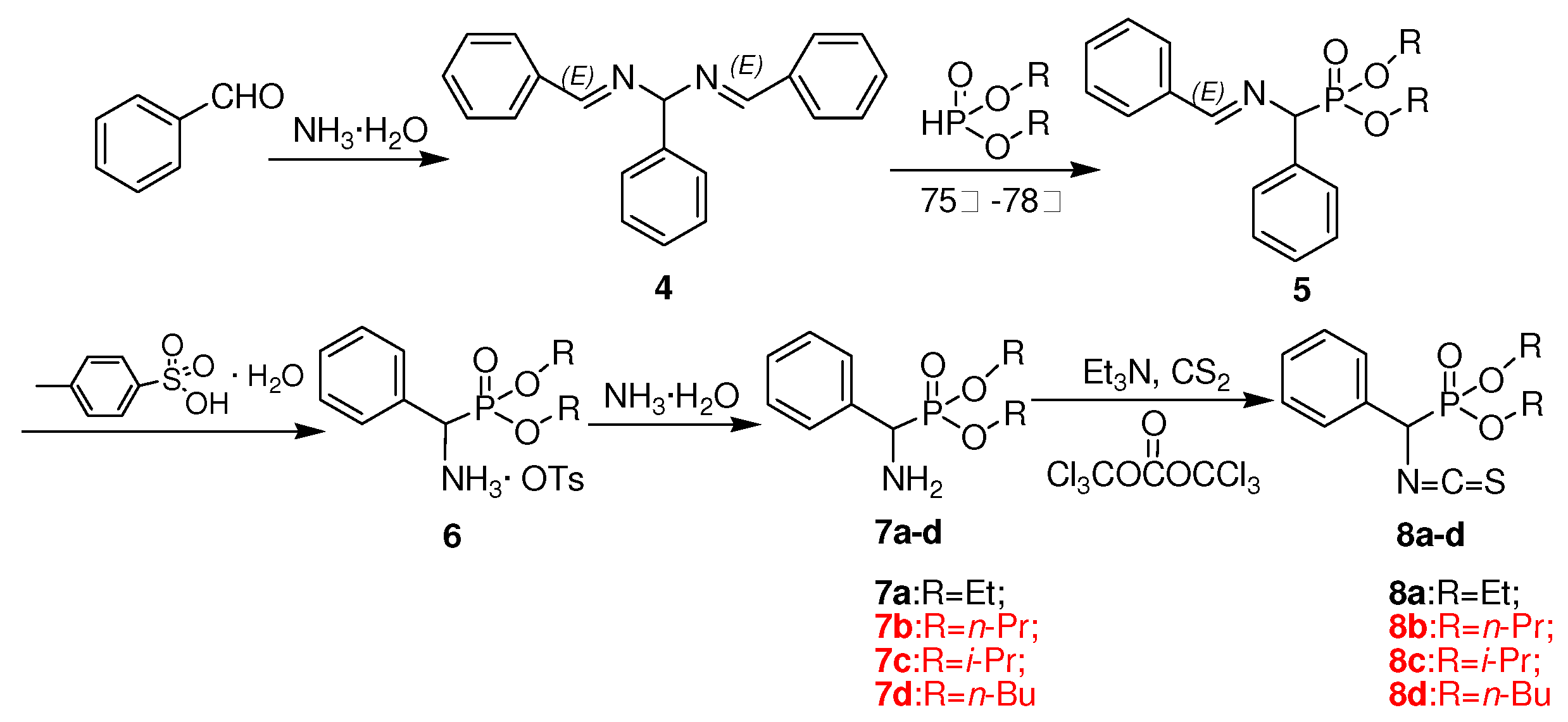
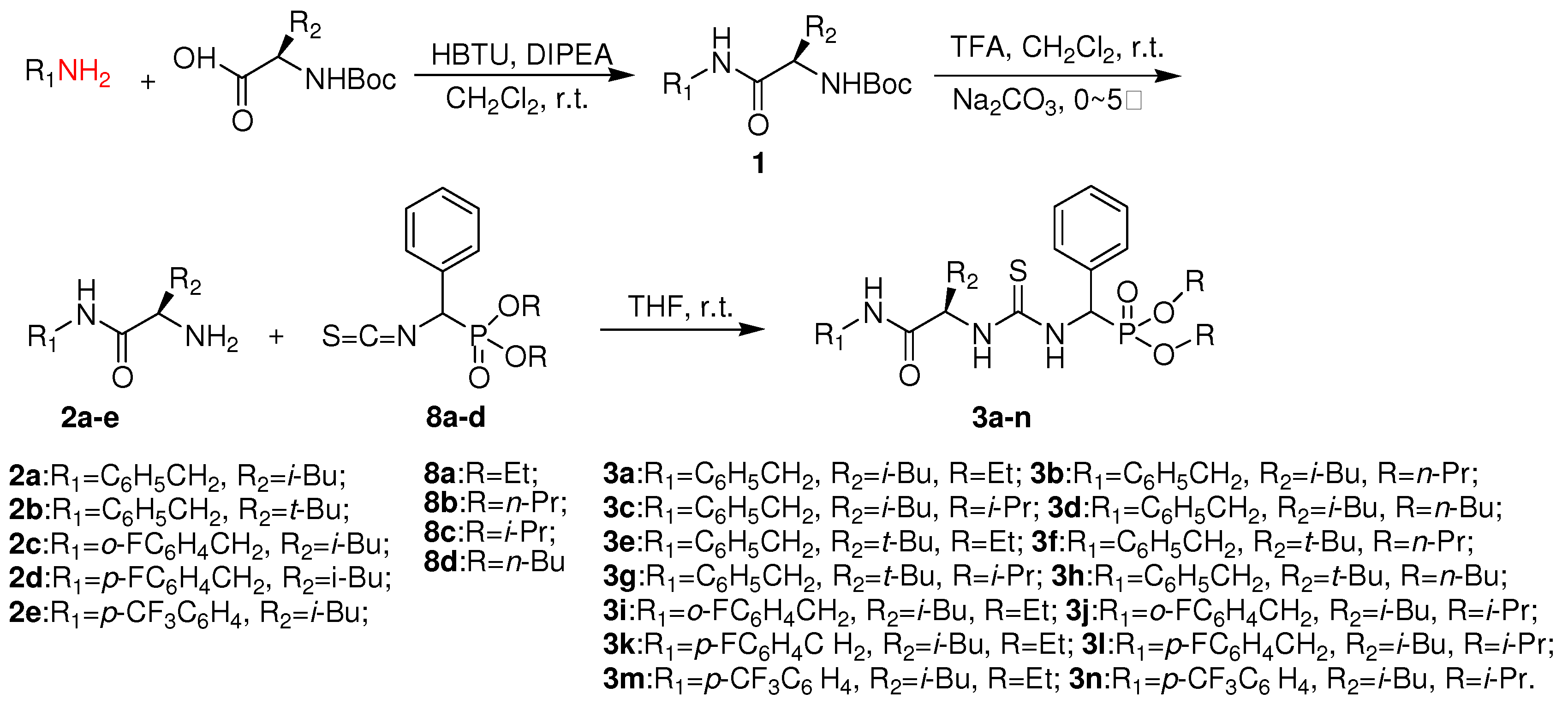
3.2. Preparation of intermediates 2a-e
L-N-Boc-leucine (2.31 g, 0.01 mol) and O-benzotriazol-1-yl-N,N,N’,N’-tetramethyluronium hexa- fluorophosphate (HBTU, 3.80 g, 0.01 mol) were loaded into an oven-dried round bottomed flask equipped with a magnetic stir bar, rubber septum, and argon inlet. Anhydrous dichloromethane (50 mL) was then added. After 3 minutes, anhydrous DIPEA (2.58 g, 0.02 mol) and arylamine (0.011 mol) were sequentially added and the reaction mixture was stirred at room temperature for 2-4 h. During the process, the state of the solution was seen to change from slightly heterogeneous to homogeneous confirming the consumption of HBTU. The reaction mixture was poured into a separatory funnel containing 1N HCl (50 mL), and was then partitioned between dichloromethane and aqueous hydrochloric acid solution. The organic layer was washed with 1N HCl (3 × 30 mL), dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The resulting light yellow oil was transferred to