Understanding the Mechanism of the Intramolecular Stetter Reaction. A DFT Study


Abstract
:
1. Introduction
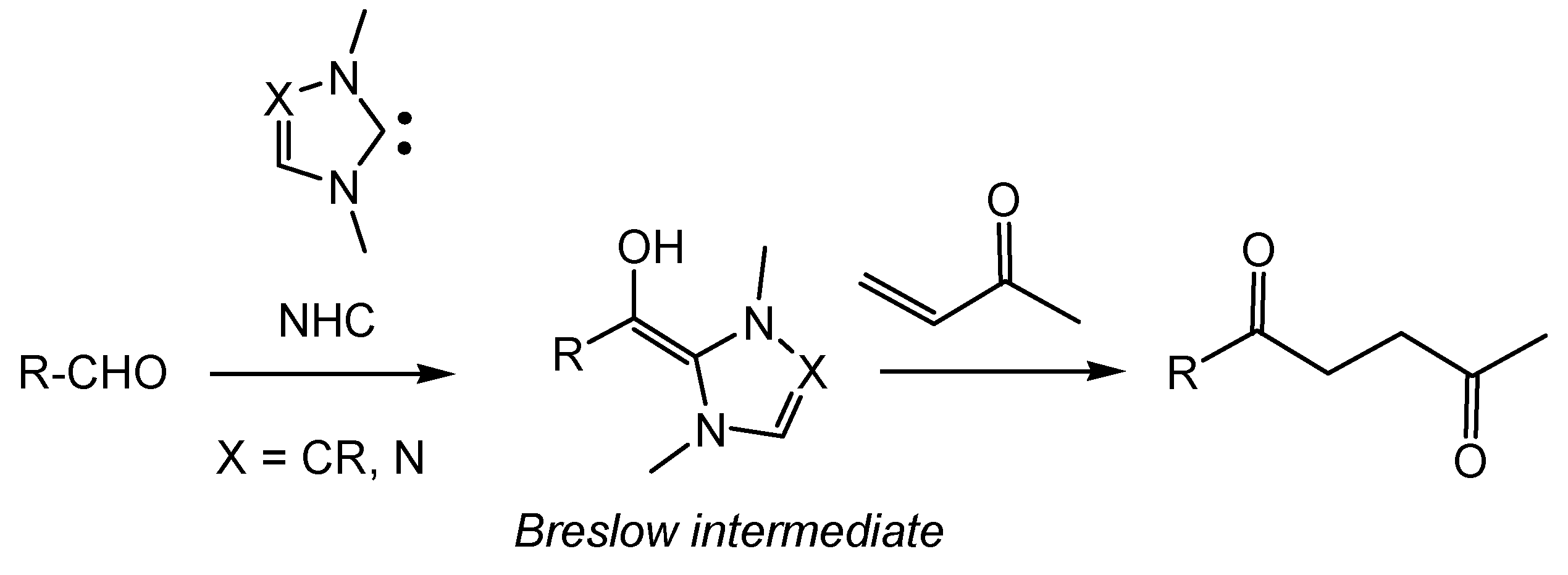
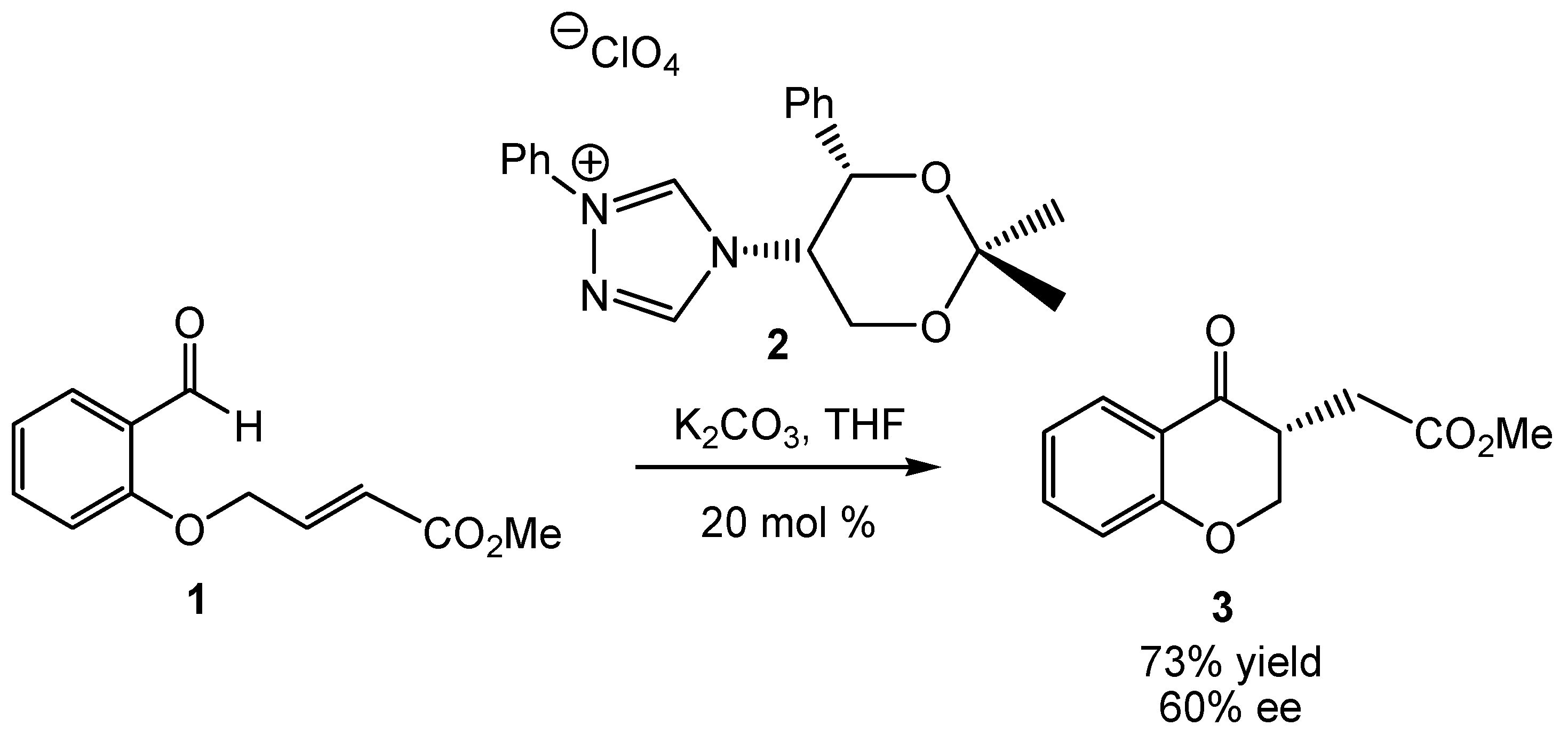

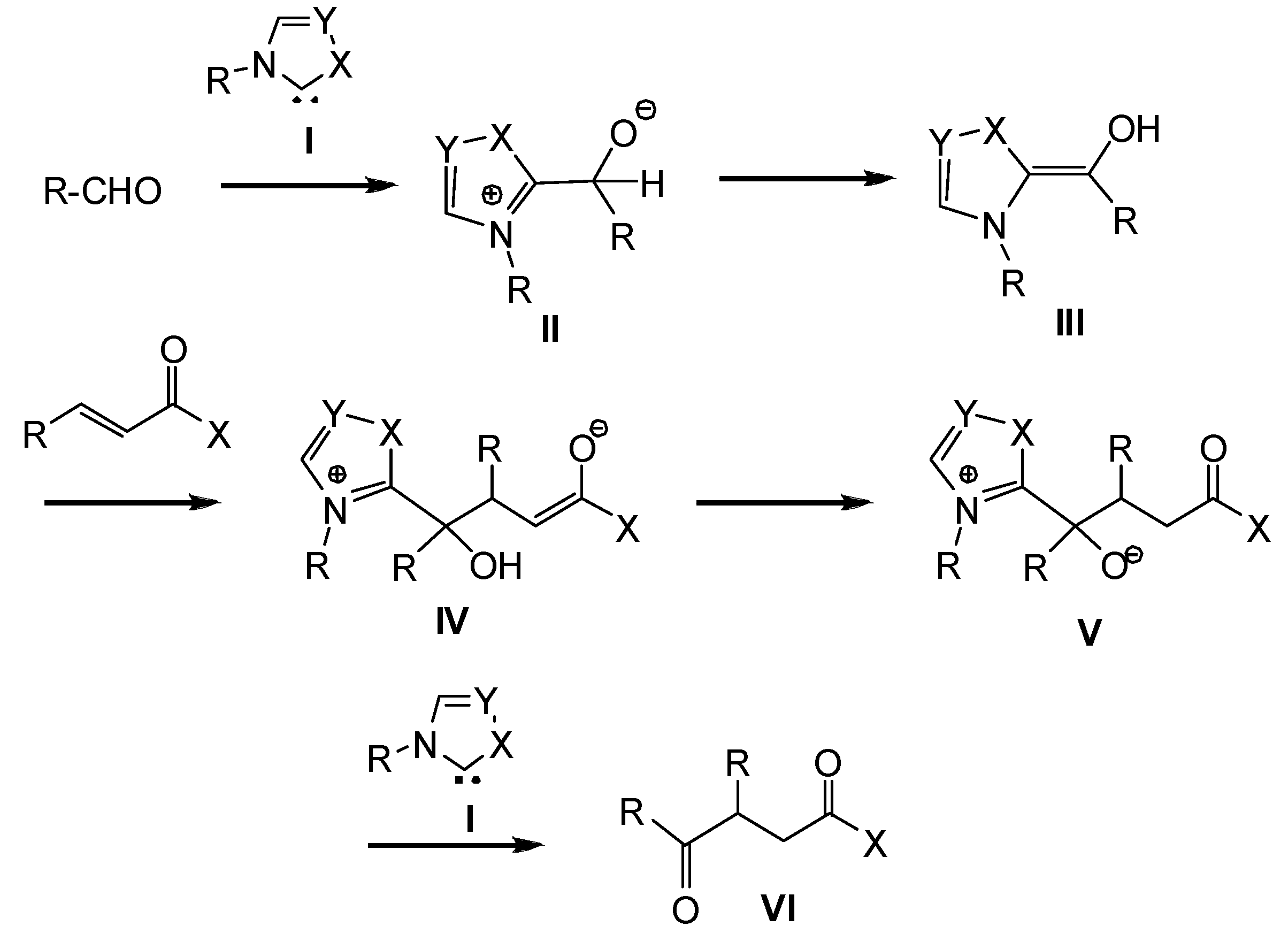

2. Computational Methods

3. Results and Discussion
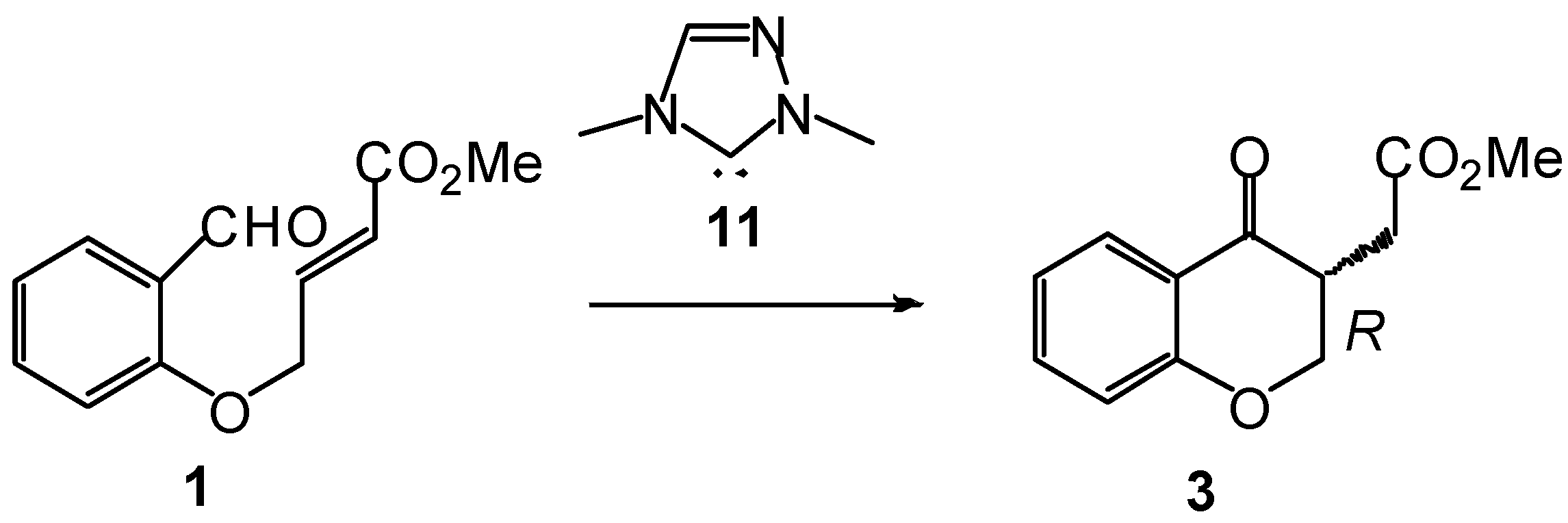
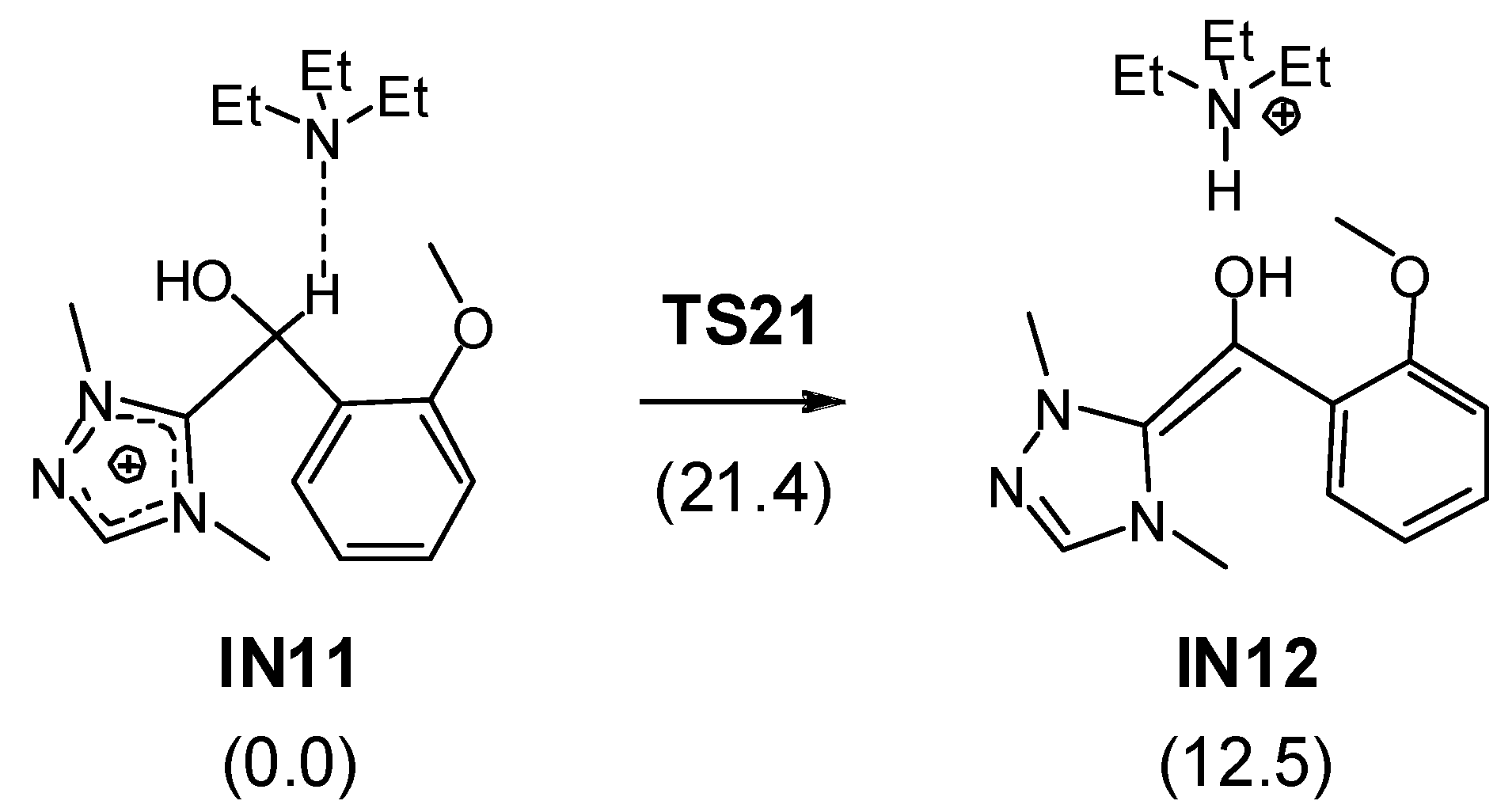
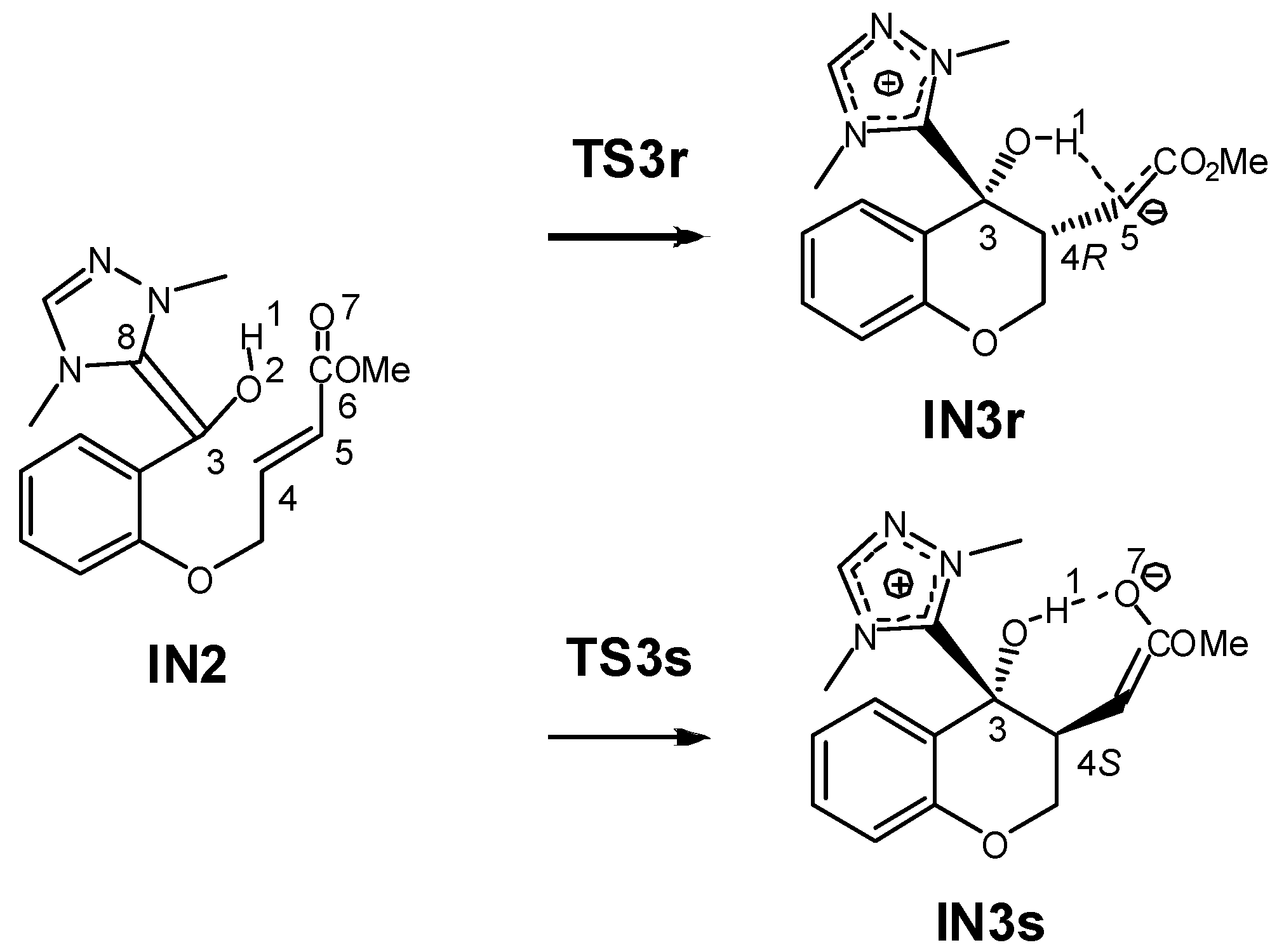
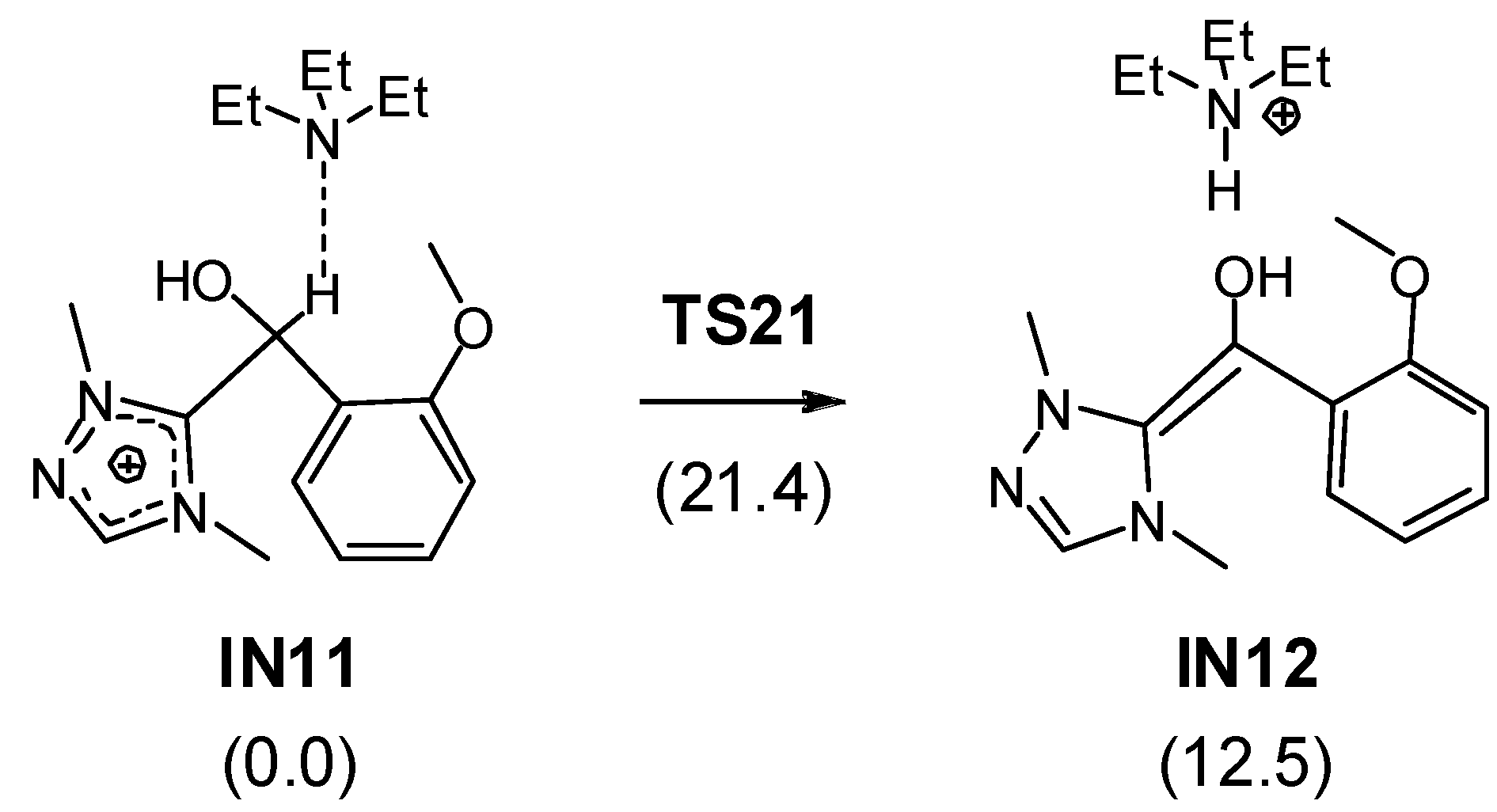
3.1. Energy and Geometrical Analysis of Stationary Points Involved in the Intramolecular Stetter Reaction of Salicylaldehyde 1
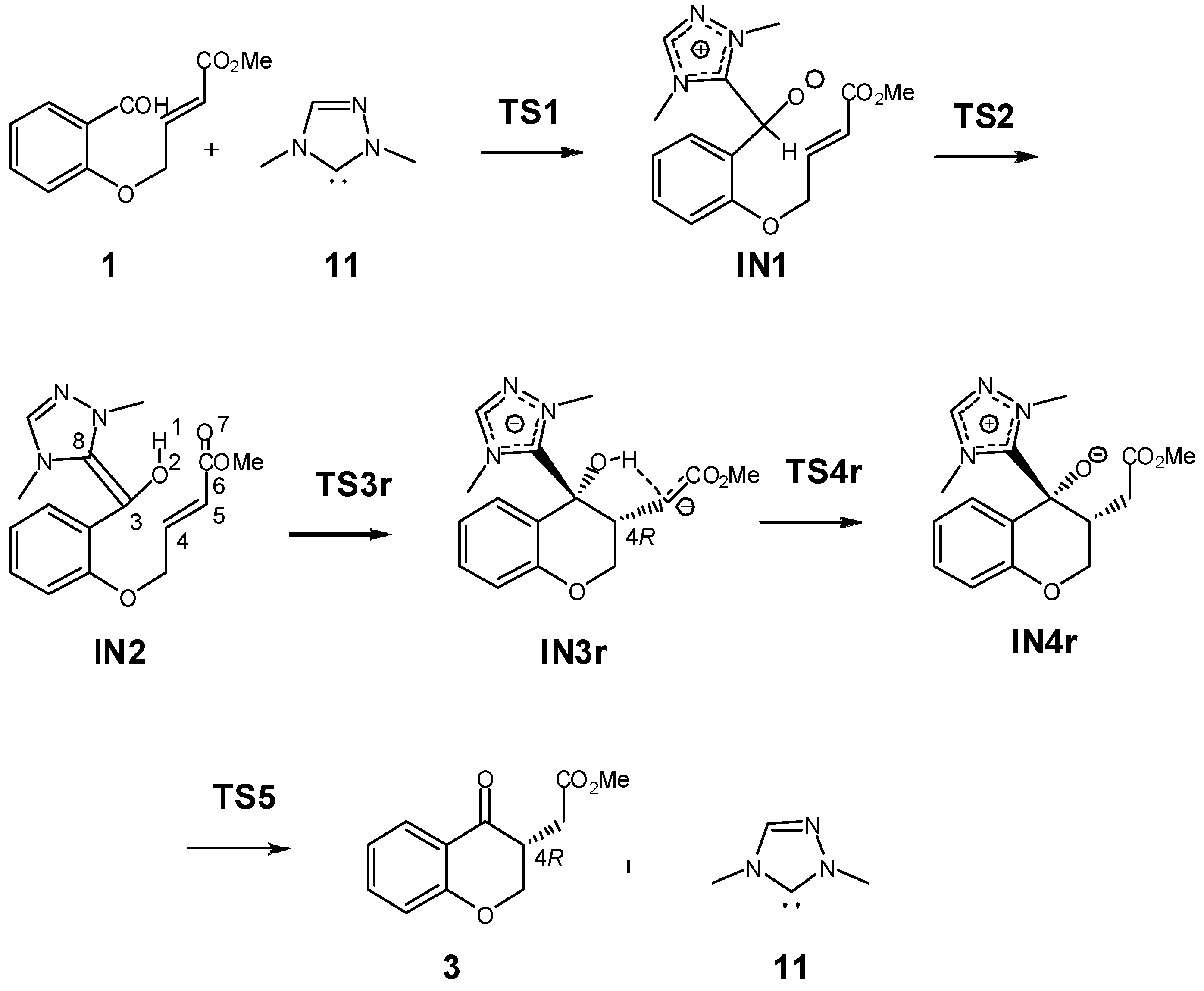

{kind=link}
{kind=link}
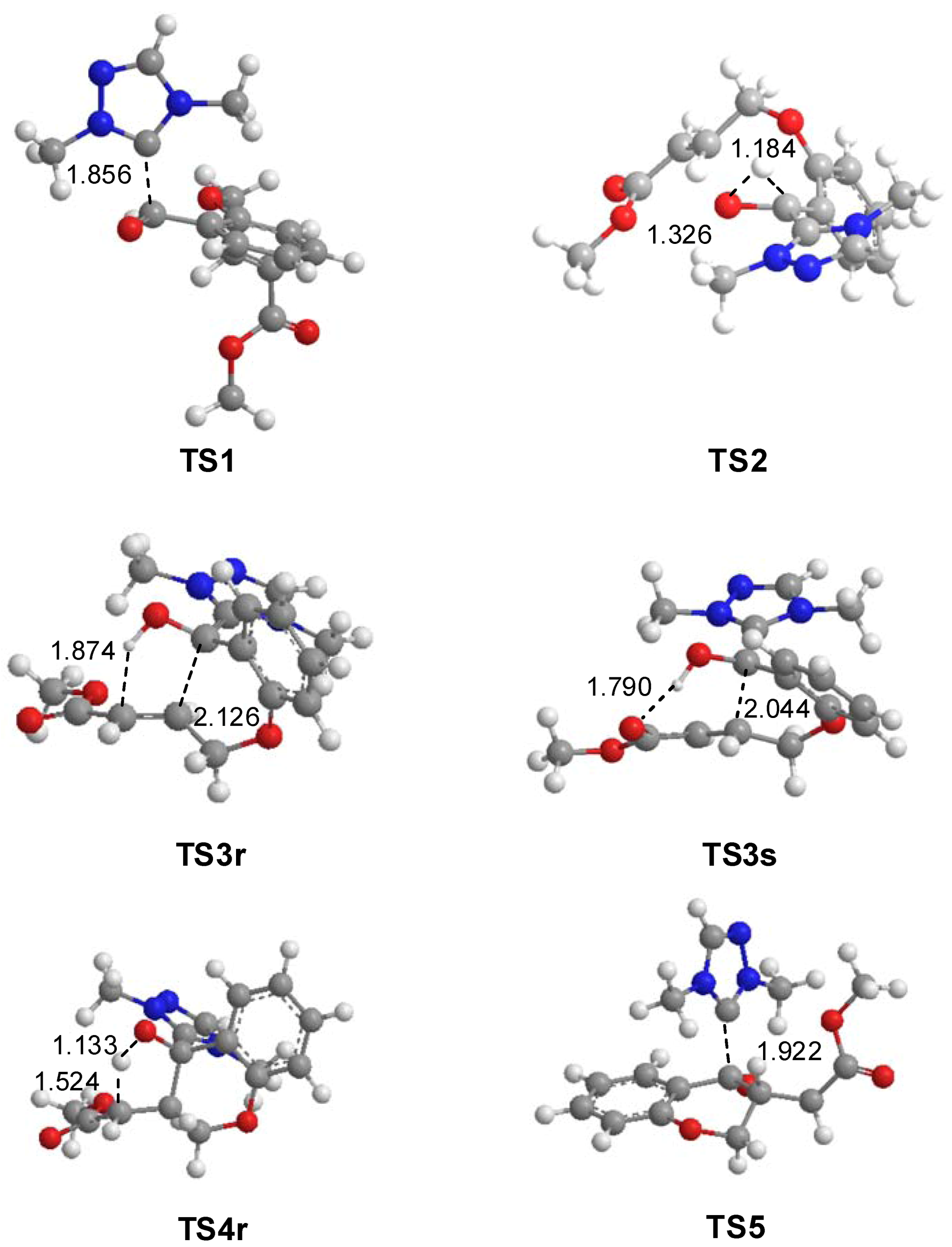{kind=link}
{kind=link}
{kind=link}
{kind=link}
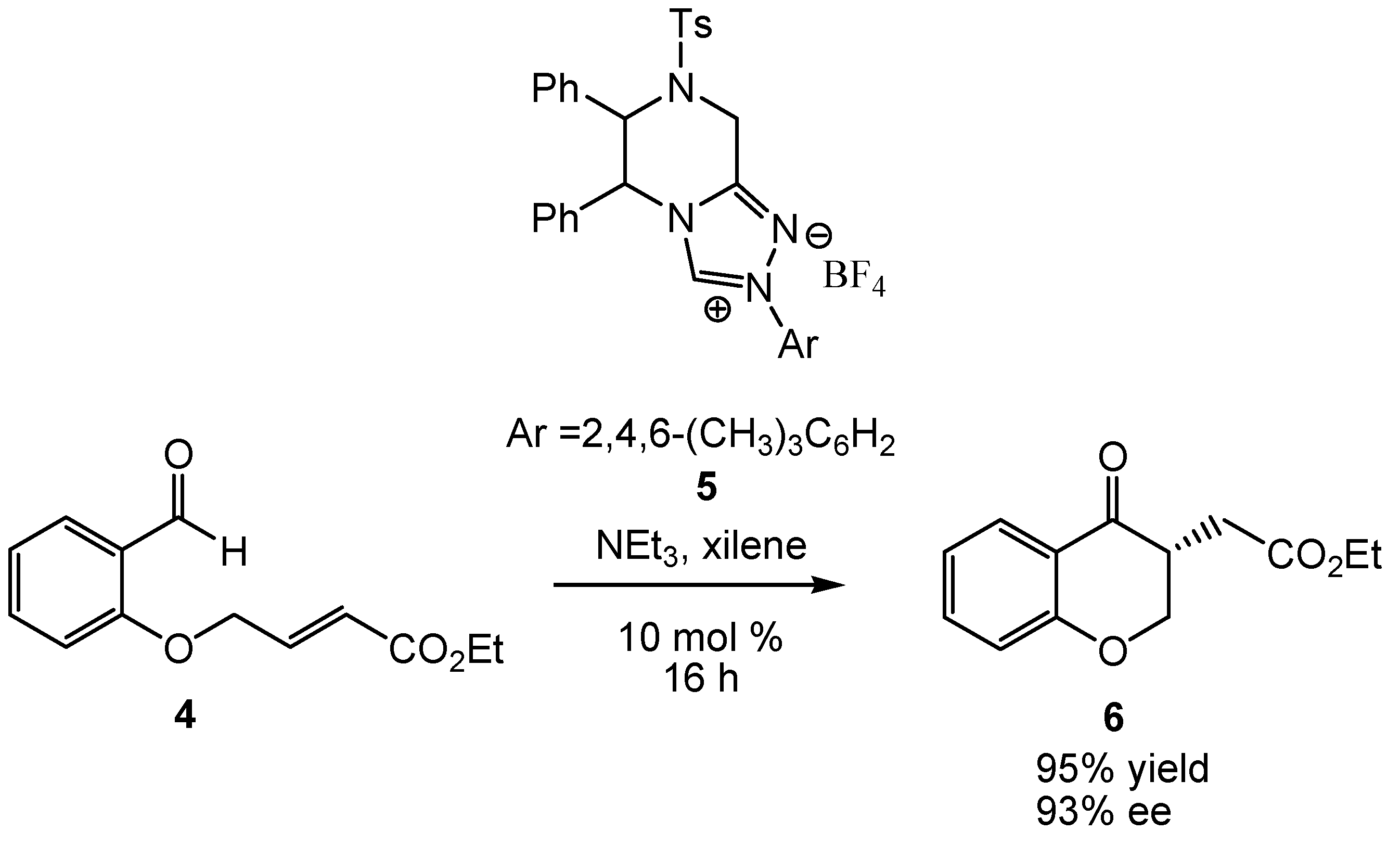{kind=link}
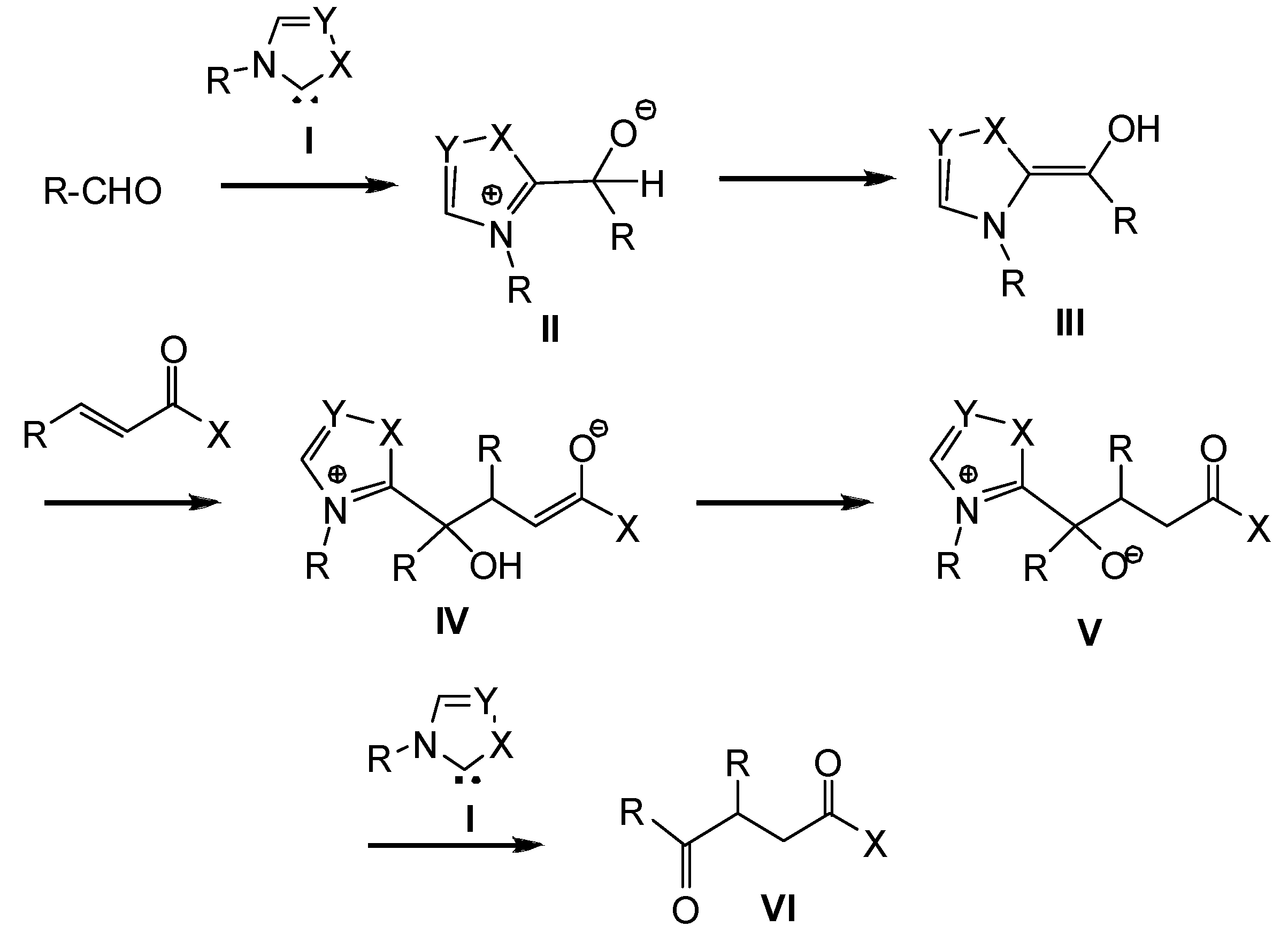{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H | ΔH | G | ΔG | |
|---|---|---|---|---|
| 1 | −765.154909 | −765.216688 | ||
| 11 | −320.722503 | −320.761367 | ||
| TS1 | −1085.864102 | 8.4 | −1085.943426 | 21.7 |
| IN1 | −1085.875090 | 1.5 | −1085.953173 | 15.6 |
| TS2 | −1085.807807 | 43.7 | −1085.885314 | 58.2 |
| IN2 | −1085.879507 | −1.3 | −1085.957544 | 12.9 |
| TS3r | −1085.873415 | 2.5 | −1085.944874 | 20.8 |
| IN3r | −1085.884019 | −4.1 | −1085.955978 | 13.9 |
| TS3s | −1085.870106 | 4.6 | −085.941886 | 22.7 |
| IN3s | −1085.884133 | −4.2 | −1085.954410 | 14.8 |
| TS4r | −1085.885839 | −5.3 | −1085.956900 | 13.3 |
| IN4r | −1085.900792 | −14.7 | −1085.973665 | 2.8 |
| IN4s | −1085.878805 | −0.9 | −1085.949454 | 17.9 |
| TS5 | −1085.896768 | −12.1 | −1085.970371 | 4.8 |
| 3 | −765.194373 | −24.8 | −765.251292 | −21.7 |

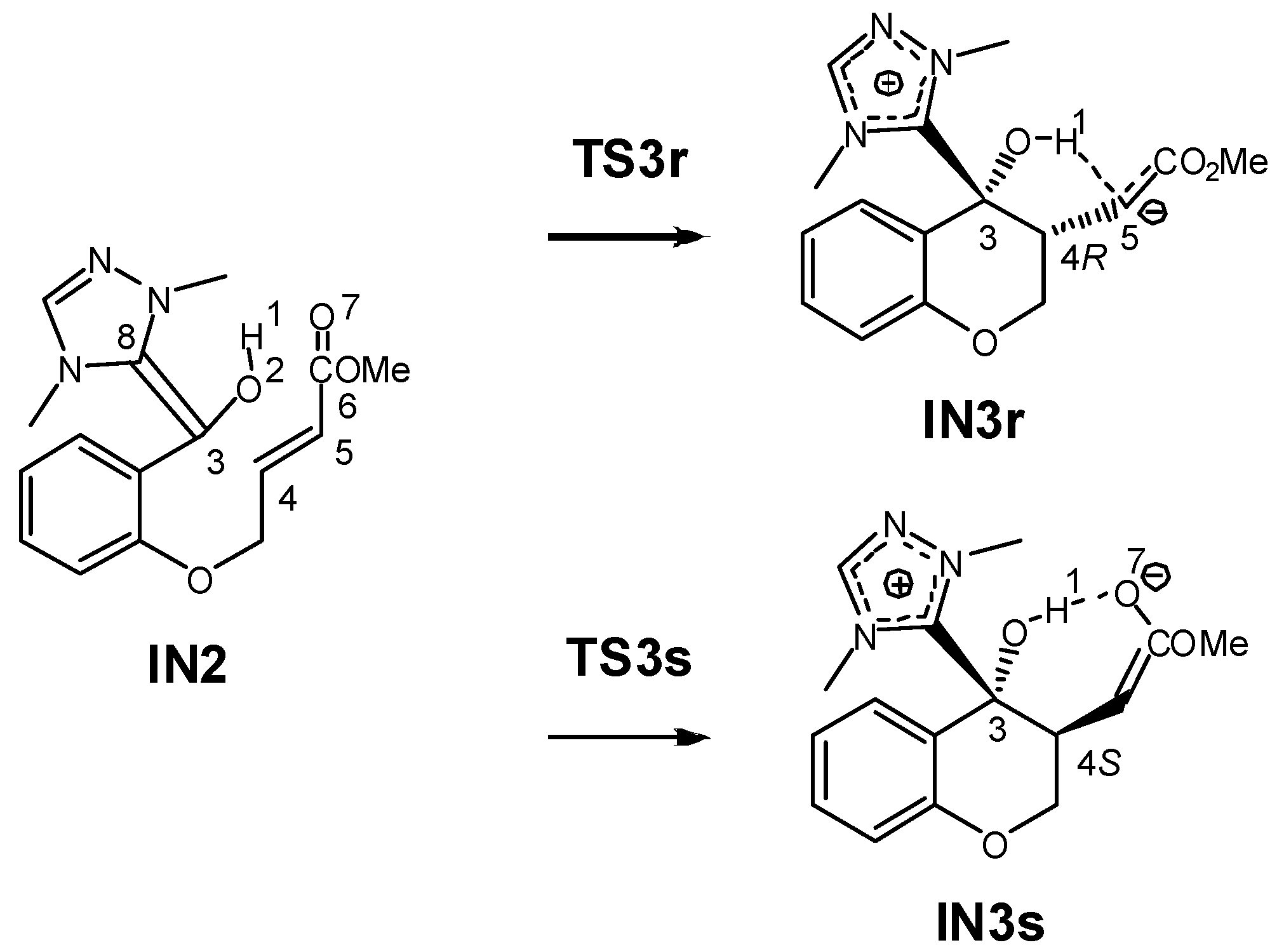

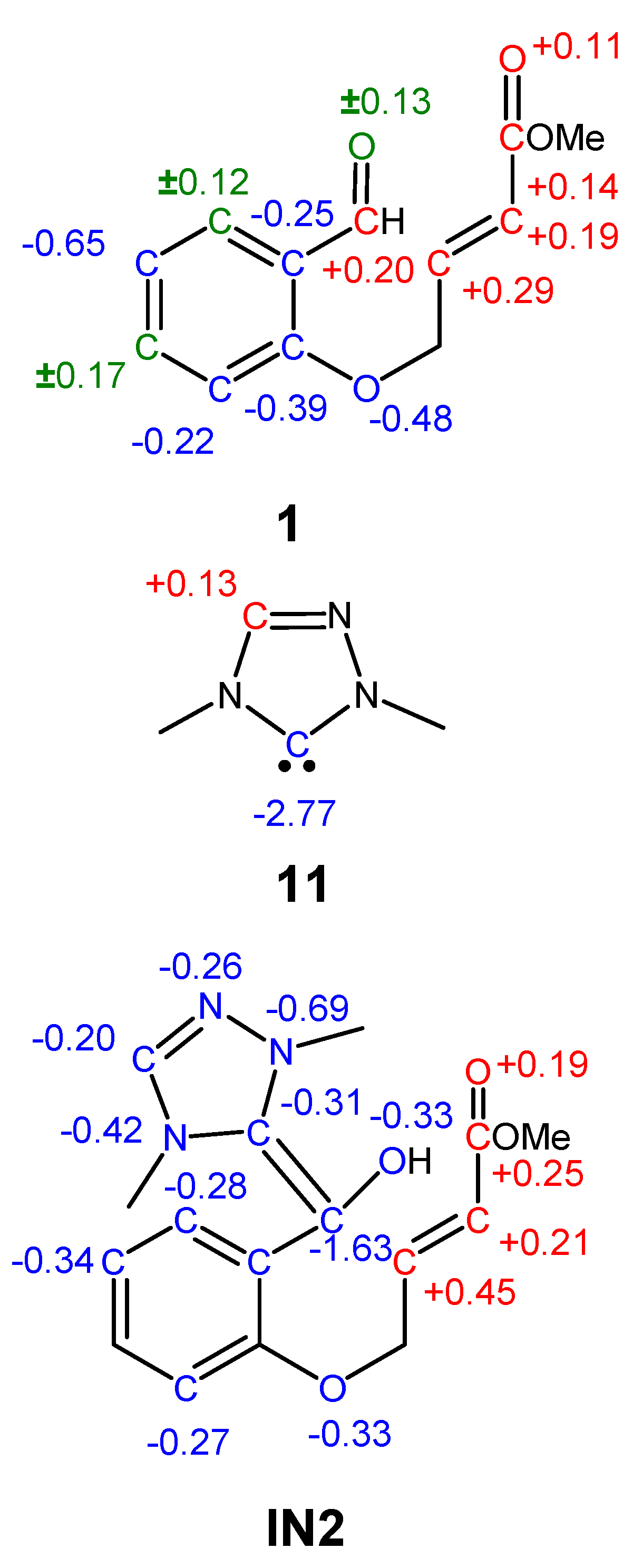
3.2. Analysis of the Reaction Based on DFT Reactivity Indices
| μ | η | ω | N | |
|---|---|---|---|---|
| Benzaldehyde | −0.1590 | 0.1923 | 1.79 | 2.18 |
| 1 | −0.1503 | 0.1752 | 1.75 | 2.65 |
| Methyl acrylate | −0.1586 | 0.2267 | 1.51 | 1.72 |
| IN2 | −0.0975 | 0.0974 | 1.33 | 5.14 |
