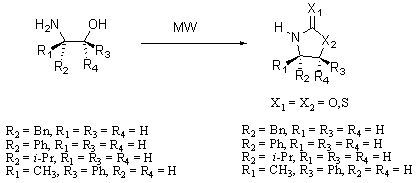
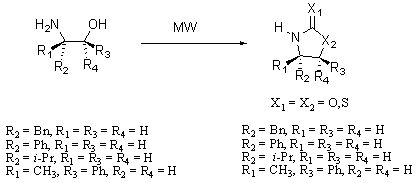
3.2. Synthesis of Oxazolidin-2-ones 2a-d
The equivalents of amino alcohols
1a-d, base and ethyl carbonate were almost the same as the ones reported in the literature [
1,
2,
3]. The reagents were placed in a 10 mL vessel in the BenchMate mode. The products were purified by column chromatography with a hexane-ethyl acetate solvent system.
(S)-4-Benzyl-1,3-oxazolidin-2-one (
2a). The aminoalcohol
1a (1.00 g, 6.61 mmol, 1.0 eq.), diethyl carbonate (1.17 g, 1.2 mL, 9.92 mmol, 1.5 eq.) and sodium methoxide (0.017 g, 0.33 mmol, 0.05 eq.) were placed in a 10 mL vessel in the microwave system and reacted under the BenchMate modality during 20 minutes at 135 °C and 100 W of power. When the reaction was completed, the resulting mixture was partitioned with a mixture of CH
2Cl
2 (15 mL) and H
2O (20 mL). The organic layer was separated and the aqueous layer was extracted with CH
2Cl
2 (2 × 15 mL). The organic extracts were dried over Na
2SO
4 and the solvent evaporated to furnish a residue which was purified through a silica gel chromatographic column eluting with hexane-ethyl acetate (3:2) to give 1.12 g (96% yield) of
2a as a slightly yellow solid. The spectroscopic data were compared with those reported in the literature [
10,
11]: m. p. = 84.5–86.5 °C (Lit. = 87–88 °C), [α]
D = −62.5 c = 1 in CHCl
3 ([α]
D(Lit.) = −63 c = 1 in CHCl
3), e.e. = 99%.
1H-NMR (400 MHz, CDCl
3): 7.36–7.17 (m, 5H), 5.69 (br, s, 1H), 4.45 (t, 1H,
J = 8.4 Hz), 4.15 (t, 1H,
J = 8.4 Hz), 4.09 (m, 1H), 2.88 (d, 2H,
J = 7.2 Hz).
13C-NMR (100 MHz, CDCl
3): 159.3, 127.4, 69.8, 54.0, 41.7 Anal. Calcd. for C
10H
11NO
2: C, 67.77; H, 6.20; N, 7.90. Found: C, 67.50; H, 6.13; N, 7.71.
(S)-4-Phenyl-1,3-oxazolidin-2-one (
2b). The aminoalcohol
1b (1.00 g, 7.29 mmol, 1.0 eq.), diethyl carbonate (1.80 g, 1.8 mL, 15.31 mmol, 2.1 eq.) and potassium carbonate (0.15 g, 1.09 mmol, 0.15 eq.) of were placed in a 10 mL vessel in the microwave system and reacted under the BenchMate modality for 20 minutes at 125 °C and 125 W of power. When the reaction was completed, the resulting mixture was dissolved in CH
2Cl
2 (30 mL) and the insoluble K
2SO
3 were filtered off with suction and brine (15 mL) was added to the organic phase. The organic layer was separated and the aqueous layer was extracted with CH
2Cl
2 (2 × 15 mL). The organic extracts were dried over Na
2SO
4 and the solvent evaporated to furnish a residue which was purified through a silica gel chromatographic column eluting with hexane-ethyl acetate (3:2) to give 1.16 g (98% yield) of
2b as a white solid. The spectroscopic data were compared with those reported in the literature [
12]: m. p. = 131–133 °C (Lit. = 129–132 °C), [α]
D = + 46.1 c=2 in CHCl
3 ([α]
D(Lit.) = +48 c=2 in CHCl
3), e.e. = 96%.
1H-NMR (200 MHz, CDCl
3): 7.33–7.23 (m, 5H), 6.44 (br, s, 1H), 4.88 (t, 1H,
J = 7.7 Hz), 4.64 (t, 1H,
J = 8.6 Hz), 4.08 (td, 1H,
J = 8.2, 1.2 Hz.
13C-NMR (50 MHz, CDCl
3): 160.4, 139.6, 129.2, 128.7, 126.0, 72.6, 56.5 Anal. Calcd. for C
9H
9NO
2: C, 66.27; H, 5.50; N, 8.58. Found: C, 66.15; H, 5.80; N, 8.64.
(S)-4-Isopropyl-1,3-oxazolidin-2-one (
2c). The aminoalcohol
1c (1.00 g, 9.69 mmol, 1.0 eq.), diethyl carbonate (1.71 g, 1.7 mL, 14.54 mmol, 1.5 eq.)and sodium methoxide (0.026 g, 0.48 mmol, 0.05 eq.) were placed in a 10 mL vessel in the microwave system and reacted under the BenchMate modality for 20 minutes at 135 °C and 125 W of power. When the reaction was completed, the resulting mixture was partitioned with a mixture of CH
2Cl
2 (10 mL) and H
2O (20 mL). The organic layer was separated and the aqueous layer was extracted with CH
2Cl
2 (2 × 10 mL). The organic extracts were dried over Na
2SO
4 and the solvent evaporated to furnish a residue which was purified through a silica gel chromatographic column eluting with hexane-ethyl acetate (3:2) to give 1.17 g (94% yield) of
2c as a slightly yellow solid. The spectroscopic data were compared with those reported in the literature [
13]: m. p. = 71.5–73 °C (Lit. = 70–73 °C), [α]
D = −18 c = 6 in EtOH ([α]
D(Lit.) = −17.0 c = 6 in EtOH), e.e. = 95%.