a 100-mL flask and redissolved in dichloromethane (30 mL). Trifluoroacetic acid (7 mL) was then added in one portion. After 2-4 h, the mixture was slowly partitioned with dichloromethane and chilled, saturated aqueous sodium carbonate solution (50 mL) was added. The aqueous layer was extracted with dichloromethane (3 × 30 mL), and the combined organic extracts were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified by thin layer chromatography (TLC) on a silica gel (developing solvent: 3-4% MeOH/CH2Cl2, V/V) to give the intermediates 2a-e.
L-2-Amino-N-benzyl-4-methylpentanamide (2a). White crystals, yield 77%, m.p. 60-62 ºC; 1H-NMR (CDC13): δ 0.95 (d, 6H, J = 3.2 Hz, 2CH3), 1.73-1.75 (m, 2H, CH2), 1.96 (br s, 2H, NH2), 2.80-2.82 (m, 1H, CH), 3.45-3.48 (m, H, CH), 4.43 (d, J = 6.3 Hz, 2H, NCH2), 7.26-7.35 (m, 5H, ArH), 7.72 (br s, 1H, NH); 13C-NMR (CDC13): δ 175.6, 138.6, 128.7, 127.8, 127.4, 53.6, 44.1, 43.2, 24.9, 21.4; IR (KBr, cm-1): v 3304, 2958, 2868, 1647; Anal. Calcd. for C13H20N2O: C 70.87, H 9.15, N 12.72; Found: C 70.62, H 9.34, N 12.86.