| 11 | −0.0964 | 0.2334 | 0.54 | 3.32 |

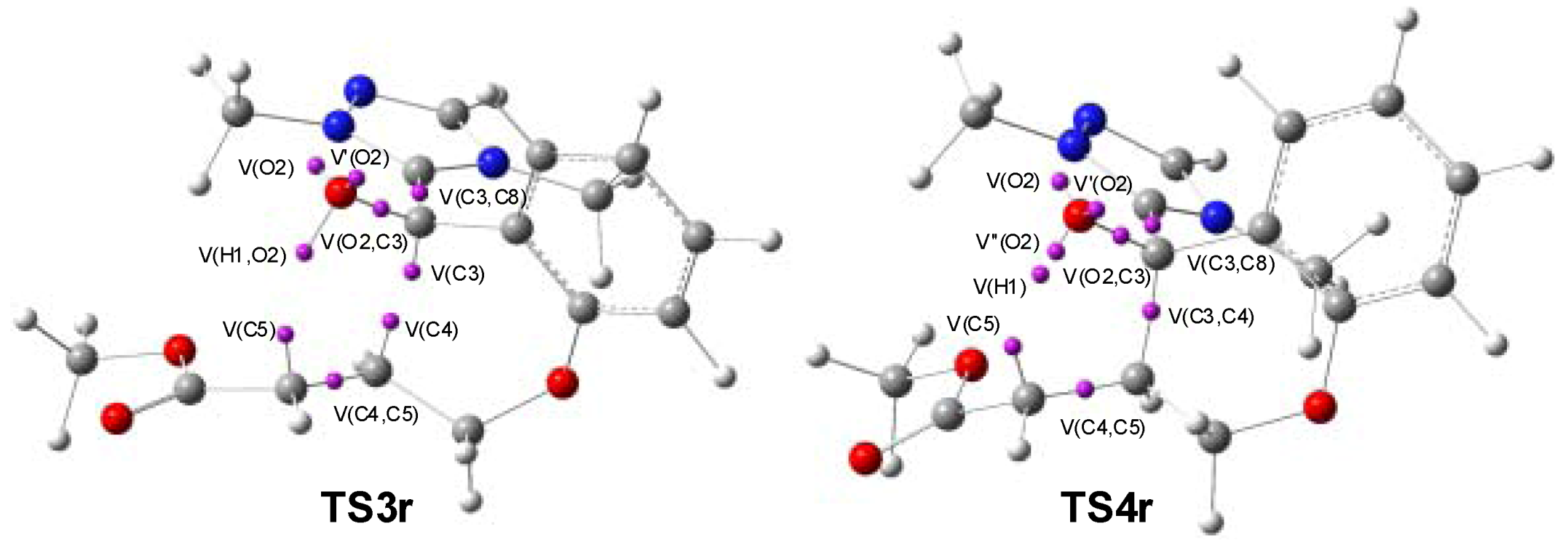
3.3. ELF Bonding Analysis along the Intramolecular Michael Addition in Breslow Intermediate IN2
| IN2 | TS3r | IN3r | TS4r | IN4r | |
|---|---|---|---|---|---|
| V(C3,C8) | 2.01 | 2.85 | 2.45 | 2.44 | 2.44 |
| V’(C3,C8) | 2.20 | ||||
| V(O2) | 2.39 | 2.44 | 2.34 | 2.44 | 2.17 |
| V’(O2) | 2.43 | 2.47 | 2.52 | 2.57 | 3.87 |
| V’’(O2) | 0.94 | ||||
| V(O2,C3) | 1.24 | 1.35 | 1.36 | 1.35 | 1.58 |
| V(C4,C5) | 1.76 | 2.79 | 2.02 | 1.99 | 1.85 |
| V’(C4,C5) | 1.72 | ||||
| V(C3,C4) | 1.04 | 1.85 | 1.86 | 1.96 | |
| V(C4) | 0.34 | ||||
| V(C5) | 0.54 | 1.17 | 1.26 | ||
| V(H1,O2) | 1.71 | 1.65 | 1.61 | ||
| V(H1) | 0.57 | ||||
| V(H1,C5) | 2.01 |
4. Conclusions
Acknowledgments
References and Notes
- Nair, V.; Vellalath, S.; Babu, B.P. Recent advances in carbon-carbon bond-forming reactions involving homoenolates generated by NHC catalysis. Chem. Soc. Rev. 2008, 37, 2691–2698. [Google Scholar]
- Enders, D.; Niemeier, O.; Henseler, A. Organocatalysis by N-heterocyclic carbenes. Chem. Rev. 2007, 107, 5606–5655. [Google Scholar]
- Marion, N.; Díez-González, S.; Nolan, S.P. N-Heterocyclic carbenes as organocatalysts. Angew. Chem. Int. Ed. 2007, 46, 2988–3000. [Google Scholar] [CrossRef]
- Chiang, P.-C.; Bode, J.W. N-Heterocyclic carbenes as organic catalysts. In N-Heterocyclic Carbenes; Díez-González, S., Ed.; The Royal Society of Chemistry: Cambridge, UK, 2011; pp. 399–435, Chapter 14. [Google Scholar]
- Moore, J.L.; Rovis, T. Asymmetric organocatalysis. Top. Curr. Chem. 2011, 291, 77–144. [Google Scholar]
- Vora, H.U.; Rovis, T. Asymmetric N-heterocyclic carbene (NHC) catalyzed acyl anion reactions. Aldrichimica Acta 2010, 44, 3–11. [Google Scholar]
- Enders, D.; Kallfass, U. An efficient nucleophilic carbene catalyst for the asymmetric benzoin condensation. N-Heterocyclic carbene-catalyzed generation of homoenolates: γ-Butyrolactones by direct annulations of enals and aldehydes. Angew. Chem. Int. Ed. 2002, 41, 1743–1745. [Google Scholar]
