Potential of the Dietary Antioxidants Resveratrol and Curcumin in Prevention and Treatment of Hematologic Malignancies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
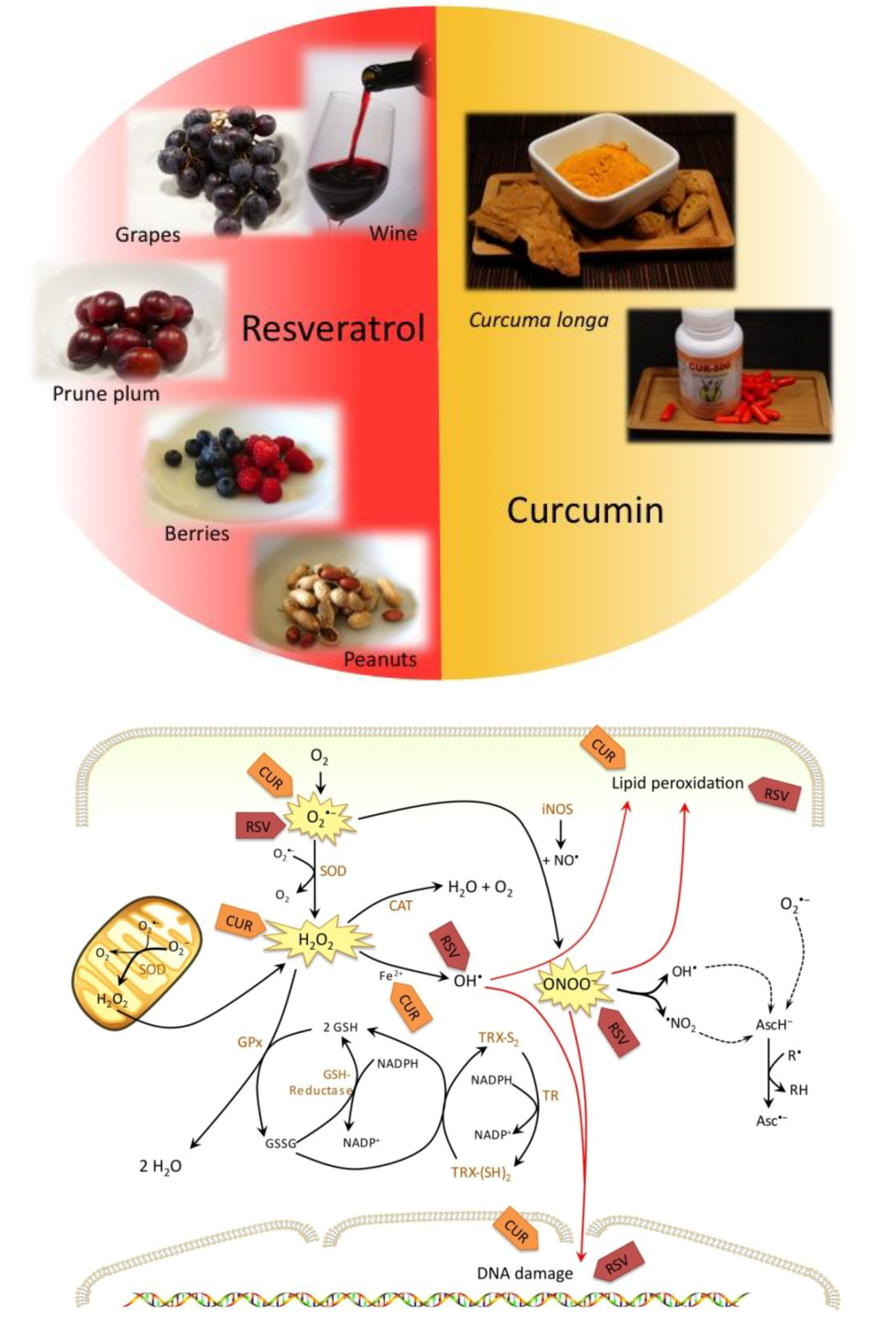
1.1. Role of Oxidative Stress and Cellular Antioxidant Defense Mechanisms
1.2. Role of Oxidative Stress in Development and Evolution of Leukemia
1.3. Natural Antioxidants in the Treatment of Hematologic Malignancies
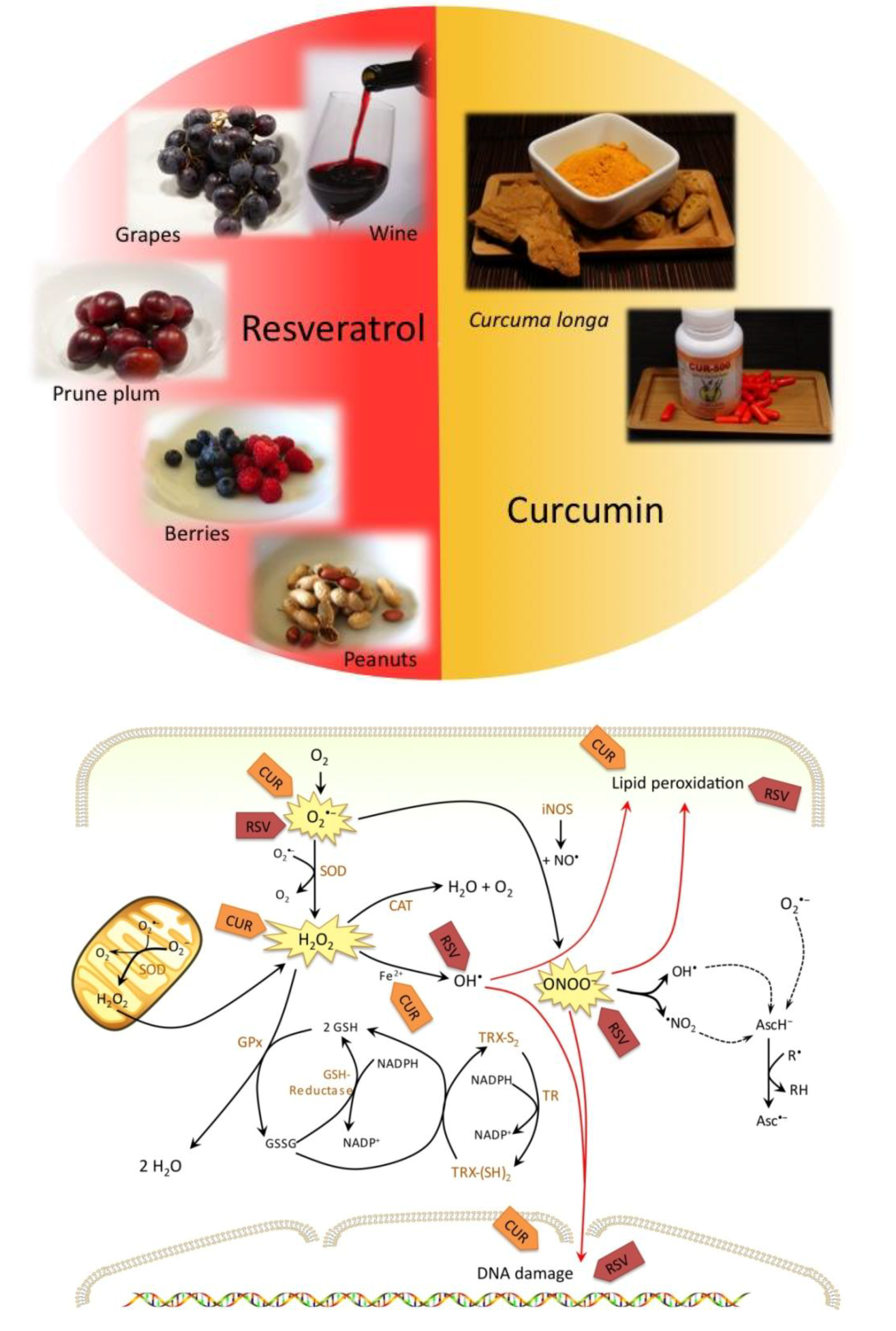
2. Resveratrol
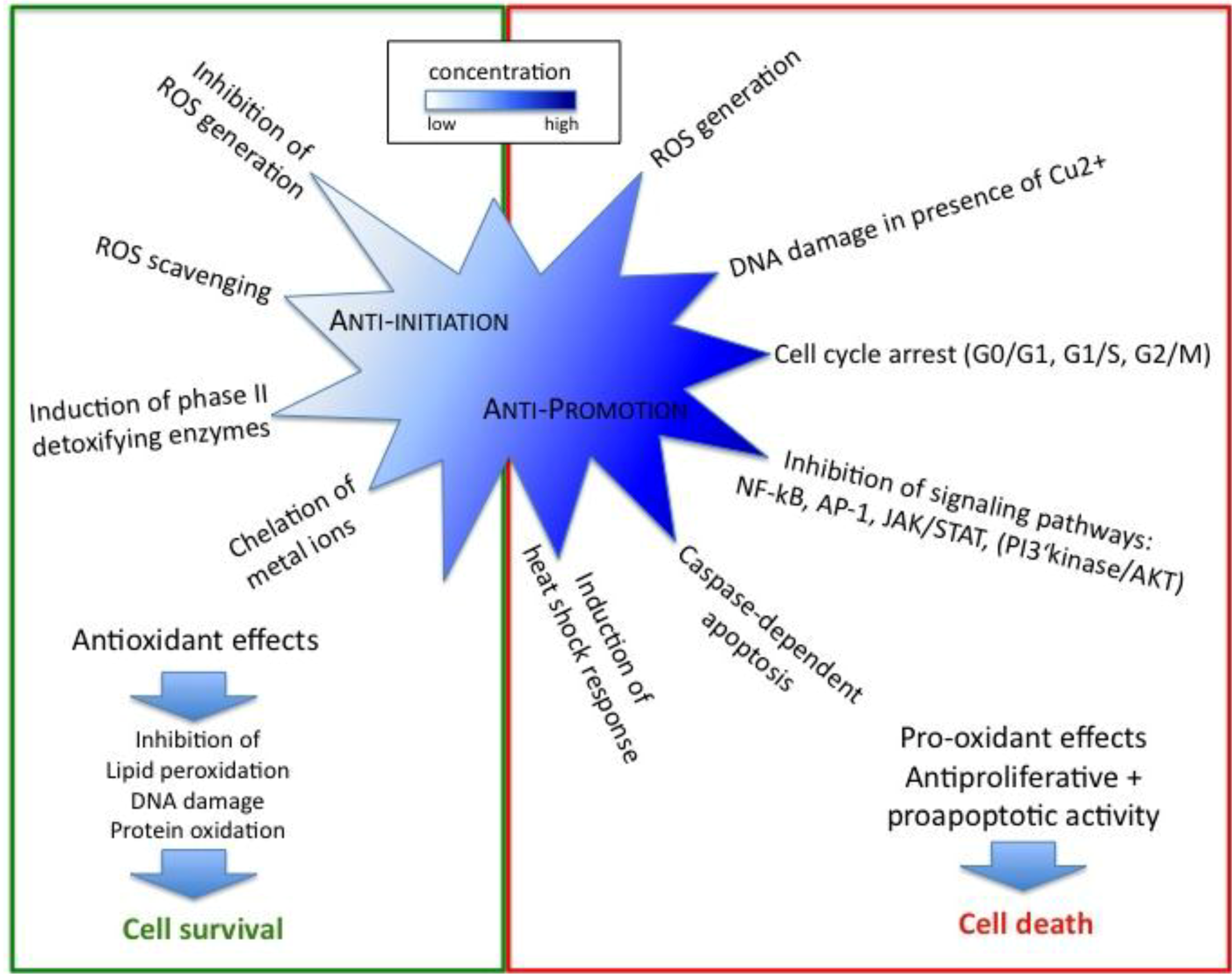
2.1. Antioxidant Effects of Resveratrol Prevent Initiation of Carcinogenesis
2.2. Regulation of Cell Cycle, Proliferation and Apoptosis by Resveratrol Affects Cancer Promotion
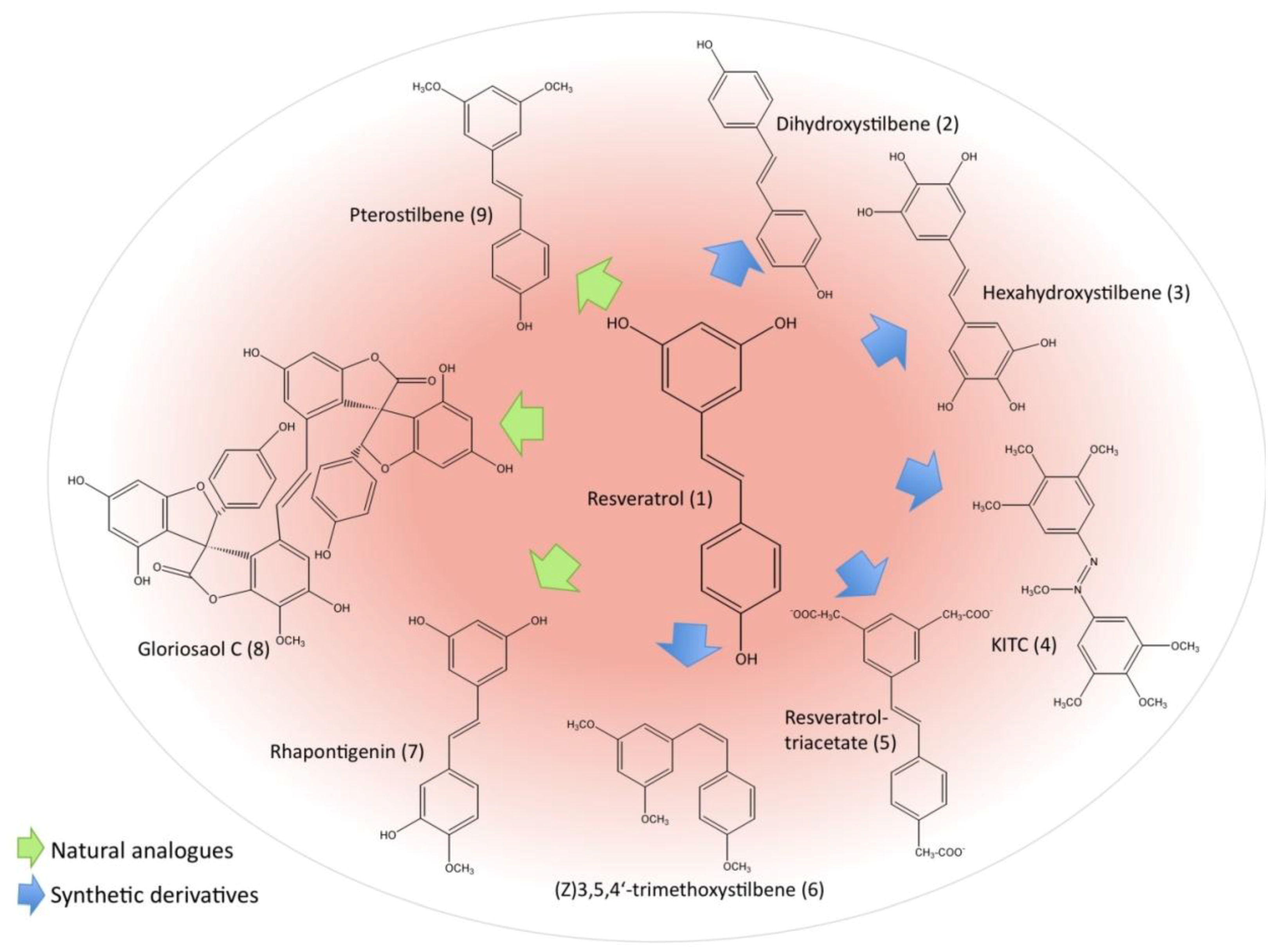
2.3. Resveratrol Derivatives and Their Potential as Anti-leukemic Agents
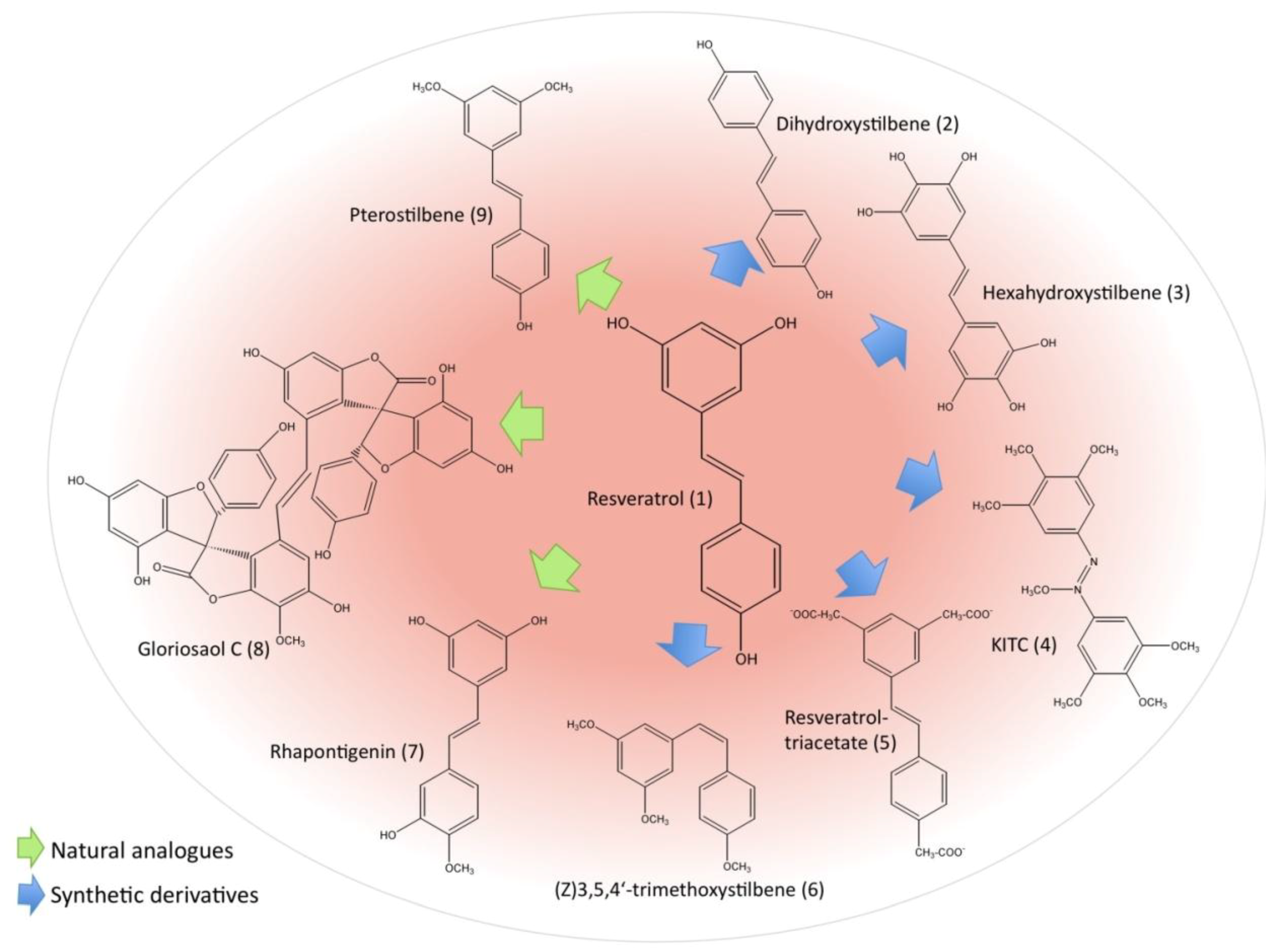
2.4. Possible Negative Effects of Resveratrol
3. Curcumin
3.1. Antioxidant Effects of Curcumin Prevent Initiation of Carcinogenesis
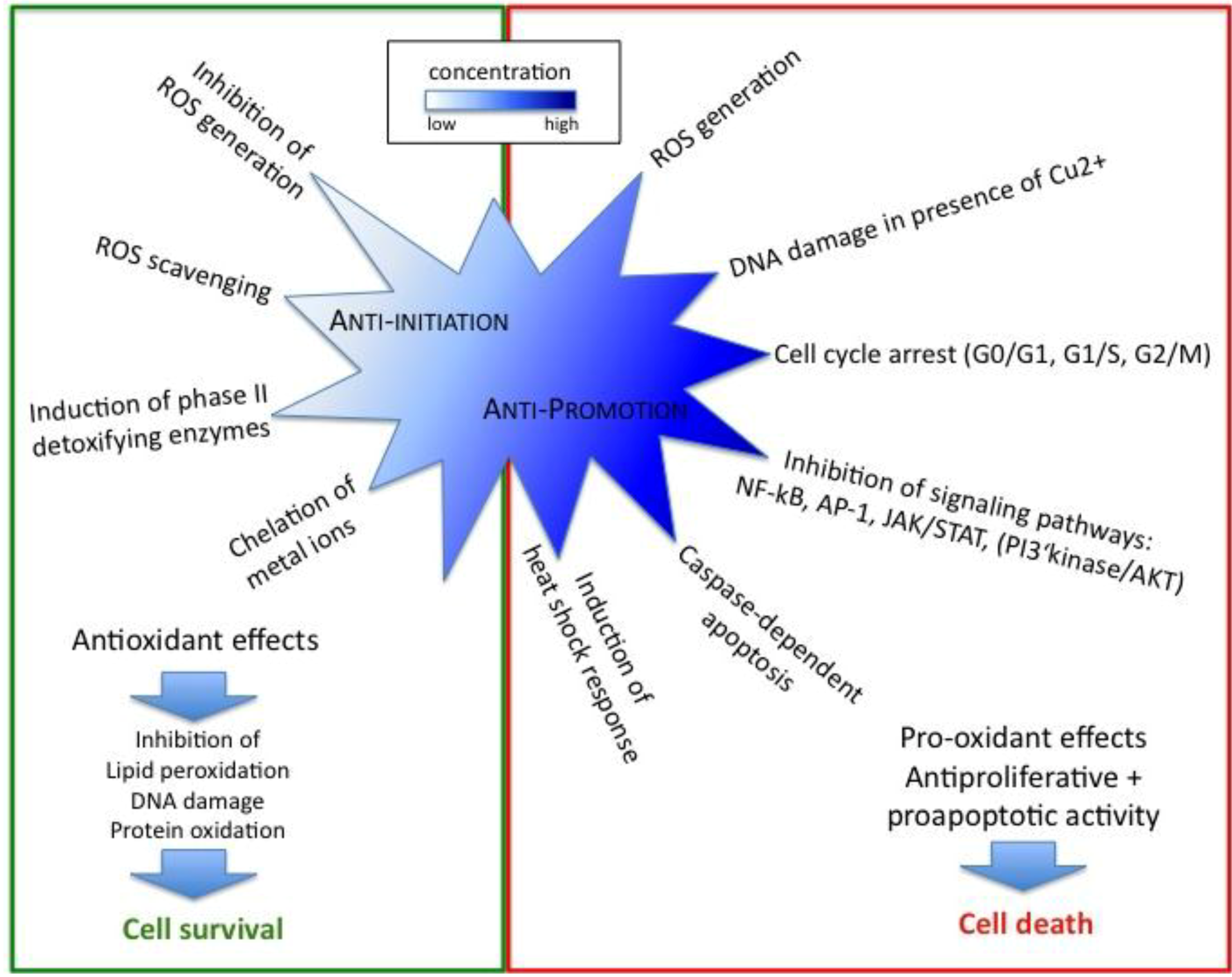
3.2. Curcumin Exerts its Anticancer Properties also as Pro-oxidant
3.3. Regulation of Cell Cycle, Proliferation and Apoptosis by Curcumin Affects Cancer Promotion
3.4. Overcoming Complications/Chemotherapy Resistance of Leukemia by Curcumin Treatment
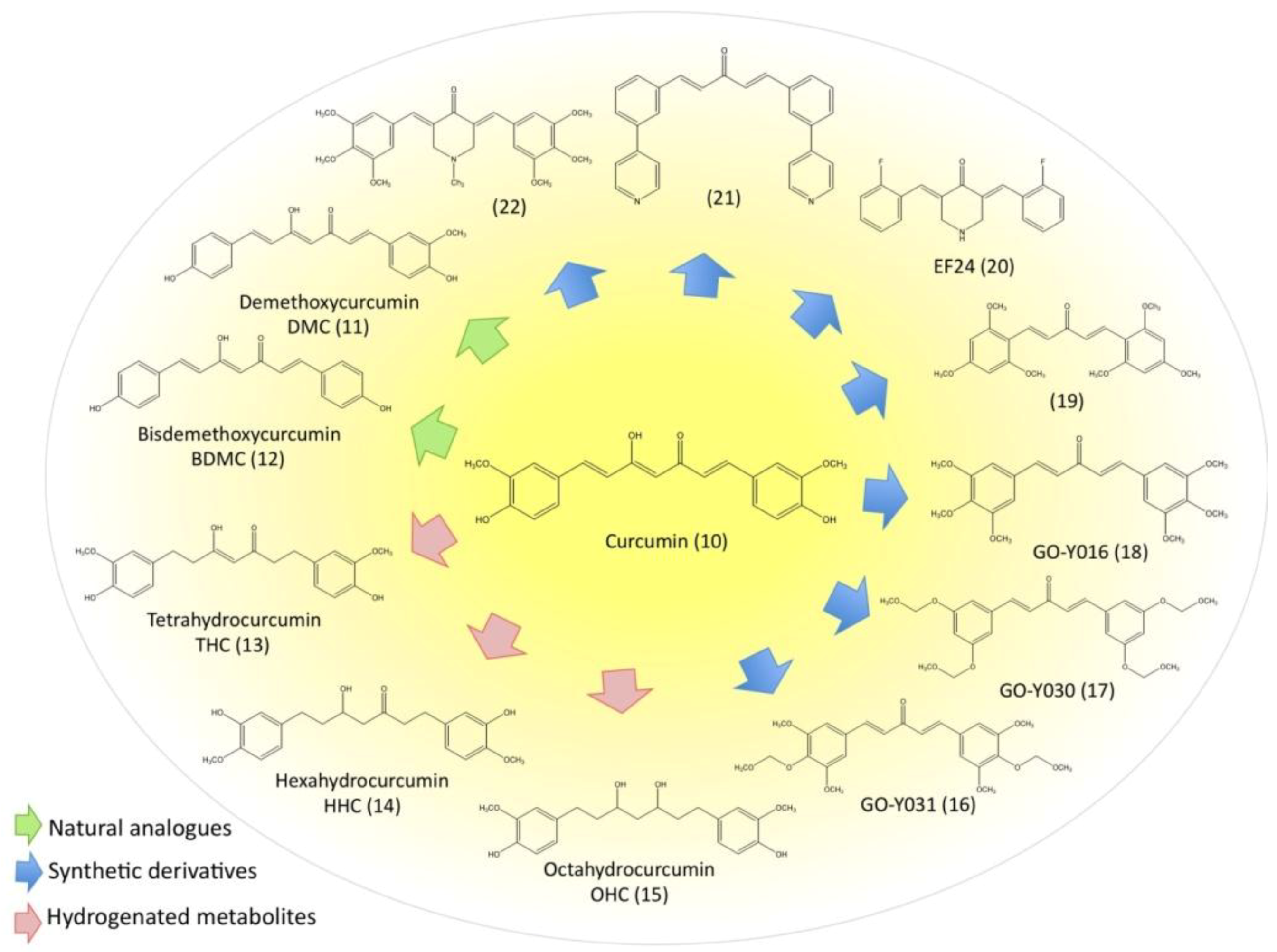
3.5. Evaluation of Natural and Synthetic Curcumin Derivatives and Other Strategies to Improve Bioavailability of Curcumin
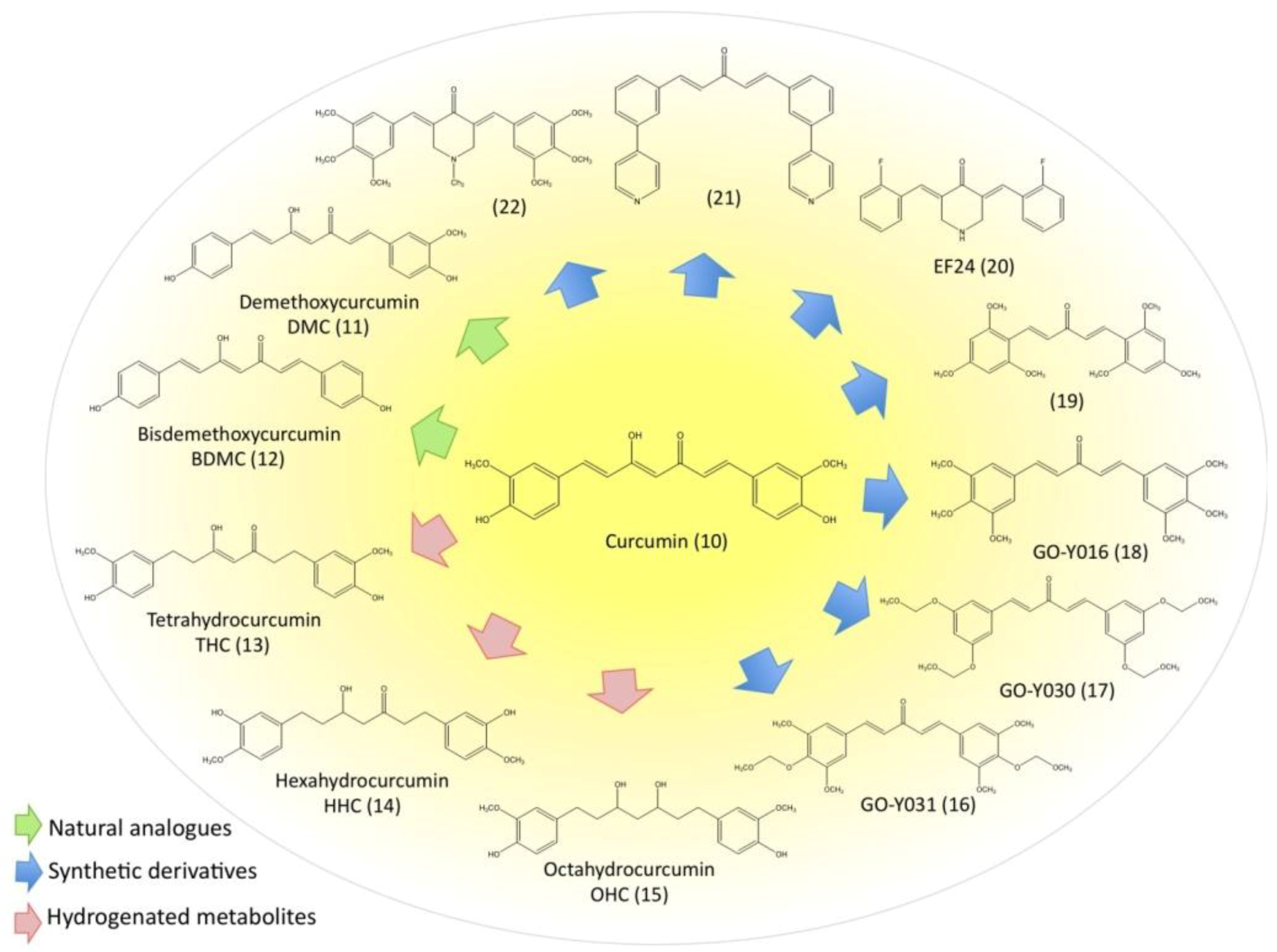
3.6. Disadvantages and Possible Negative Effects of Curcumin
4. Conclusions
Acknowledgements
References
- Debatin, K.M. Role of apoptosis in congenital hematologic disorders and bone marrow failure. Rev. Clin. Exp. Hematol. 2003, 7, 57–71. [Google Scholar]
- Gregory, T.K.; Wald, D.; Chen, Y.; Vermaat, J.M.; Xiong, Y.; Tse, W. Molecular prognostic markers for adult acute myeloid leukemia with normal cytogenetics. J. Hematol. Oncol. 2009, 2, 23. [Google Scholar]