1H-NMR (400 MHz, CDCl
3): 7.18 (br, s, 1H), 4.44 (dd, 1H,
J = 8.8, 8.8 Hz), 4.10 (dd, 1H,
J = 8.8, 6.8 Hz), 3.62 (dddd, 1H,
J = 8.8, 6.8, 2.4, 0.8 Hz), 1.72 (dh, 1H,
J = 6.8, 6.8 Hz), 0.96 (d, 3H,
J = 6.8 Hz), 0.90 (d, 3H,
J = 6.8 Hz).
13C-NMR (100 MHz, CDCl
3): 160.0, 68.6, 58.6, 32.8, 18.1, 17.7 Anal. Calcd. for C
6H
11NO
2: C, 55.79; H, 8.57; N, 10.84. Found: C, 55.70; H, 8.49; N, 10.81.
(4R, 5S)-(+)-4-methyl-5-phenyl-1,3-oxazolidin-2-one (
2d). The aminoalcohol
1d (0.50 g, 3.30 mmol, 1.0 eq.), diethyl carbonate (0.58 g, 0.60 mL, 4.95 mmol, 1.5 eq.) and sodium methoxide (0.008 g, 0.16 mmol, 0.05 eq.) were placed in a 10 mL vessel in the microwave system and reacted under the BenchMate modality during 15 minutes at 135 °C and 145 W of power. When the reaction was completed, the resulting mixture was partitioned with a mixture of CH
2Cl
2 (20 mL) and H
2O (20 mL). The organic layer was separated and the aqueous layer was extracted with CH
2Cl
2 (2 × 10 mL). The organic extracts were dried over Na
2SO