- Sohn, S.S.; Rosen, E.L.; Bode, J.W. N-Heterocyclic carbene-catalyzed generation of homoenolates: γ-Butyrolactones by direct annulations of enals and aldehydes. J. Am. Chem. Soc. 2004, 126, 14370–14371. [Google Scholar] [CrossRef]
- Burstein, C.; Glorius, F. Organocatalyzed conjugate umpolung of α,β-unsaturated aldehydes for the synthesis of γ-butyrolactones. Angew. Chem. Int. Ed. 2004, 6205–6208. [Google Scholar] [CrossRef]
- Hachisu, Y.; Bode, J.W.; Suzuki, K. Catalytic intramolecular crossed aldehyde-ketonebenzoin reactions: A novel synthesis of functionalized preanthraquinones. J. Am. Chem. Soc. 2003, 125, 8432–8433. [Google Scholar]
- Takikawa, H.; Suzuki, K. Modified chiral triazolium salts for enantioselective benzoin cyclization of enolizable keto-aldehydes: Synthesis of (+)-Sappanone B. Org. Lett. 2007, 9, 2713–2716. [Google Scholar] [CrossRef]
- Takikawa, H.; Hachisu, Y.; Bode, J.W.; Suzuki, K. Catalytic enantioselective crossed aldehyde-ketone benzoin cyclization. Angew. Chem. Int. Ed. 2006, 45, 3492–3494. [Google Scholar] [CrossRef]
- Enders, D.; Niemeier, O.; Balensiefer, T. Asymmetric intramolecular crossed-benzoin reactions by N-heterocyclic carbene catalysis. Angew. Chem. Int. Ed. 2006, 45, 1463–1467. [Google Scholar] [CrossRef]
- Burstein, C.; Tschan, S.; Xie, X.L.; Glorius, F. N-heterocyclic carbene-catalyzed conjugate umpolung for the synthesis of gamma-butyrolactones. Synthesis 2006, 2418–2439. [Google Scholar]
- Schrader, W.; Handayani, P.P.; Burstein, C.; Glorius, F. Investigating organocatalytic reactions: Mass spectrometric studies of a conjugate umpolung reaction. Chem. Commun. 2007, 716–718. [Google Scholar]
- Hirano, K.; Piel, I.; Glorius, F. Diastereoselective synthesis of trifluoromethylated γ-butyrolactones via N-heterocyclic carbene-catalyzed conjugated umpolung of α,β-unsaturated aldehydes. Adv. Synth. Catal. 2008, 350, 984–988. [Google Scholar] [CrossRef]
- Nair, V.; Vellalath, S.; Poonoth, M.; Mohan, R.; Suresh, E. N-Heterocyclic carbene catalyzed reaction of enals and 1,2-dicarbonyl compounds: Stereoselective synthesis of spiro γ-butyrolactones. Org. Lett. 2006, 8, 507–509. [Google Scholar] [CrossRef]