L-2-Aamino-N-benzyl-3,3-dimethylbutanamide (2b). White crystals, yield 83%, m.p. 53-54 ºC, 1H-NMR (CDC13, 500 MHz): δ 0.99 (s, 9H, 3CH3), 1.56 (br s, 2H, NH2), 3.11 (s, 1H, CH), 4.42 (d, J = 3.45 Hz, 2H, NCH2), 7.25-7.31 (m, 5H, ArH), 7.78 (br s, 1H, NH); 13C-NMR (CDC13, 125 MHz): δ 173.6, 138.6, 128.7, 128.0, 127.5, 64.5, 43.2, 34.3, 26.9; IR (KBr, cm-1): v 3306, 2946, 1650; Anal. Calcd. for C13H20N2O: C 70.87, H 9.15, N 12.72; Found: C 70.71, H 9.23, N 12.36.
L-N-(2-Fluorobenzyl)-2-amino-4-methylpentanamide (2c). Colorless viscous liquid, yield 68%; 1H-NMR (CDC13): δ 0.99 (d, 6H, J = 4.6 Hz, 2CH3), 1.23 (t, 2H, J = 7.2 Hz, CH2), 1.65 (br s, 2H, NH2), 1.76-1.84 (m, 1H, CH), 3.52-3.55 (m, H, CH), 4.23 (d, J = 5.3 Hz, 2H, NCH2), 7.05-7.33(m, 4H, ArH), 8.43 (br s, 1H, NH); 13C-NMR (CDC13): δ 174.2, 159.7, 131.4, 128.8, 115.3, 124.1, 53.2, 43.5, 34.6, 24.6, 21.3; 19F-NMR (CDC13): δ -115.5; IR (KBr, cm-1) v: 3308, 2960, 2873, 1649; Anal. Calcd. for C13H19FN2O: C 65.52, H 8.04, N 11.76; Found: C 65.36, H 8.32, N 11.65.
L-N-(4-Fluorobenzyl)-2-amino-4-methylpentanamide (2d). Colorless liquid, yield 73%, n25D 1.5055; 1H-NMR (CDC13): δ 0.90 (d, 6H, J = 3.4 Hz, 2CH3), 1.63-1.72 (m, 2H, CH2), 2.25 (br s, 2H, NH2), 2.76-2.79 (m, 1H, CH), 3.35-3.38 (m, 2H, CH), 4.34 (d, J = 5.15 Hz, 2H, NCH2), 6.96-7.22 (m, 4H, ArH), 7.98 (br s, 1H, NH); 13C-NMR (CDC13): δ 174.7, 161.5, 137.4, 128.6, 115.2, 52.9, 44.5, 42.8, 24.7, 21.4; 19F-NMR (CDC13): δ -115.3; IR (KBr, cm-1): v 3309, 2954, 2870, 1645, 1504, 1223; Anal. Calcd. for C13H19FN2O: C 65.52, H 8.04, N 11.76; Found: C 65.33, H 8.26, N 11.51.
L-2-Amino-4-methyl-N-(4-(trifluoromethyl)phenyl)pentanamide (2e). Colorless liquid, yield 56%, n25D 1.4750; 1H-NMR (CDC13): δ 0.95 (d, 6H, J = 6.3 Hz, 2CH3), 1.43-1.47 (m, 1H, CH), 1.75-1.79 (m, 1H, CH2), 2.51 (br s, 2H, NH2), 3.53 (d, 1H,J = 4.0 Hz, , NCH), 7.54-7.78(d, 4H, J = 8.6 Hz, ArH), 9.99 (br s, 1H, NH); 13C-NMR (CDC13): δ 174.7, 141.3, 125.9, 125.6, 123.3, 119.1, 54.0, 43.7, 24.8, 21.4; 19F-NMR (CDC13): δ -62.0; IR (KBr, cm-1): v 3462, 3269, 2953, 1628, 1506, 1323, 1115, 1067; Anal. Calcd. for C13H17F3N2O: C 56.93, H 6.25, N 10.21; Found: C 57.18, H 6.47, N 10.39.