4 and the solvent evaporated to furnish a residue which was purified through a silica gel chromatographic column eluting with hexane-ethyl acetate (3:2) to give 0.50 g (87% yield) of
2d as a white solid. The spectroscopic data were compared with those reported in the literature [
9,
10]: m. p. = 119–120 °C (Lit. = 121–123 °C), [α]
D = +162 c = 1.9 in CHCl
3 ([α]
D(Lit.) = +168 c = 2 in CHCl
3), e.e. = 96%.
1H-NMR (200 MHz, CDCl
3): 7.40–7.26 (m, 5H), 6.25 (br, s, 1H), 5.71 (d, 1H,
J = 8.0 Hz), 4.21 (m, 1H), 0.81 (d, 3H,
J = 6.6 Hz).
13C-NMR (50 MHz, CDCl
3): 159.6, 135.0, 128.6, 126.0, 81.2, 52.6, 17.8 Anal. Calcd. for C
10H
11NO
2: C, 67.77; H, 6.20; N, 7.90. Found: C, 67.61; H, 6.18; N, 7.82.
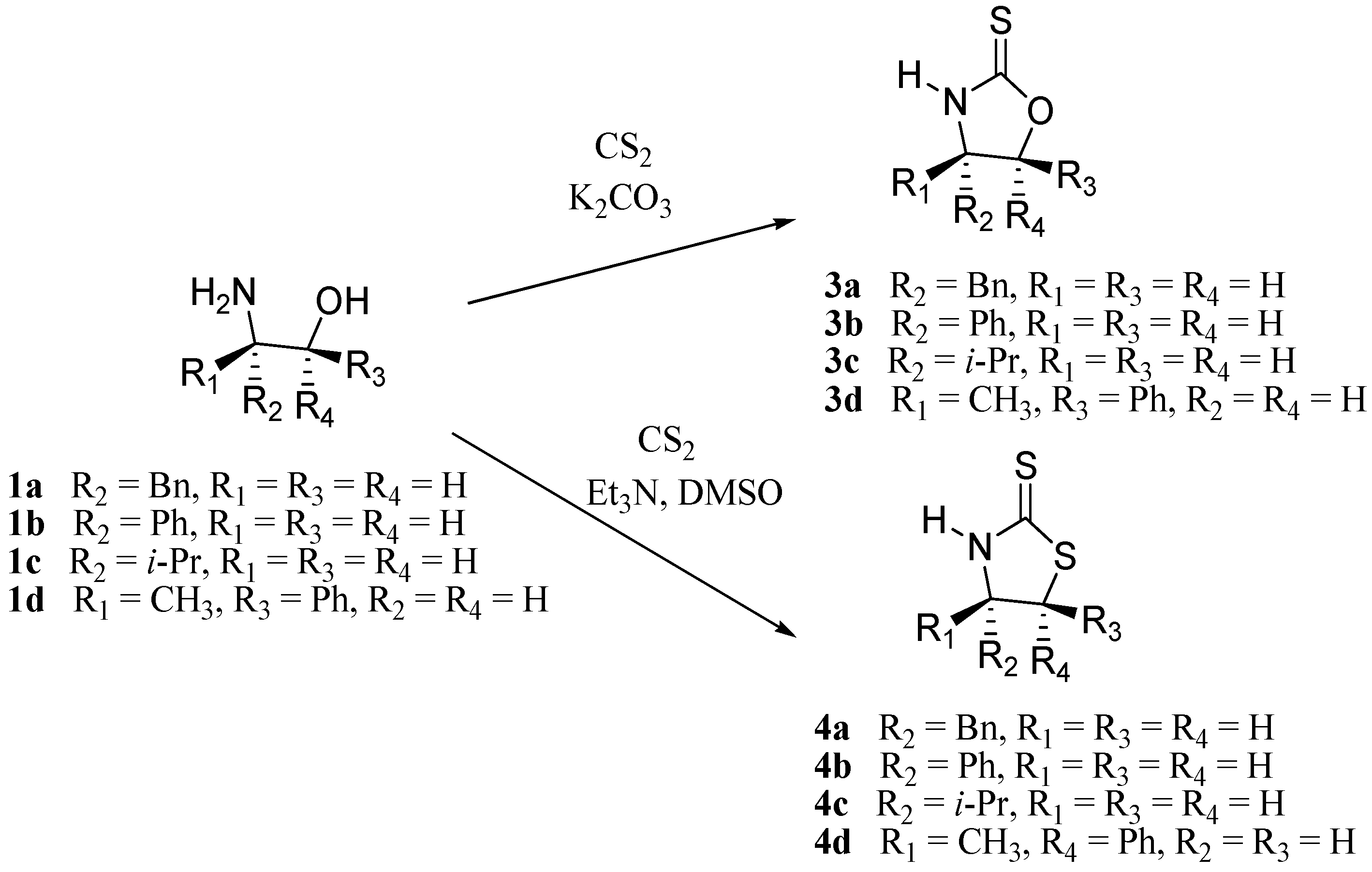
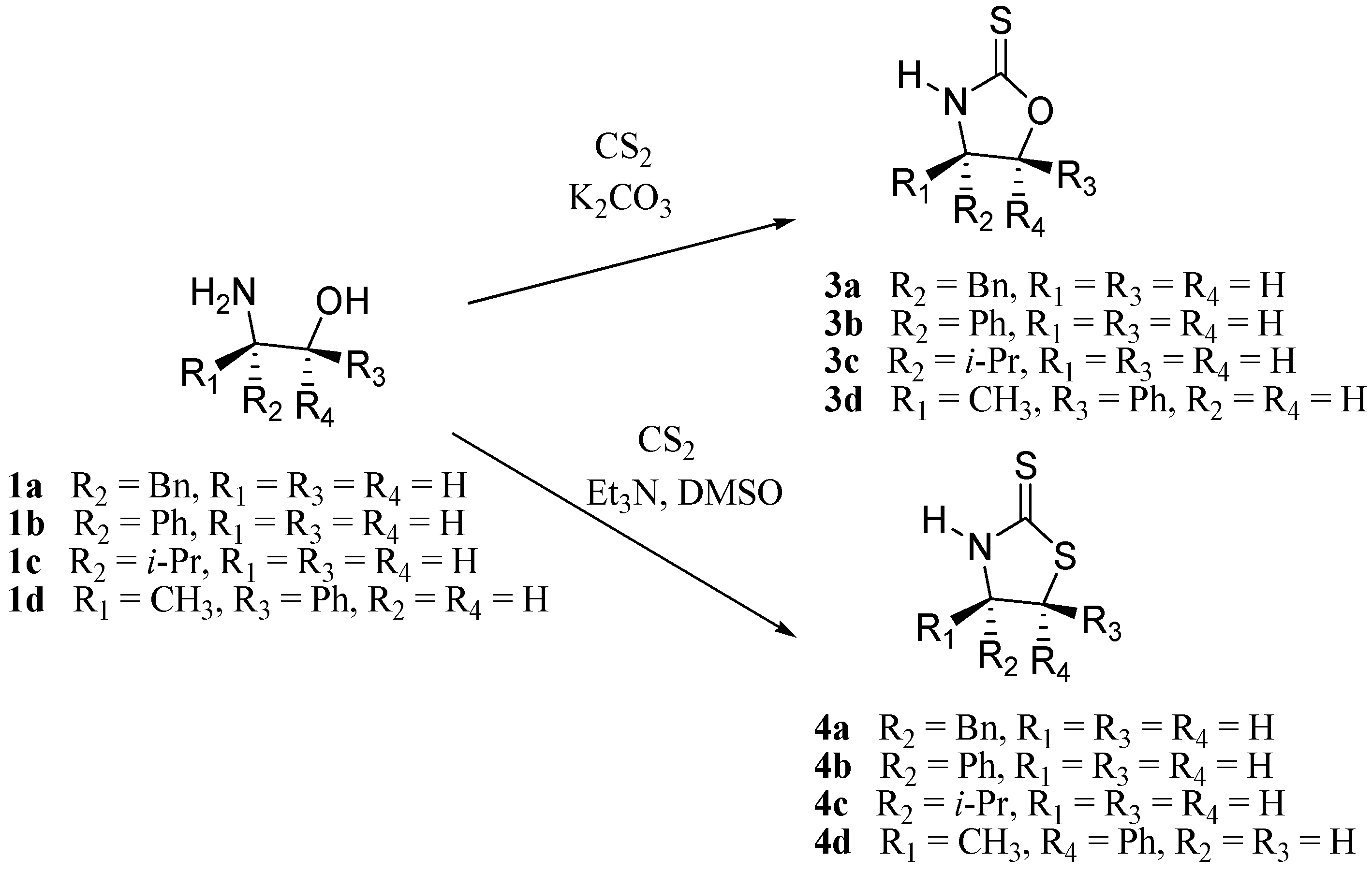
3.3. Synthesis of Oxazolidine-2-thiones 3a-d
In this case were used two different procedures. Compounds
3a and
3b were prepared using the same reagents and proportions reported by Wu. [
16] Compounds
3c-d were prepared according to the method described by LeCorre [
17] with minor variations.