- Chan, A.; Scheidt, K.A. Hydroacylation of activated ketones catalyzed by N-heterocyclic carbenes. J. Am. Chem. Soc. 2006, 128, 4558–4559. [Google Scholar] [CrossRef]
- Mennen, S.M.; Gipson, J.D.; Kim, Y.R.; Miller, S. Thiazolylalanine-derived catalysts for enantioselective intermolecular aldehyde-imine cross-couplings. J. Am. Chem. Soc. 2005, 127, 1654–1655. [Google Scholar]
- Li, G.-Q.; Dai, L.-X.; You, S.-L. Thiazolium-derived carbene-catalyzed cross-coupling of aldehydes with unactivated imines. Chem. Commun. 2007, 852–854. [Google Scholar]
- He, M.; Bode, J.W. Catalytic synthesis of γ-lactams via direct annulations of enals and N-sulfonylimines. Org. Lett. 2005, 7, 3131–3134. [Google Scholar]
- Rommel, M.; Fukuzumi, T.; Bode, J.W. Cyclic ketimines as superior electrophiles for NHC-catalyzed homoenolate additions with broad scope and low catalyst loadings. J. Am. Chem. Soc. 2008, 130, 17266–17267. [Google Scholar]
- Christmann, M. New developments in the asymmetric Stetter reaction. Angew. Chem. Int. Ed. 2005, 44, 2632–2634. [Google Scholar] [CrossRef]
- Read de Alaniz, J.; Rovis, T. The catalytic asymmetric intramolecular Stetter seaction. Synlett 2009, 1189–1207. [Google Scholar]
- Stetter, H. Catalyzed addition of aldehydes to activated double bonds—A new synthetic approach. Angew. Chem. Int. Ed. Engl. 1976, 15, 639–647. [Google Scholar]
- Stetter, H.; Schreckenberg, M. A new method for addition of aldehydes to activated double bonds. Angew. Chem. Int. Ed. Engl. 1973, 12, 81. [Google Scholar]
- Stetter, H.; Kuhlmann, H. The catalyzed nucleophilic addition of aldehydes to electrophilic double bonds. Org. React. 1991, 40, 407–496. [Google Scholar]
- Enders, D.; Breuer, K.; Runsink, J.; Teles, J.H. The first asymmetric intramolecular Stetter reaction. Preliminary communication. Helv. Chim. Acta 1996, 79, 1899–1902. [Google Scholar] [CrossRef]