- Olsson, I.; Bergh, G.; Ehinger, M.; Gullberg, U. Cell differentiation in acute myeloid leukemia. Eur. J. Haematol. 1996, 57, 1–16. [Google Scholar]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative stress and cell signalling. Curr. Med. Chem. 2004, 11, 1163–1182. [Google Scholar] [CrossRef]
- Carr, A.C.; McCall, M.R.; Frei, B. Oxidation of LDL by myeloperoxidase and reactive nitrogen species: reaction pathways and antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1716–1723. [Google Scholar] [CrossRef]
- Halliwell, B. Tell me about free radicals, doctor: a review. J. R. Soc. Med. 1989, 82, 747–752. [Google Scholar]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef]
- Dargel, R. Lipid peroxidation – a common pathogenetic mechanism? Exp. Toxicol. Pathol. 1992, 44, 169–181. [Google Scholar] [CrossRef]
- Kasai, H. DNA damage by oxygen radicals and carcinogenesis. Gan To Kagaku Ryoho 1989, 16, 459–465. [Google Scholar]
- Perera, M.I.; Betschart, J.M.; Virji, M.A.; Katyal, S.L.; Shinozuka, H. Free radical injury and liver tumor promotion. Toxicol. Pathol. 1987, 15, 51–59. [Google Scholar]
- Sahu, S.C. Oncogenes, oncogenesis, and oxygen radicals. Biomed. Environ. Sci. 1990, 3, 183–201. [Google Scholar]
- Sun, Y. Free radicals, antioxidant enzymes, and carcinogenesis. Free Radic. Biol. Med. 1990, 8, 583–599. [Google Scholar] [CrossRef]
- Valko, M.; Izakovic, M.; Mazur, M.; Rhodes, C.J.; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell. Biochem. 2004, 266, 37–56. [Google Scholar]
- Cotgreave, I.A.; Moldeus, P.; Orrenius, S. Host biochemical defense mechanisms against prooxidants. Annu. Rev. Pharmacol. Toxicol. 1988, 28, 189–212. [Google Scholar] [CrossRef]