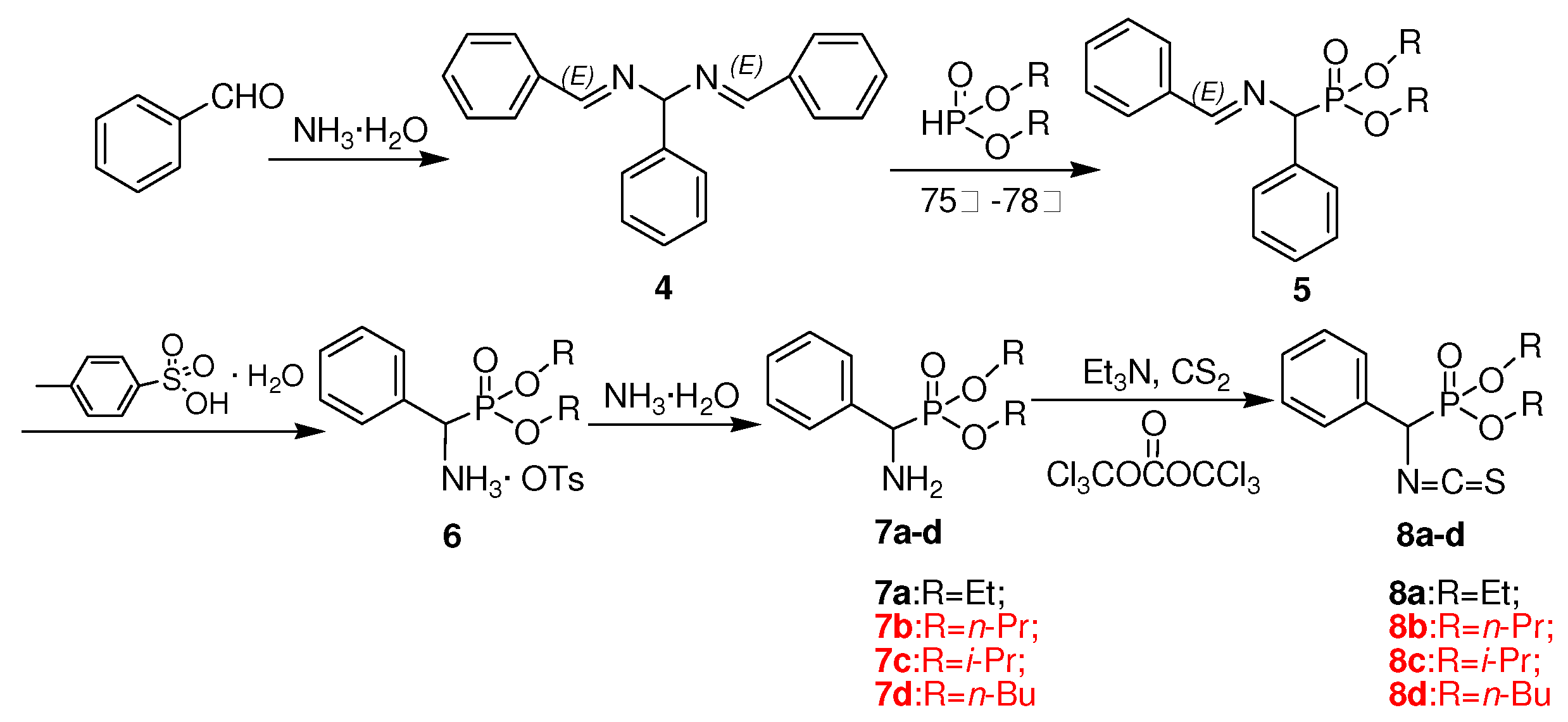
3.4. Preparation of title chiral thioureas 3a-n
A solution of O,O’-dialkylisothiocyanato(phenyl)methylphosphonate 8 (1 mmol) in tetrahydrofuran (5 mL) was stirred, followed by dropwise addition of chiral amine 2a-e (1.1 mmol). The stirring was continued for 0.5 h at 23 ºC, the solvent was evaporated and the crude product was purified by preparative TLC using a mixture of ethyl acetate and n-hexane (V:V = 1:1) as developing solvent to give title compound 3a-n.
Diethyl (3-(L-1-benzylamino-4-methyl-1-oxopentan-2-yl)thioureido)(phenyl)methylphosphonate (3a). White solid, yield 94%; m.p. 43-45 ºC; [α]20D-17.9º (c 0.1, acetone); 1H-NMR (CDC13, 500 MHz): δ 0.79 (d, 6H, J = 6.3 Hz, 2CH3), 1.06 (t, 6H, J = 5.75 Hz, 2CH3), 1.24-1.46(m, 2H, CH2), 1.65-1.72 (m, 1H, CH), 3.68-4.07 (m, 2H, NCH2), 4.12-4.49 (m, 4H, 2OCH2) , 5.07 (d, 1H, J = 6.7 Hz, CH), 6.28 (s, 1H, PCH), 6.45 (s, 1H, NH), 6.95-7.48 (m, 10H, ArH), 7.91 (s, 1H, NH), 8.82 (br s, 1H, NH); 13C-NMR (CDCl3, 125 MHz): δ 183.1, 172.5, 139.2, 136.1, 128.5, 127.6, 126.9, 64.3, 58.1, 54.3, 44.5, 41.4, 26.4, 23.7, 16.3; 31P- NMR (CDCl3, 200 MHz):δ 21.8; IR (KBr, cm-1): v 3284, 3065, 2938, 1659, 1560, 1216, 1012; Anal. Calcd for C25H36N3O4PS: C 59.39, H 7.18, N 8.31; Found C 59.57, H 7.33, N 8.15.