- Kerr, M.S.; Read de Alaniz, J.; Rovis, T. A highly enantioselective catalytic intramolecular Stetter reaction. J. Am. Chem. Soc. 2002, 124, 10298–10299. [Google Scholar] [CrossRef]
- Read de Alaniz, J.; Rovis, T. A highly enantio- and diastereoselective catalytic intramolecular Stetter reaction. J. Am. Chem. Soc. 2005, 127, 6284–6289. [Google Scholar] [CrossRef]
- Liu, Q.; Rovis, T. Asymmetric synthesis of hydrobenzofuranones via desymmetrization of cyclohexadienones using the intramolecular Stetter reaction. J. Am. Chem. Soc. 2006, 128, 2552–2553. [Google Scholar]
- Read de Alaniz, J.; Kerr, M.S.; Moore, J.L.; Rovis, T. Scope of the asymmetric intramolecular Stetter reaction catalyzed by chiral nucleophilic triazolinylidene carbenes. J. Org. Chem. 2008, 73, 2033–2040. [Google Scholar] [CrossRef]
- DiRocco, D.A.; Rovis, T. Catalytic asymmetric intermolecular Stetter reaction of enals with nitroalkenes: Enhancement of catalytic efficiency through bifunctional additives. J. Am. Chem. Soc. 2011, 133, 10402–10405. [Google Scholar]
- Jia, M.-Q.; Li, Y.; Rong, Z.-Q.; You, S.-L. Synthesis of (1R, 2R)-DPEN-derived triazolium salts and their application in asymmetric intramolecular Stetter reactions. Org. Biomol. Chem. 2011, 9, 2072–2074. [Google Scholar]
- Moore, J.L.; Silvestri, A.P.; Read de Alaniz, J.; DiRocco, D.A.; Rovis, T. Mechanistic investigation of the enantioselective intramolecular Stetter reaction: Proton transfer is the first irreversible step. Org. Lett. 2011, 13, 1742–1745. [Google Scholar]
- Hawkes, K.J.; Yates, B.F. The mechanism of the Stetter reaction—A DFT study. Eur. J. Org. Chem. 2008, 5563–5570. [Google Scholar] [CrossRef]
- He, J.; Tang, S.; Liu, J.; Su, Y.; Pan, X.; She, X. N-Heterocyclic carbene catalyzed intramolecular nucleophilic addition of carbonyl anion equivalents to enol ethers. Tetrahedron 2008, 64, 8797–8800. [Google Scholar]
- Um, J.M.; DiRocco, D.A.; Noey, E.L.; Rovis, T.; Houk, K.N. Quantum mechanical investigation of the effect of catalyst fluorination in the intermolecular asymmetric stetter reaction. J. Am. Chem. Soc. 2011, 133, 11249–11254. [Google Scholar]