- Masella, R.; Di Benedetto, R.; Vari, R.; Filesi, C.; Giovannini, C. Novel mechanisms of natural antioxidant compounds in biological systems: involvement of glutathione and glutathione-related enzymes. J. Nutr. Biochem. 2005, 16, 577–586. [Google Scholar] [CrossRef]
- Klatt, P.; Lamas, S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur. J. Biochem. 2000, 267, 4928–4944. [Google Scholar] [CrossRef]
- Finkel, T. Redox-dependent signal transduction. FEBS Lett. 2000, 476, 52–54. [Google Scholar] [CrossRef]
- Flohe, L. Glutathione peroxidase: fact and fiction. Ciba Found Symp 1978, 65, 95–122. [Google Scholar]
- Klaunig, J.E.; Kamendulis, L.M. The role of oxidative stress in carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 239–267. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Nakamura, H.; Nakamura, K.; Yodoi, J. Redox regulation of cellular activation. Annu. Rev. Immunol. 1997, 15, 351–369. [Google Scholar] [CrossRef]
- Kojo, S. Vitamin C: basic metabolism and its function as an index of oxidative stress. Curr. Med. Chem. 2004, 11, 1041–1064. [Google Scholar] [CrossRef]
- Al-Gayyar, M.M.; Eissa, L.A.; Rabie, A.M.; El-Gayar, A.M. Measurements of oxidative stress status and antioxidant activity in chronic leukaemia patients. J. Pharm. Pharmacol. 2007, 59, 409–417. [Google Scholar]
- Oltra, A.M.; Carbonell, F.; Tormos, C.; Iradi, A.; Saez, G.T. Antioxidant enzyme activities and the production of MDA and 8-oxo-dG in chronic lymphocytic leukemia. Free Radic. Biol. Med. 2001, 30, 1286–1292. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell. Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Senturker, S.; Karahalil, B.; Inal, M.; Yilmaz, H.; Muslumanoglu, H.; Gedikoglu, G.; Dizdaroglu, M. Oxidative DNA base damage and antioxidant enzyme levels in childhood acute lymphoblastic leukemia. FEBS Lett. 1997, 416, 286–290. [Google Scholar]