Dipropyl (3-(L-1-benzylamino-4-methyl-1-oxopentan-2-yl)thioureido)(phenyl)methylphosphonate (3b). Colorless viscous liquid, yield 89%; [α]20D-15.8º (c 0.1, acetone); 1H-NMR (CDC13): δ 0.81-1.04 (m, 12H, 4CH3), 1.27-1.45 (m, 6H, 3CH2), 1.67-1.70 (m, 1H, CH), 3.71-4.09 (m, 2H, NCH2), 4.12-4.53 (m, 4H, 2OCH2), 5.10 (d, 1H, J = 6.4 Hz, CH), 6.31 (s, 1H, PCH), 6.48 (s, 1H, NH), 6.94-7.49(m, 10H, ArH), 7.95 (s, 1H, NH), 9.07 (br s, 1H, NH); 13C-NMR (CDC13): δ 183.5, 172.3, 138.4, 135.6, 128.8, 127.1, 126.7, 68.2, 62.5, 54.4, 43.8, 41.3, 26.5, 24.2, 22.7, 10.1; 31P-NMR (CDCl3): δ 21.5; IR (KBr, cm-1): 3276, 3081, 2927, 1663, 1564, 1219, 1009 cm-1; Anal. Calcd for C27H40N3O4PS: C 60.77, H 7.55, N 7.87; Found: C 60.52, H 7.81, N 7.69.
Diisopropyl (3-(L-1-benzylamino-4-methyl-1-oxopentan-2-yl)thioureido)(phenyl)methylphosphonate (3c). White solid, yield 95%; m.p. 51-53 ºC; [α]20D-14.2º (c 0.1, acetone); 1H-NMR (CDC13): δ 0.82 (d, 18H, J = 6.3 Hz, 6CH3), 1.42-1.59 (m, 2H, CH2), 1.76-1.89 (m, H, CH), 4.26-4.48 (m, 2H, NCH2), 4.69-4.78 (m, 2H, 2OCH), 5.09 (d, 1H, J = 5.75 Hz, CH), 6.27 (s, 1H, PCH), 7.06-7.46 (m, 10H, ArH), 7.81 (s, 1H, NH), 8.73 (br s, 1H, NH); 13C-NMR (CDC13): δ 183.9, 172.0, 138.2, 135.5, 128.7, 128.5, 127.6, 127.2, 73.2, 56.4, 55.9, 43.5, 40.2, 38.7, 24.3, 23.1; 31P-NMR (CDCl3): δ 22.8; IR (KBr, cm-1): v 3285, 3063, 2929, 1674, 1558, 1215, 1014; Anal. Calcd for C27H40N3O4PS: C 60.77, H 7.55, N 7.87; Found: C 60.63, H 7.24, N 7.66.
Dibutyl (3-(L-1-benzylamino-4-methyl-1-oxopentan-2-yl)thioureido)(phenyl)methylphosphonate (3d). Colorless viscous liquid, yield 86%; [α]20D-16.4º (c 0.1, acetone); 1H-NMR (CDC13): δ 0.80-0.95 (m, 12H, 4CH3), 1.20-1.43 (m, 10H, 5CH2), 1.55-1.72 (m, 1H, CH), 3.66-4.05 (m, 2H, NCH2), 4.11-4.55 (m, 4H, 2OCH2), 5.04 (d, 1H, J = 6.3 Hz, CH), 6.27 (s, 1H, PCH), 6.43(s, 1H, NH), 6.92-7.44 (m, 10H, ArH), 7.83 (s, 1H, NH), 8.70 (br s, 1H, NH); 13C-NMR (CDC13): δ 184.5, 170.6, 138.2, 135.6, 128.9, 128.6, 127.8, 67.7, 65.8, 55.5, 43.4, 34.8, 32.2, 26.9, 18.8, 13.7; 31P-NMR (CDCl3): δ 22.1; IR (KBr, cm-1): v 3269, 3082, 2953, 1670, 1550, 1220, 1024; Anal. Calcd for C29H44N3O4PS: C, 62.01; H, 7.90; N, 7.48; Found: C 62.28, H 7.67, N 7.72.