- Domingo, L.R.; Aurell, M.J.; Arnó, M. Understanding the mechanism of the N-heterocyclic carbene-catalyzed ring-expansion of 4-formyl-[beta]-lactams to succinimide derivatives. Tetrahedron 2009, 65, 3432–3440. [Google Scholar] [CrossRef]
- Domingo, L.R.; Zaragozá, R.J.; Arnó, M. Understanding the mechanism of stereoselective synthesis of cyclopentenes via N-heterocyclic carbene catalyzed reactions of enals with enones. Org. Biomol. Chem. 2010, 8, 4884–4891. [Google Scholar] [CrossRef]
- Domingo, L.R.; Zaragozá, R.J.; Arnó, M. Understanding the cooperative NHC/LA catalysis for stereoselective annulation reactions with homoenolates. A DFT study. Org. Biomol. Chem. 2011, 9, 6616–6622. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar]
- Schlegel, H.B. Advanced series in physical chemistry. In Modern Electronic Structure Theory; Yarkony, D.R., Ed.; World Scientific Publishing: Singapore, 1994; Volume 2. [Google Scholar]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar]
- Tomasi, J.; Persico, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar]
- Simkin, B.Y.; Sheikhet, I. Quantum Chemical and Statistical Theory of Solutions-A Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar]
- Savin, A.; Becke, A.D.; Flad, J.; Nesper, R.; Preuss, H.; Vonschnering, H.G. A new look at electron localization. Angew. Chem. Int. Ed. Engl. 1991, 30, 409–412. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar]
- Silvi, B. The synaptic order: A key concept to understand multicenter bonding. J. Mol. Struct. 2002, 614, 3–10. [Google Scholar]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian03; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar]
- Domingo, L.R.; Pérez, P. The nucleophilicity N index in organic chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. [Google Scholar]
- Kohn, W.; Sham, L. Self-consistent equations including exchange and correlation effects. J. Phys. Rev. 1965, 140, 1133–1138. [Google Scholar]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the local electrophilicity of organic molecules. Understanding the regioselectivity on Diels-Alder reactions. J. Phys. Chem. 2002, 106, 6871–6875. [Google Scholar]
