [Google Scholar]
- Iwasaki, F.; Yasui, M.; Yoshida, S.; Nishiyama, H.; Shimamoto, S.; Matsumura, N. Crystal and Molecular Structures of Novel Metal-Carbene Complexes III. Rh-Carbene Complexes and Cu Complex. B. Chem. Soc. Jpn. 1996, 69, 2759–2770. [Google Scholar]
- Guisado-Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Crystalline 1H-1,2,3-triazol-5-ylidenes: New Stable Mesoionic Carbenes (MICs). Angew. Chem. Int. Ed. 2010, 49, 4759–4762. [Google Scholar] [CrossRef]
- Huynh, H.V.; Jothibasu, R. Dipalladium Bis(μ-isopropylthiolato) Complexes with a [Pd2S2] Core Supported by N-Heterocyclic Carbenes. Organometallics 2007, 26, 6852–6856. [Google Scholar] [CrossRef]
- Yuan, D.; Huynh, H.V. 1,2,3-Triazolin-5-ylidenes: Synthesis of Hetero-bis(carbene) Pd(II) Complexes, Determination of Donor Strengths, and Catalysis. Organometallics 2012, 31, 405–412. [Google Scholar] [CrossRef]
- Dunleavy, J.K. Sulfur as a Catalyst Poison. Platinum Metals Rev. 2006, 50, 110. [Google Scholar] [CrossRef]
- Evans, D.A.; Campos, K.R.; Tedrow, J.S.; Michael, F.E.; Gagné, M.R. Application of Chiral Mixed Phosphorus/Sulfur Ligands to Palladium-Catalyzed Allylic Substitutions. J. Am. Chem. Soc. 2000, 122, 7905–7920. [Google Scholar] [CrossRef]
- Barbaro, P.; Currao, A.; Herrmann, J.; Nesper, R.; Pregosin, P.S.; Salzmann, R. Chiral P,S-Ligands Based on β-D-Thioglucose Tetraacetate. Palladium(II) Complexes and Allylic Alkylation. Organometallics 1996, 15, 1879–1888. [Google Scholar]
- Herrmann, J.; Pregosin, P.S.; Salzmann, R. Palladium π-Allyl Chemistry of New P,S Bidentate Ligands. Selective but Variable Dynamics in the Isomerization of the η3-C3H5 and η3-PhCHCHCHPh π-Allyl Ligands. Organometallics 1995, 14, 3311–3318. [Google Scholar]
- Pàmies, O.; Diéguez, M.; Net, G.; Ruiz, A.; Claver, C. Synthesis and Coordination Chemistry of Novel Chiral P,S-Ligands with a Xylofuranose Backbone: Use in Asymmetric Hydroformylation and Hydrogenation. Organometallics 2000, 19, 1488–1496. [Google Scholar] [CrossRef]
- Guimet, E.; Diéguez, M.; Ruiz, A.; Claver, C. Asymmetric Hydrogenation of Prochiral Olefins Catalysed by Furanoside Thioether-Phosphinite Rh(I) and Ir(I) Complexes. Dalton Trans. 2005, 2557–2562. [Google Scholar]
- Evans, D.A.; Michael, F.E.; Tedrow, J.S.; Campos, K.R. Application of Chiral Mixed Phosphorus/Sulfur Ligands to Enantioselective Rhodium-Catalyzed Dehydroamino Acid Hydrogenation and Ketone Hydrosilylation Processes. J. Am. Chem. Soc. 2003, 125, 3534–3543. [Google Scholar]
- Hauptman, E.; Fagan, P.J.; Marshall, W. Synthesis of Novel (P,S) Ligands Based on Chiral Nonracemic Episulfides. Use in Asymmetric Hydrogenation. Organometallics 1999, 18, 2061–2073. [Google Scholar] [CrossRef]
- Dervisi, A.; Koursarou, D.; Ooi, L.; Horton, P.N.; Hursthouse, M.B. Synthesis and Characterization of Benzyl Phosphino-Thioether and -Thiolato Pd(II) Complexes and Their Applications in Suzuki Coupling Reactions. Dalton Trans. 2006, 5717–5724. [Google Scholar]
- Huynh, H.V.; Han, Y.; Ho, J.H.H.; Tan, G.K. Palladium(II) Complexes of a Sterically Bulky, Benzannulated N-Heterocyclic Carbene with Unusual Intramolecular C-H···Pd and Ccarbene···Br Interactions and Their Catalytic Activities. Organometallics 2006, 25, 3267–3274. [Google Scholar] [CrossRef]