(S)-4-Benzyl-1,3-oxazolidine-2-thione (
3a). In a 50 mL vessel provided with a condenser, the amino alcohol
1a (1.00 g, 6.61 mmol, 1.0 eq.), K
2CO
3 (0.45 g, 3.30 mmol, 0.5 eq.), commercially available anhydrous ethanol (5 mL) and CS
2 (1.00 g, 13.23 mmol, 0.8 mL, 2.0 eq.) were placed in the reactor under the Open Vessel modality with 50 °C and 50 W of power during 15 seconds. When the reaction system reached a 50 °C temperature, the microwave irradiation was stopped and a 35% H
2O
2 aq. solution (0.96 g, 9.92 mmol, 0.85 mL, 1.5 eq.) were added from the top of the condenser taking care the temperature of the reaction system did not overpassed from 50 °C. When the addition was completed, the insoluble K
2SO
3 were filtered off with suction. The filtrate was diluted with EtOAc (50 mL), washed with water (3 × 15 mL) and brine (3 × 15 mL). The organic layer was separated, dried over Na
2SO
4 and evaporated to yield a residue which was purified through a silica gel chromatographic column eluting with a hexane-ethyl acetate solvent system (6:4) to give 1.26 g of
3a (99% yield) obtained as an oil. The spectroscopic data were compared with those reported in the literature [
17]: [α]
D = −91.4 c = 1.87 in CHCl
3 ([α]
D(Lit.) = −93.03 c= 1.88 in CHCl
3), e.e. = 98%.
1H NMR (200 MHz, CDCl
3): 7.98 (br, s, 1H), 7.39–7.15 (m, 5H), 4.68 (dd, 1H,
J = 8.5, 8.5 Hz), 4.35 (m, 2H,
J = 8.4 Hz), 2.92 (d, 2H,
J = 6.6 Hz).
13C NMR (50 MHz, CDCl
3): 189.5, 135.2, 129.2, 129.0, 127.6, 74.9, 58.0, 40.6 Anal. Calcd. for C
10H
11NOS: C, 62.19; H, 5.69; N, 7.25. Found: C, 61.92; H, 5.60; N, 7.23.
(S)-4-Phenyl-1,3-oxazolidine-2-thione (
3b). In a 50 mL vessel provided with a condenser, the amino alcohol
1b (1.0 g, 7.28 mmol, 1.0 eq.), K
2CO
3 (0.50 g, 3.64 mmol, 0.5 eq.), commercially available anhydrous ethanol (5 mL) and CS
2 (1.11 g, 14.57 mmol, 0.9 mL, 2.0 eq.) were placed in the reactor under the Open Vessel modality with 50 °C and 50 W of power during 15 seconds. When the reaction system reached a 50 °C temperature, the microwave irradiation was stopped and a 35% H
2O
2 aq. solution (0.37 g, 10.92 mmol, 0.96 mL, 1.5 eq.) were added from the top of the condenser taking care the temperature of the reaction system did not overpassed from 50 °C. When the addition was completed, the insoluble K
2SO
3 were filtered off with suction. The filtrate was diluted with EtOAc (50 mL), washed with water (3 × 15 mL) and brine (3 × 15 mL). The organic layer was separated, dried over Na
2SO
4 and evaporated to yield a residue which was purified through a silica gel chromatographic column eluting with a hexane-ethyl acetate solvent system (6:4) to give 1.29 g of
3b (99% yield) obtained as a slightly yellow solid. The spectroscopic data were compared with those reported in the literature [
16,
17]: m. p.= 118–120 °C, (Lit. = 121–122 °C), [α]
D = +76.02 c = 0.2 in CHCl
3 ([α]
D(Lit.) = +77 c = 0.2 in CHCl
3), o. p. = 97%.
1H NMR (200 MHz, CDCl
3): 8.23 (br, s, 1H), 7.35–7.19 (m, 5H), 5.04 (dd, 1H,
J = 6.6, 9.2 Hz), 4.92 (t, 1H,
J = 9.2 Hz), 4.40 (dd, 1H,
J = 6.6, 8.4 Hz).
13C NMR (50 MHz, CDCl
3): 189.8, 138.0, 129.4, 129.2, 126.3, 77.8, 60.3 Anal. Calcd. for C
9H
9NOS: C, 60.30; H, 5.02; N, 7.81. Found: C, 60.27; H, 5.01; N, 7.69.