- Marnett, L.J. Lipid peroxidation-DNA damage by malondialdehyde. Mutat. Res. 1999, 424, 83–95. [Google Scholar] [CrossRef]
- Farber, C.M.; Kanganis, D.N.; Liebes, L.F.; Silber, R. Antioxidant enzymes in lymphocytes from normal subjects and patients with chronic lymphocytic leukaemia: increased glutathione peroxidase activity in CLL B lymphocytes. Br. J. Haematol. 1989, 72, 32–35. [Google Scholar] [CrossRef]
- Zhou, F.L.; Zhang, W.G.; Wei, Y.C.; Meng, S.; Bai, G.G.; Wang, B.Y.; Yang, H.Y.; Tian, W.; Meng, X.; Zhang, H.; Chen, S.P. Involvement of oxidative stress in the relapse of acute myeloid leukemia. J. Biol. Chem. 2010, 285, 15010–15015. [Google Scholar]
- Battisti, V.; Maders, L.D.; Bagatini, M.D.; Santos, K.F.; Spanevello, R.M.; Maldonado, P.A.; Brule, A.O.; Araujo Mdo, C.; Schetinger, M.R.; Morsch, V.M. Measurement of oxidative stress and antioxidant status in acute lymphoblastic leukemia patients. Clin. Biochem. 2008, 41, 511–518. [Google Scholar] [CrossRef]
- Zhou, F.; Zhang, W.; Wei, Y.; Zhou, D.; Su, Z.; Meng, X.; Hui, L.; Tian, W. The changes of oxidative stress and human 8-hydroxyguanine glycosylase1 gene expression in depressive patients with acute leukemia. Leuk. Res. 2007, 31, 387–393. [Google Scholar] [CrossRef]
- Madej, J.A.; Kaszubkiewicz, C.; Radzanowska, G. Superoxide dismutase, catalase and glutathione peroxidase activities in mice with natural lymphocytic leukemia. Pol Arch Weter 1988, 28, 25–34. [Google Scholar]
- Chang, E.T.; Zheng, T.; Weir, E.G.; Borowitz, M.; Mann, R.B.; Spiegelman, D.; Mueller, N.E. Aspirin and the risk of Hodgkin's lymphoma in a population-based case-control study. J. Natl. Cancer Inst. 2004, 96, 305–315. [Google Scholar] [CrossRef]
- Wang, W.H.; Huang, J.Q.; Zheng, G.F.; Lam, S.K.; Karlberg, J.; Wong, B.C. Non-steroidal anti-inflammatory drug use and the risk of gastric cancer: a systematic review and meta-analysis. J. Natl. Cancer Inst. 2003, 95, 1784–1791. [Google Scholar]