- Gu, S.; Xu, H.; Zhang, N.; Chen, W. Triazole-Functionalized N-Heterocyclic Carbene Complexes of Palladium and Platinum and Efficient Aqueous Suzuki-Miyaura Coupling Reaction. Chem. Asian J. 2010, 5, 1677–1686. [Google Scholar] [CrossRef]
- Bedford, R.B.; Blake, M.E.; Butts, C.P.; Holder, D. The Suzuki Coupling of Aryl Chlorides in TBAB-Water Mixtures. Chem. Commun. 2003, 466–467. [Google Scholar]
- Türkmen, H.; Can, R.; Çetinkaya, B. Aqueous-Phase Suzuki-Miyaura Cross-Coupling Reactions Catalyzed by Pd-NHC Complexes. Dalton Trans. 2009, 7039–7044. [Google Scholar]
- Rosner, T.; Bars, J.L.; Pfaltz, A.; Blackmond, D.G. Kinetic Studies of Heck Coupling Reactions Using Palladacycle Catalysts: Experimental and Kinetic Modeling of the Role of Dimer Species. J. Am. Chem. Soc. 2001, 123, 1848–1855. [Google Scholar]
- Huynh, H.V.; Ho, J.H.H.; Neo, T.C.; Koh, L.L. Solvent-Controlled Selective Synthesis of a trans-Configured Benzimidazolin-2-ylidene Palladium(II) Complex and Investigations of Its Heck-Type Catalytic Activity. J. Organomet. Chem. 2005, 690, 3854–3860. [Google Scholar] [CrossRef]
- Müller, T.E.; Hultzsch, K.C.; Yus, M.; Foubelo, F.; Tada, M. Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev. 2008, 108, 3795–3892. [Google Scholar] [CrossRef]
- Müller, T.E.; Beller, M. Metal-Initiated Amination of Alkenes and Alkynes. Chem. Rev. 1998, 98, 675–703. [Google Scholar] [CrossRef]
- Huynh, H.V.; Seow, H.X. Synthesis and Structural Characterization of Palladium Dicarbene Complexes Bearing Labile Co-Ligands. Aust. J. Chem. 2009, 62, 983–987. [Google Scholar] [CrossRef]
- Gischig, S.; Togni, A. PdII Complexes of Tridentate PCP N-Heterocyclic Carbene Ligands: Structural Aspects and Application in Asymmetric Hydroamination of Cyano Olefins. Eur. J. Inorg. Chem. 2005, 4745–4754. [Google Scholar] [CrossRef]
- Dash, C.; Shaikh, M.M.; Butcher, R.J.; Ghosh, P. A Comparison between Nickel and Palladium Precatalysts of 1,2,4-Triazole Based N-Heterocyclic Carbenes in Hydroamination of Activated Olefins. Dalton Trans. 2010, 39, 2515–2524. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yuan, D.; Huynh, H.V. Sulfur-Functionalized N-Heterocyclic Carbene Complexes of Pd(II): Syntheses, Structures and Catalytic Activities. Molecules 2012, 17, 2491-2517. https://doi.org/10.3390/molecules17032491
Yuan D, Huynh HV. Sulfur-Functionalized N-Heterocyclic Carbene Complexes of Pd(II): Syntheses, Structures and Catalytic Activities. Molecules. 2012; 17(3):2491-2517. https://doi.org/10.3390/molecules17032491
Chicago/Turabian StyleYuan, Dan, and Han Vinh Huynh. 2012. "Sulfur-Functionalized N-Heterocyclic Carbene Complexes of Pd(II): Syntheses, Structures and Catalytic Activities" Molecules 17, no. 3: 2491-2517. https://doi.org/10.3390/molecules17032491
APA StyleYuan, D., & Huynh, H. V. (2012). Sulfur-Functionalized N-Heterocyclic Carbene Complexes of Pd(II): Syntheses, Structures and Catalytic Activities. Molecules, 17(3), 2491-2517. https://doi.org/10.3390/molecules17032491