- Pérez, P.; Domingo, L.R.; Duque-Noreña, M.; Chamorro, E. A condensed-to-atom nucleophilicity index. An application to the director effects on the electrophilic aromatic substitutions. J. Mol. Struct. (Theochem) 2009, 895, 86–91. [Google Scholar]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Contreras, R.; Fuentealba, P.; Galván, M.; Pérez, P. A direct evaluation of regional Fukui functions in molecules. Chem. Phys. Lett. 1999, 304, 405–413. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Duley, S.; Domingo, L.R. Understanding local electrophilicity/nucleophilicity activation through a single reactivity difference index. Org. Biomol. Chem. 2012, in press. [Google Scholar]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Pérez, P. A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J. Mol. Struct. 2008, 865, 68–72. [Google Scholar]
- Soto-Delgado, J.; Domingo, L.R.; Contreras, R. Quantitative characterization of group electrophilicity and nucleophilicity for intramolecular Diels-Alder reactions. Org. Biomol. Chem. 2010, 8, 3678–3683. [Google Scholar] [CrossRef]
- Soto-Delgado, J.; Aizman, A.; Domingo, L.R.; Contreras, R. Invariance of electrophilicity of independent fragments. Application to intramolecular Diels-Alder reactions. Chem. Phys. Lett. 2010, 499, 272–277. [Google Scholar]
- Berski, S.; Andrés, J.; Silvi, B.; Domingo, L.R. The joint use of catastrophe theory and electron localization function to characterize molecular mechanisms. A density functional study of the Diels-Alder reaction. J. Phys. Chem. A 2003, 107, 6014–6024. [Google Scholar]
- Polo, V.; Andrés, J.; Castillo, R.; Berski, S.; Silvi, B. Understanding the molecular mechanism of the 1,3-dipolar cycloaddition between fulminic acid and acetylene in terms of the electron localization function and catastrophe theory. Chem. Eur. J. 2004, 10, 5165–5172. [Google Scholar] [CrossRef]