- Garber, K. Aspirin for cancer chemoprevention: still a headache? J. Natl. Cancer Inst. 2004, 96, 252–253. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Inflammation and oncogenesis: a vicious connection. Curr. Opin. Genet. Dev. 2010, 20, 65–71. [Google Scholar]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Bartsch, H.; Nair, J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch. Surg. 2006, 391, 499–510. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar]
- Karin, M.; Greten, F.R. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef]
- Hung, L.M.; Chen, J.K.; Huang, S.S.; Lee, R.S.; Su, M.J. Cardioprotective effect of resveratrol, a natural antioxidant derived from grapes. Cardiovasc Res. 2000, 47, 549–555. [Google Scholar] [CrossRef]
- Langcake, P.; Pryce, R.J. The production of resveratrol by Vitis vinifera and other members of the Vitaseae as a response to infection or injury. Physiol. Plant Pathol. 1976, 9, 77–86. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Bhardwaj, A.; Aggarwal, R.S.; Seeram, N.P.; Shishodia, S.; Takada, Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res. 2004, 24, 2783–2840. [Google Scholar]
- Lee, S.K.; Zhang, W.; Sanderson, B.J. Selective growth inhibition of human leukemia and human lymphoblastoid cells by resveratrol via cell cycle arrest and apoptosis induction. J. Agric. Food Chem. 2008, 56, 7572–7577. [Google Scholar] [CrossRef]
- Harikumar, K.B.; Kuttan, G.; Kuttan, R. Inhibition of progression of erythroleukemia induced by Friend virus in BALB/c mice by natural products--berberine, curcumin and picroliv. J. Exp. Ther. Oncol. 2008, 7, 275–284. [Google Scholar]
- Kimura, Y.; Okuda, H.; Arichi, S. Effects of stilbenes on arachidonate metabolism in leukocytes. Biochim. Biophys. Acta. 1985, 834, 275–278. [Google Scholar]