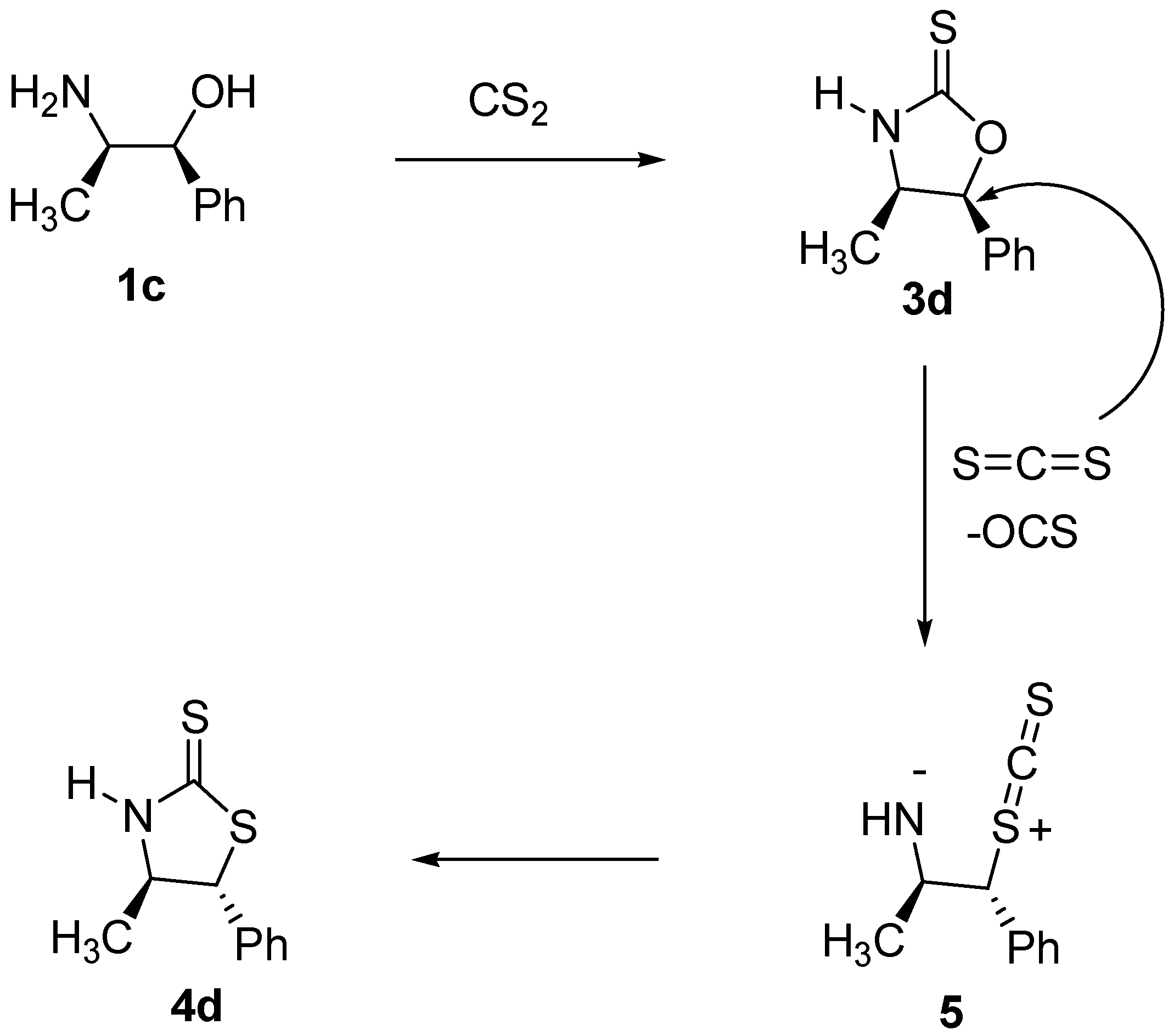
(S)-4-Isopropyl-1,3-oxazolidine-2-thione (
3c). In a 25 mL vessel, the amino alcohol
1c (0.50 g, 4.48 mmol, 1.0 eq.), K
2CO
3 (0.66 g, 4.48 mmol, 1.0 eq.) and CS
2 (0.55 g, 7.27 mmol, 0.43 mL, 1.5 eq.) were placed in the reactor
. The reaction mixture was stirred in the BenchMate modality with 50 °C and 50 W of power for 10 min. When the reaction was completed, the resulting mixture was partitioned with a mixture of CH
2Cl
2 (15 mL) and H
2O (20 mL). The organic layer was separated and the aqueous layer was extracted with CH
2Cl
2 (2 × 15 mL). The organic extracts were dried over Na
2SO
4 and the solvent evaporated to furnish a residue which was purified through a silica gel chromatographic column eluting with a hexane-ethyl acetate solvent system (65:35) to give 0.57 g of
3c (89% yield) obtained as a slightly yellow solid. The spectroscopic data were compared with those reported in the literature [
17]: m. p. = 42–43 °C (Lit. = 45–46 °C), [α]
D = −20 c = 0.4 in CHCl
3 ([α]
D(Lit.) = −22 c = 0.4 in CHCl
3), e.e. = 90%.
1H NMR (200 MHz, CDCl
3): 7.14 (br, s, 1H), 4.44 (t, 1H,
J = 8.8 Hz), 4.09 (dd, 1H,
J = 8.4, 5.9 Hz), 3.62 (dd, 1H,
J = 15.0, 7.0 Hz), 1.71 (m, 1H), 0.96 (d, 3H,
J = 6.6 Hz), 0.90 (d 3H,
J = 6.6 Hz).