Diethyl (3-(L-1-benzylamino-3,3-dimethyl-1-oxobutan-2-yl)thioureido)(phenyl)methylphosphonate (3e). White crystals, yield 98%; m.p. 157-159 ºC; [α]20D+15.5º (c 0.1, acetone); 1H-NMR (CDC13): δ 0.98 (s, 9H, C(CH3)3), 1.07 (t, 6H, J = 6.9 Hz, 2CH3), 3.75-4.06 (m, 2H, NCH2), 4.10-4.46 (m, 4H, 2OCH2), 4.93 (d, 1H, J = 8.6 Hz, CH(t-Bu)), 6.26 (s, 1H, PCH), 6.47 (s, 1H, NH), 7.15-7.47 (m, 10H, ArH), 7.92 (s, 1H, NH), 9.03 (br s, 1H, NH); 13C-NMR (CDCl3): δ 184.3, 170.7, 138.2, 135.4, 128.7, 128.0, 127.4, 66.3, 64.1, 55.4, 43.4, 34.8, 26.9; 16.5; 31P-NMR (CDCl3): δ 21.7; IR (KBr, cm-1): v 3292, 3068, 2965, 2867, 1669, 1562, 1221, 1013; Anal. Calcd for C25H36N3O4PS: C 59.39, H 7.18, N 8.31; Found: C 59.52, H 7.34, N 8.12.
Dipropyl (3-(L-1-benzylamino-3,3-dimethyl-1-oxobutan-2-yl)thioureido)(phenyl)methylphosphonate (3f). White crystals, yield 93%; m.p. 171-172 ºC; [α]20D+9.6º (c 0.1, acetone); 1H-NMR (CDC13): δ 0.76 (t, 6H, J = 6.85 Hz, 2CH3), 1.03 (s, 9H, 3CH3), 1.42-1.74 (m, 4H, 2CH2), 3.70-3.95 (m, 2H, NCH2), 4.09-4.47 (m, 4H, 2OCH2), 4.94 (d, 1H, J = 8.6 Hz, CH(t-Bu)), 6.25 (s, 1H, PCH), 6.53 (s, 1H, NH), 7.12-7.47 (m, 10H, ArH), 7.94 (s, 1H, NH), 9.11 (br s, 1H, NH); 13C-NMR (CDC13): δ 184.4, 170.6, 138.0, 135.2, 128.7, 128.4, 127.5, 69.4, 65.8, 55.3, 43.3, 34.7, 26.8, 24.0, 9.9; 31P-NMR (CDCl3): δ 21.5; IR (KBr, cm-1): v 3288, 3056, 2952, 2873, 1677, 1543, 1218, 1012; Anal. Calcd for C27H40N3O4PS: C 60.77, H 7.55, N 7.87; Found: C 60.52, H 7.71, N 7.59.
Diisopropyl (3-(L-1-benzylamino-3,3-dimethyl-1-oxobutan-2-yl)thioureido)(phenyl)methylphos- phonate (3g). White crystals, yield 96%; m.p. 176-177 ºC; [α]20D+7.2º (c 0.1, acetone); 1H-NMR (CDC13, 500 MHz): δ 1.01 (s, 9H, 3CH3), 1.19 (d, 12H, J = 6.3 Hz, 4CH3), 4.22-4.46 (m, 2H, NCH2), 4.63-4.84 (m, 2H, 2OCH), 4.96 (d, 1H, J = 7.45 Hz, CH(t-Bu)), 6.32 (s, 1H, PCH), 6.38 (s, 1H, NH), 7.11-7.52 (m, 10H, ArH), 7.80 (s, 1H, NH), 9.05 (br s, 1H, NH); 13C-NMR (CDC13, 125 MHz): δ 184.6, 170.7, 138.3, 135.8, 128.9, 128.6, 127.4, 73.1, 66.4, 55.9, 43.2, 34.6, 27.0, 24.3; 31P-NMR(CDCl3, 200 MHz): δ 19.7; IR (KBr, cm-1): v 3286, 3057, 2963, 2861, 1672, 1548, 1224, 1011; Anal. Calcd for C27H40N3O4PS: C 60.77, H 7.55, N 7.87; Found: C 60.91, H 7.34, N 7.48.
Dibutyl (3-(L-1-benzylamino-3,3-dimethyl-1-oxobutan-2-yl)thioureido)(phenyl)methylphosphonate (3h). White crystals, yield 91%; m.p. 122-124 ºC; [α]20D+12.1º (c 0.1, acetone); 1H-NMR (CDC13): δ 0.79 (t, 6H, J = 7.45 Hz, 2CH3), 0.96 (s, 9H, 3CH3), 1.16-1.71 (m, 8H, 2 CH2CH2), 3.71-4.01 (m, 2H, NCH2), 4.06-4.51 (m, 4H, 2OCH2), 4.92 (d, 1H, J = 8.6 Hz, CH(t-Bu)), 6.25 (s, 1H, PCH), 6.46 (s, 1H, NH), 7.13-7.46 (m, 10H, ArH), 7.92 (s, 1H, NH), 9.04 (br s, 1H, NH); 13C-NMR (CDC13): δ 184.5, 170.6, 138.2, 135.6, 128.9, 128.6, 127.8, 67.7, 65.8, 55.5, 43.4, 34.8, 32.2, 26.9, 18.8, 13.7; 31P-NMR (CDCl3): δ 21.5; IR (KBr, cm-1): v 3283, 3065, 2954, 1671, 1552, 1216, 1013; Anal. Calcd for C29H44N3O4PS: C, 62.01; H, 7.90; N, 7.48; Found: C 62.24, H 7.63, N 7.69.