- Rotondo, S.; Rajtar, G.; Manarini, S.; Celardo, A.; Rotillo, D.; de Gaetano, G.; Evangelista, V.; Cerletti, C. Effect of trans-resveratrol, a natural polyphenolic compound, on human polymorphonuclear leukocyte function. Br. J. Pharmacol. 1998, 123, 1691–1699. [Google Scholar] [CrossRef]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; Moon, R.C.; Pezzuto, J.M. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef]
- Surh, Y. Molecular mechanisms of chemopreventive effects of selected dietary and medicinal phenolic substances. Mutat. Res. 1999, 428, 305–327. [Google Scholar]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef]
- Dreher, D.; Junod, A.F. Role of oxygen free radicals in cancer development. Eur. J. Cancer 1996, 32A, 30–38. [Google Scholar] [CrossRef]
- Jang, D.S.; Kang, B.S.; Ryu, S.Y.; Chang, I.M.; Min, K.R.; Kim, Y. Inhibitory effects of resveratrol analogs on unopsonized zymosan-induced oxygen radical production. Biochem. Pharmacol. 1999, 57, 705–712. [Google Scholar] [CrossRef]
- Burkitt, M.J.; Duncan, J. Effects of trans-resveratrol on copper-dependent hydroxyl-radical formation and DNA damage: evidence for hydroxyl-radical scavenging and a novel, glutathione-sparing mechanism of action. Arch. Biochem. Biophys. 2000, 381, 253–263. [Google Scholar] [CrossRef]
- Shamon, L.A.; Chen, C.; Mehta, R.G.; Steele, V.; Moon, R.C.; Pezzuto, J.M. A correlative approach for the identification of antimutagens that demonstrate chemopreventive activity. Anticancer Res. 1994, 14, 1775–1778. [Google Scholar]
- Prochaska, H.J.; Santamaria, A.B. Direct measurement of NAD(P)H:quinone reductase from cells cultured in microtiter wells: a screening assay for anticarcinogenic enzyme inducers. Anal. Biochem. 1988, 169, 328–336. [Google Scholar] [CrossRef]