13C NMR (50 MHz, CDCl
3): 160.7, 68.7, 58.5, 32.8, 18.1, 17.8 Anal. Calcd. for C
6H
11NOS: C, 49.67; H, 7.58; N, 9.65. Found: C, 49.50; H, 7.51; N, 9.53.
(4R, 5S)-(+)-4-methyl-5-phenyl-1,3-oxazolidine-2-thione (
3d). In a 25 mL vessel, the amino alcohol
1d (0.5 g, 3.30 mmol, 1.0 eq.), K
2CO
3 (0.45 g, 3.30 mmol, 1.0 eq.) and CS
2 (0.37g, 4.95 mmol, 0.3 mL, 1.5 eq.) were placed in the reactor
. The reaction mixture was stirred in the BenchMate modality with 50 °C and 50 W of power for 10 min. When the reaction was over, the resulting mixture was partitioned with a mixture of CH
2Cl
2 (15 mL) and brine (20 mL). The organic layer was separated and the aqueous layer was extracted with CH
2Cl
2 (2 × 15 mL). The organic extracts were dried over Na
2SO
4 and the solvent evaporated. The product was purified by a silica gel chromatographic column eluting with a hexane-ethyl acetate solvent system (6:4) to give 0.52 g of
3d (82% yield) obtained as a slightly yellow solid. The spectroscopic data were compared with those reported in the literature [
17]. m. p. = 93–93 °C (Lit. = 95–97 °C), [α]
D = +215.01 c = 0.44 in CHCl
3 ([α]
D(Lit.) = +219.2 c = 0.44 in CHCl
3), e.e. = 98%.
1H NMR (200 MHz, CDCl
3): 8.50 (br, s, 1H), 7.19–7.38 (m, 5H), 5.90 (d, 1H,
J = 8.4 Hz), 4.44 (dq, 1H,
J = 6.4, 8.4 Hz), 0.81 (d, 3H,
J = 6.4 Hz).
13C NMR (50 MHz, CDCl
3): 188.7, 133.4, 128.7, 128.3, 126.0, 87.9, 55.83, 15.9 Anal. Calcd. for C
10H
11NOS: C, 62.19; H, 5.69; N, 7.25. Found: C, 61.98; H, 5.52; N, 7.17.
3.4. Synthesis of Thiazolidine-2-thiones 4a-d
These compounds were prepared according to the method described by LeCorre [
17] without inorganic basic media.
(S)-4-Benzyl-1,3-thiazolidine-2-thione (
4a). In a 10 mL reaction vessel were placed the amino alcohol
1a (1.00 g, 6.61 mmol, 1.0 eq.), Et
3N (1.67 g, 16.53 mmol, 2.3 mL, 2.5 eq.), CS
2 (1.51 g, 19.84 mmol, 1.2 mL, 3.0 eq.) and DMSO (0.3 mL). Under the BenchMate modality
4a was prepared in 60 minutes with 100 °C and 40 W of power. When the reaction was completed, 30 mL of water were added and the resulting mixture extracted with ethyl acetate. The organic layer was separated, dried over Na
2SO
4 and evaporated to yield a residue which was purified by a silica gel chromatographic column using a hexane-ethyl acetate (7:3) solvent system to give 1.27 g of
4a (92% yield) obtained as a slightly yellow solid. The spectroscopic data were compared with those reported in the literature [
17]: m. p. = 82–83 °C (Lit. = 84–85 °C), [α]
D = −120.01 c = 0.96 in CHCl
3 ([α]
D(Lit.) = −122 c = 1 in CHCl
3), e.e. = 98%.
1H NMR (200 MHz, CDCl
3): 8.01 (br, s, 1H), 7.41–7.17 (m, 5H), 4.45 (q, 1H,
J = 7.2 Hz), 3.61 (dd, 1H,
J = 11.2, 7.7 Hz), 3.33 (dd, 1H,
J = 11.0, 7.0 Hz), 3.0 (d, 2H,
J = 7.4 Hz).
13C NMR (50 MHz, CDCl
3): 201.0, 135.9, 129.3, 129.1, 127.6, 65.2, 40.4, 38.4 Anal. Calcd. for C
10H
11NS
2: C, 57.37; H, 5.25; N, 6.68. Found: C, 57.25; H, 5.20; N, 6.54.
(S)-4-Phenyl-1,3-thiazolidine-2-thione (
4b). In a 10 mL reaction vessel were placed the amino alcohol
1b (1.00 g, 7.29 mmol, 1.0 eq.), Et
3N (1.84 g, 18.22 mmol, 2.5 mL, 2.5 eq.), CS
2 (1.66 g, 21.87 mmol, 1.3 mL, 3.0 eq.) and DMSO (0.3 mL). Under the BenchMate modality
4b was prepared in 90 minutes with 100 °C and 40 W of power. When the reaction was completed, 50 mL of water were added and the resulting mixture extracted with ethyl acetate. The organic layer was separated, dried over Na
2SO
4 and evaporated to yield a residue which was purified by a silica gel chromatographic column using a hexane-ethyl acetate (6:4) solvent system to give 1.26 g of
4b (89% yield) obtained as a yellow solid. The spectroscopic data were compared with those reported in the literature [
17]: m. p. = 122–123 °C (Lit. = 125–126 °C), [α]
D = +205.03 c = 0.9 in CHCl
3 ([α]
D(Lit.) = +207 c = 1 in CHCl
3), e.e. = 99%.