Diethyl (3-(L-1-(2-fluorobenzylamino)-4-methyl-1-oxopentan-2-yl)thioureido)(phenyl)methyl-phosphonate (3i). Colorless viscous liquid, yield 92%; [α]20D-22.3º (c 0.1, acetone); 1H-NMR (CDCl3): δ 0.85 (d, 6H, J = 6.8 Hz, 2CH3), 1.11 (t, 6H, J = 5.7 Hz, 2CH3), 1.31-1.53(m, 2H, CH2), 1.67-1.74 (m, 1H, CH), 3.67-3.96 (m, 2H, NCH2), 4.26-4.47 (m, 4H, 2OCH2) , 5.20 (d, 1H, J = 6.7 Hz, CH), 6.29 (s, 1H, PCH), 6.32 (s, 1H, NH), 6.79-7.45 (m, 9H, ArH), 7.86 (s, 1H, NH), 8.57 (br s, 1H, NH); 13C-NMR (CDCl3): δ 183.5, 171.4, 159.8, 136.3, 131.3, 128.6, 127.2, 126.5, 124.1, 115.2, 63.9, 58.2, 54.4, 41.6, 34.1, 26.1, 23.3, 16.4; 31P-NMR (CDCl3):δ 22.7; 19F-NMR (CDCl3):δ -118.6; IR (KBr, cm-1): v 3290, 3085, 2977, 1662, 1557, 1216, 1025; Anal. Calcd for C25H35FN3O4PS: C 57.35, H 6.74, N 8.03; Found: C 57.53, H 6.55, N 8.28.
Diisopropyl (3-(L-1-(2-fluorobenzylamino)-4-methyl-1-oxopentan-2-yl)thioureido)(phenyl)methyl-phosphonate (3j). Colorless viscous liquid, yield 88%; [α]20D-17.2º (c 0.1, acetone); 1H-NMR (CDCl3): δ0.87 (d, 18H, J = 6.4 Hz, 6CH3), 1.36-1.43 (m, 2H, CH2), 1.51-1.62 (m, H, CH), 3.75-4.16 (m, 2H, NCH2), 4.53-4.71 (m, 2H, 2OCH), 5.23 (d, 1H, J = 6.8 Hz, CH), 6.27 (s, 1H, PCH), 6.30 (s, 1H, NH), 6.82-7.43 (m, 9H, ArH), 7.74 (s, 1H, NH), 8.42 (br s, 1H, NH); 13C-NMR (CDCl3):δ 183.7, 171.2, 159.5, 136.4, 131.5, 128.8, 127.0, 126.7, 124.2, 115.3, 73.0, 59.1, 55.4, 41.4, 34.3, 26.6, 23.5, 16.3; 31P-NMR (CDCl3):δ 20.3; 19F-NMR (CDCl3):δ -118.8; IR (KBr, cm-1): v 3275, 3067, 2961, 1668, 1554, 1227, 1015; Anal. Calcd for C27H39FN3O4PS: C 58.78, H 7.13, N 7.62; Found: C 58.53, H 7.32, N 7.34.