- Zhang, Y.; Kensler, T.W.; Cho, C.G.; Posner, G.H.; Talalay, P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc. Natl. Acad. Sci. USA 1994, 91, 3147–3150. [Google Scholar] [CrossRef]
- Talalay, P. Mechanisms of induction of enzymes that protect against chemical carcinogenesis. Adv. Enzyme Regul. 1989, 28, 237–250. [Google Scholar]
- Chen, C.Y.; Jang, J.H.; Li, M.H.; Surh, Y.J. Resveratrol upregulates heme oxygenase-1 expression via activation of NF-E2-related factor 2 in PC12 cells. Biochem. Biophys. Res. Commun. 2005, 331, 993–1000. [Google Scholar] [CrossRef]
- Wasserman, W.W.; Fahl, W.E. Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. USA 1997, 94, 5361–5366. [Google Scholar] [CrossRef]
- Paine, A.; Eiz-Vesper, B.; Blasczyk, R.; Immenschuh, S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 2010. [Google Scholar] [CrossRef]
- Chan, M.M.; Mattiacci, J.A.; Hwang, H.S.; Shah, A.; Fong, D. Synergy between ethanol and grape polyphenols, quercetin, and resveratrol, in the inhibition of the inducible nitric oxide synthase pathwy. Biochem. Pharmacol. 2000, 60, 1539–1548. [Google Scholar] [CrossRef]
- Moreno, J. J. Resveratrol modulates arachidonic acid release, prostaglandin synthesis, and 3T6 fibroblast growth. J Pharmacol. Exp. Ther. 2000, 294, 333–338. [Google Scholar]
- Basu, A.K.; Marnett, L.J. Unequivocal demonstration that malondialdehyde is a mutagen. Carcinogenesis 1983, 4, 331–333. [Google Scholar]
- Yau, T.M. Mutagenicity and cytotoxicity of malonaldehyde in mammalian cells. Mech. Ageing Dev. 1979, 11, 137–144. [Google Scholar] [CrossRef]
- Pandya, G.A.; Moriya, M. 1,6-ethenodeoxyadenosine, a DNA adduct highly mutagenic in mammalian cells. Biochemistry 1996, 35, 11487–11492. [Google Scholar] [CrossRef]
- Tadolini, B.; Juliano, C.; Piu, L.; Franconi, F.; Cabrini, L. Resveratrol inhibition of lipid peroxidation. Free Radic. Res. 2000, 33, 105–114. [Google Scholar] [CrossRef]