1H NMR (200 MHz, CDCl
3): 8.07 (br, s, 1H), 7.41–7.35 (m, 5H), 5.32 (t, 1H,
J = 8.1 Hz), 3.84 (dd, 1H,
J = 11.0, 8.0 Hz), 3.48 (dd, 1H,
J = 11.0, 8.0 Hz).
13C NMR (50 MHz, CDCl
3): 201.0, 137.4, 128.8, 128.7, 125.7, 66.9, 44.0 Anal. Calcd. for C
9H
9NS
2: C, 55.34; H, 4.60; N, 7.16. Found: C, 55.20; H, 4.51; N, 7.08.
(S)-4-Isopropyl-1,3-thiazolidine-2-thione (
4c). In a 10 mL reaction vessel were placed the amino alcohol
1c (0.50 g, 4.84 mmol, 1.0 eq.), Et
3N (1.22 g, 12.11 mmol, 1.7 mL, 2.5 eq.), CS
2 (1.10 g, 14.54 mmol, 0.9 mL, 3.0 eq.) and DMSO (0.3 mL). Under the BenchMate modality
4c was prepared in 110 minutes with 110 °C and 100 W of power. When the reaction was completed, 40 mL of water were added and the resulting mixture extracted with ethyl acetate. The organic layer was separated, dried over Na
2SO
4 and evaporated to yield a residue which was purified by a silica gel chromatographic column using a hexane-ethyl acetate (6:4) solvent system to give 0.70 g of
4c (90% yield) obtained as a yellow solid. The spectroscopic data were compared with those reported in the literature [
17]: m. p. = 67–68 °C (Lit. = 69–71 °C), [α]
D = −35.1 c = 1 in CHCl
3 ([α]
D(Lit.) = −37 c = 1 in CHCl
3), e.e. = 94%.
1H NMR (200 MHz, CDCl
3): 8.54 (br, s, 1H), 4.09 (m, 1H), 3.50 (dd, 1H,
J = 7.9, 10.8 Hz), 3.24 (dd, 1H,
J = 7.9, 10.8 Hz), 1.97 (m, 1H), 0.99 (d, 3H,
J = 7.0 Hz), 0.97 (d, 3H,
J = 7.0 Hz).
13C NMR (50 MHz, CDCl
3): 200.5, 70.1, 35.5, 31.7, 18.5, 18.2 Anal. Calcd. for C
6H
11NS
2: C, 44.67; H, 6.82; N, 8.68. Found: C, 44.60; H, 6.87; N, 8.58.
(4R, 5R)-(+)-4-methyl-5-phenyl-1,3-thiazolidine-2-thione (
4d). In a 10 mL reaction vessel were placed the amino alcohol
1d (0.50 g, 3.30 mmol, 1.0 eq.), Et
3N (0.83 g, 8.26 mmol, 1.15 mL, 2.5 eq.), CS
2 (0.75 g, 9.91 mmol, 0.6 mL, 3.0 eq.) and DMSO (0.3 mL). Under the BenchMate modality
4d was prepared in 120 minutes with 110 °C and 100 W of power. When the reaction was completed, 40 mL of water were added and the resulting mixture extracted with ethyl acetate. The organic layer was separated, dried over Na
2SO
4 and evaporated to yield a residue which was purified by a silica gel chromatographic column using a hexane-ethyl acetate (6:4) solvent system to give 0.45 g of
4d (65% yield) obtained as a yellow solid. The spectroscopic data were compared with those reported in the literature [
17,
19,
20,
21,
22,
23,
24]: m. p. = 72–73 °C (Lit. = 75–77 °C), [α]
D = +30.7 c = 1 in CHCl
3 ([α]
D(Lit.) = +33 c = 1 in CHCl
3), e.e. = 93%.
1H NMR (200 MHz, CDCl
3): 7.89 (br, s, 1H), 7.45–7.22 (m, 5H), 4.68 (d, 1H,
J = 8.8 Hz), 4.01 (m, 1H), 1.43 (d, 3H,
J = 6.6 Hz).
13C NMR (50 MHz, CDCl
3): 199.1, 136.9, 129.3, 129.1, 128.1, 74.8, 53.2, 18.5 Anal. Calcd. for C
10H
11NS
2: C, 57.32; H, 4.78; N, 6.69. Found: C, 57.32; H, 4.74; N, 6.50.