- Domingo, L.R.; Picher, M.T.; Arroyo, P.; Sáez, J.A. 1,3-Dipolar cycloadditions of electrophilically activated benzonitrile N-oxides. Polar cycloaddition versus oxime formation. J. Org. Chem. 2006, 71, 9319–9330. [Google Scholar]
- Berski, S.; Andrés, J.; Silvi, B.; Domingo, L.R. New findings on the Diels-Alder reactions. An analysis based on the bonding evolution theory. J. Phys. Chem. A 2006, 110, 13939–13947. [Google Scholar]
- Polo, V.; Andrés, J.; Berski, S.; Domingo, L.R.; Silvi, B. Understanding reaction mechanisms in organic chemistry from catastrophe theory applied to the electron localization function topology. J. Phys. Chem. A 2008, 112, 7128–7136. [Google Scholar]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the high reactivity of the azomethineylides in [3+2] cycloaddition reactions. Lett. Org. Chem. 2010, 7, 432–439. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. Understanding the electronic reorganization along the nonpolar [3+2] cycloaddition reactions of carbonyl ylides. J. Org. Chem. 2011, 76, 373–379. [Google Scholar]
- Sample Availability: Cartesian coordinates of all stationary points are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Domingo, L.R.; Zaragozá, R.J.; Saéz, J.A.; Arnó, M. Understanding the Mechanism of the Intramolecular Stetter Reaction. A DFT Study. Molecules 2012, 17, 1335-1353. https://doi.org/10.3390/molecules17021335
Domingo LR, Zaragozá RJ, Saéz JA, Arnó M. Understanding the Mechanism of the Intramolecular Stetter Reaction. A DFT Study. Molecules. 2012; 17(2):1335-1353. https://doi.org/10.3390/molecules17021335
Chicago/Turabian StyleDomingo, Luis R., Ramón J. Zaragozá, Jose A. Saéz, and Manuel Arnó. 2012. "Understanding the Mechanism of the Intramolecular Stetter Reaction. A DFT Study" Molecules 17, no. 2: 1335-1353. https://doi.org/10.3390/molecules17021335
APA StyleDomingo, L. R., Zaragozá, R. J., Saéz, J. A., & Arnó, M. (2012). Understanding the Mechanism of the Intramolecular Stetter Reaction. A DFT Study. Molecules, 17(2), 1335-1353. https://doi.org/10.3390/molecules17021335