Diethyl (3-(L-1-(4-fluorobenzylamino)-4-methyl-1-oxopentan-2-yl)thioureido)(phenyl)methylphos-phonate (3k). White solid, yield 94%; m.p. 31-32 ºC; [α]20D-16.7º (c 0.1, acetone); 1H-NMR (CDCl3): δ 0.82 (d, 6H, J = 6.5 Hz, 2CH3), 1.09 (t, 6H, J = 5.8Hz, 2CH3), 1.27-1.52(m, 2H, CH2), 1.66-1.71 (m, 1H, CH), 3.71-3.99 (m, 2H, NCH2), 4.14-4.43 (m, 4H, 2OCH2) , 5.20 (d, 1H, J = 6.7 Hz, CH), 6.29 (s, 1H, PCH), 6.37 (s, 1H, NH), 6.74-7.45 (m, 9H, ArH), 8.01 (s, 1H, NH), 8.74 (br s, 1H, NH); 13C-NMR (CDCl3): δ 183.6, 171.7, 160.9, 137.1, 135.8, 128.7, 127.1, 126.6, 115.4, 64.1, 58.5, 54.2, 44.2, 41.3, 26.3, 23.6, 16.5; 31P-NMR (CDCl3):δ 22.2; 19F-NMR (CDCl3):δ -115.1 ppm; IR (KBr, cm-1): v 3285, 3080, 2963, 1676, 1551, 1223, 1020 cm-1; Anal. Calcd for C25H35FN3O4PS: C 57.35, H 6.74, N 8.03; Found: C 57.59, H 6.48, N 7.96
Diisopropyl (3-(L-1-(4-fluorobenzylamino)-4-methyl-1-oxopentan-2-yl)thioureido)(phenyl)methyl-phosphonate (3l). White solid, yield 90%; m.p. 43-44 ºC; [α]20D-13.3º (c 0.1, acetone); 1H-NMR (CDCl3): δ 0.84 (d, 18H, J = 6.3 Hz, 6CH3), 1.39-1.57 (m, 2H, CH2), 1.64-1.69 (m, H, CH), 4.21-4.43 (m, 2H, NCH2), 4.63-4.72 (m, 2H, 2OCH), 5.19 (d, 1H, J = 5.8 Hz, CH), 6.25 (s, 1H, PCH), 6.35 (s, 1H, NH), 6.95-7.40 (m, 9H, ArH), 7.98 (s, 1H, NH), 8.71 (br s, 1H, NH); 13C-NMR (CDC13): δ 183.7, 172.1, 161.2, 137.3, 135.6, 128.8, 127.4, 126.8, 115.3, 73.3, 59.6, 55.7, 43.4, 40.8, 26.4, 24.5, 23.2; 31P-NMR (CDCl3): δ 21.9; 19F-NMR (CDCl3):δ -115.4; IR (KBr, cm-1): v 3287, 3072, 2968, 1678, 1548, 1220, 1008; Anal. Calcd for C27H39FN3O4PS: C 58.78, H 7.13, N 7.62; Found: C 58.64, H 7.35, N 7.41.
Diethyl (3-(L-4-methyl-1-oxo-1-(4-(trifluoromethyl)phenylamino)pentan-2-yl)thioureido)(phenyl)-methylphosphonate (3m). Colorless viscous liquid, yield 84%; [α]20D-8.9º (c 0.1, acetone); 1H-NMR (CDCl3): δ0.94 (d, 6H, J = 5.7 Hz, 2CH3), 1.13 (t, 6H, J = 6.9 Hz, 2CH3), 0.99-1.02(m, H, CH), 1.66-1.72 (m, 2H, CH2), 3.92-4.13 (m, 4H, 2OCH2), 5.03-5.11 (m, 1H, CH), 6.31 (s, 1H, PCH), 7.31-7.79 (m, 9H, ArH), 10.07 (s, 1H, NH); 13C-NMR (CDCl3): δ 183.8, 174.2, 141.7, 136.9, 128.4, 127.0, 126.5, 126.1, 125.7, 123.3, 119.8, 73.4, 58.2, 54.1, 41.4, 26.2, 23.4, 21.8; 31P NMR (CDCl3): δ 22.3; 19F-NMR (CDCl3): δ -63.4; IR (KBr, cm-1): v 3467, 3280, 2968, 1648, 1552, 1218, 1026; Anal. Calcd for C25H33F3N3O4PS: C 53.66, H 5.94, N 7.51; Found: C 53.74, H 6.17, N 7.22.
Diisopropyl(3-(L-4-methyl-1-oxo-1-(4-(trifluoromethyl)phenylamino)pentan-2-yl)thioureido) (phenyl -methylphosphonate (3n). Colorless viscous liquid, yield 79%; [α]20D-10.5º (c 0.1, acetone); 1H-NMR (CDCl3): δ 0.98 (d, 18H, J = 6.4 Hz, 6CH3), 1.36-1.42 (m, 1H, CH), 1.63-1.68 (m, H, CH2), 4.17-4.43 (m, 2H, 2OCH), 5.06-5.16 (m, 1H, CH), 6.33 (s, 1H, PCH), 7.29-7.74 (m, 9H, ArH), 10.12 (br s, 1H, NH); 13C-NMR (CDC13): δ 183.9, 174.4, 141.5, 137.2, 128.5, 127.1, 126.8, 126.2, 125.8, 123.5, 119.4, 73.6, 58.5, 54.2, 41.3, 26.1, 23.2, 21.7; 31P-NMR (CDCl3): δ 22.5; 19F-NMR (CDCl3): δ -63.2; IR (KBr, cm-1): v 3465, 3276, 2962, 1645, 1546, 1212, 1028; Anal. Calcd for C27H37F3N3O4PS: C 55.19, H 6.35, N 7.15; Found: C 55.26, H 6.71, N 6.97.




{kind=link}
{kind=link}