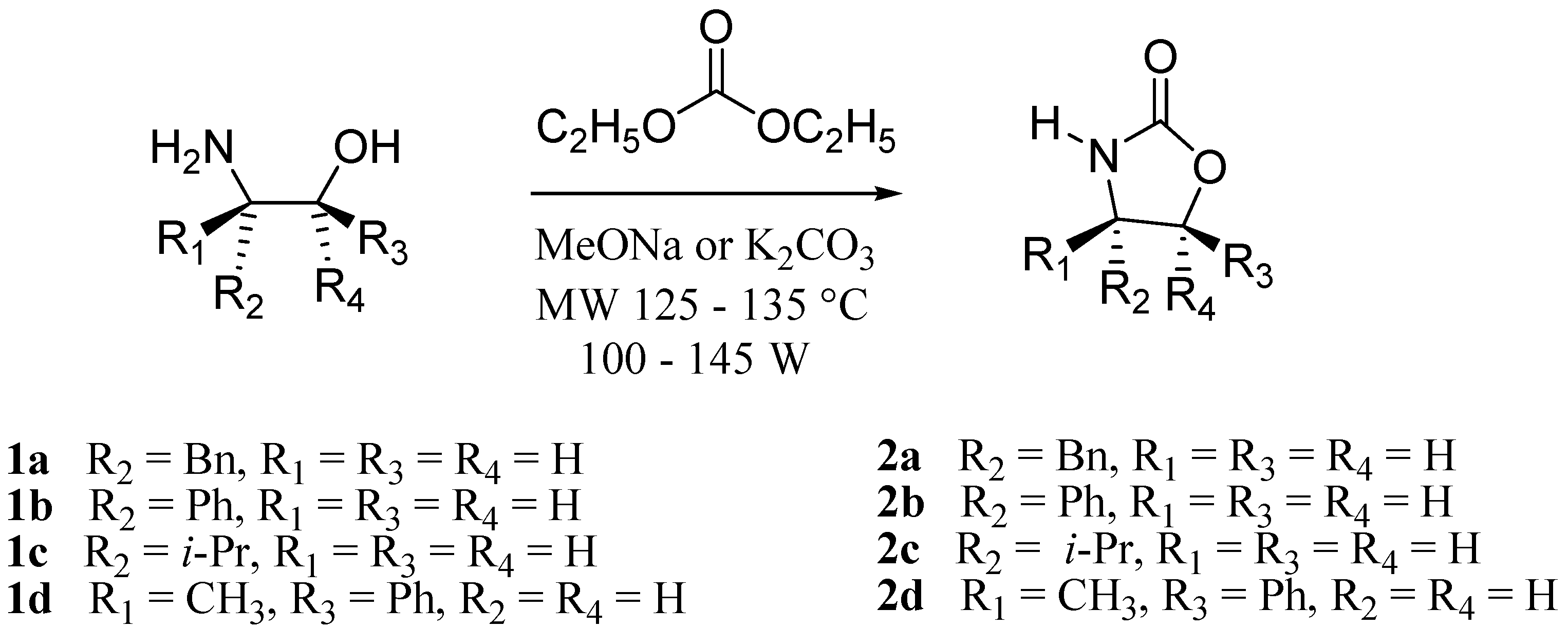

{kind=link}
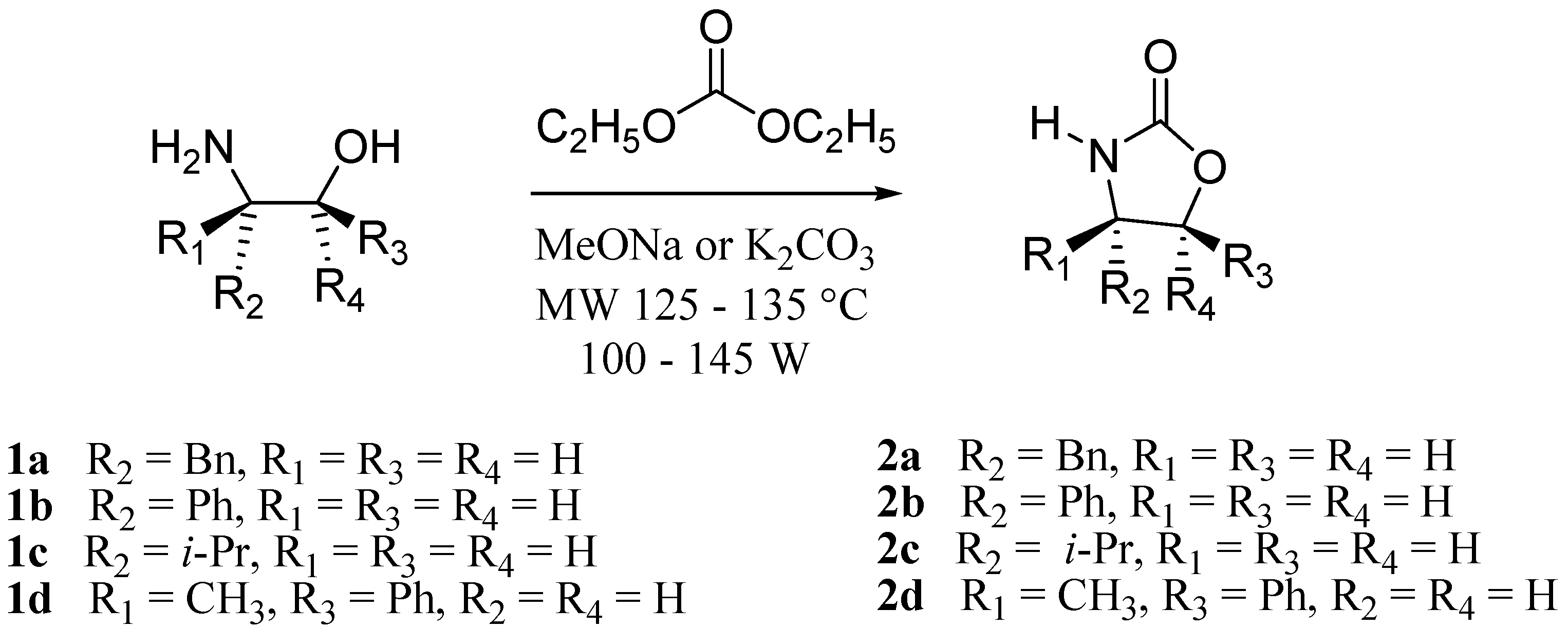{kind=link}
{kind=link}
{kind=link}