- Murcia, M.A.; Martinez-Tome, M. Antioxidant activity of resveratrol compared with common food additives. J. Food Prot. 2001, 64, 379–384. [Google Scholar]
- Stojanovic, S.; Sprinz, H.; Brede, O. Efficiency and mechanism of the antioxidant action of trans-resveratrol and its analogues in the radical liposome oxidation. Arch. Biochem. Biophys. 2001, 391, 79–89. [Google Scholar] [CrossRef]
- Belguendouz, L.; Fremont, L.; Linard, A. Resveratrol inhibits metal ion-dependent and independent peroxidation of porcine low-density lipoproteins. Biochem. Pharmacol. 1997, 53, 1347–1355. [Google Scholar] [CrossRef]
- Brune, M.; Rossander, L.; Hallberg, L. Iron absorption and phenolic compounds: importance of different phenolic structures. Eur. J. Clin. Nutr. 1989, 43, 547–557. [Google Scholar]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar]
- De Salvia, R.; Festa, F.; Ricordy, R.; Perticone, P.; Cozzi, R. Resveratrol affects in a different way primary versus fixed DNA damage induced by H(2)O(2) in mammalian cells in vitro. Toxicol. Lett. 2002, 135, 1–9. [Google Scholar]
- Gautam, S.C.; Xu, Y.X.; Dumaguin, M.; Janakiraman, N.; Chapman, R.A. Resveratrol selectively inhibits leukemia cells: a prospective agent for ex vivo bone marrow purging. Bone Marrow Transplant 2000, 25, 639–645. [Google Scholar] [CrossRef]
- Ahmad, A.; Farhan Asad, S.; Singh, S.; Hadi, S.M. DNA breakage by resveratrol and Cu(II): reaction mechanism and bacteriophage inactivation. Cancer Lett. 2000, 154, 29–37. [Google Scholar] [CrossRef]
- Heiss, E.H.; Schilder, Y.D.; Dirsch, V.M. Chronic treatment with resveratrol induces redox stress- and ataxia telangiectasia-mutated (ATM)-dependent senescence in p53-positive cancer cells. J. Biol. Chem. 2007, 282, 26759–26766. [Google Scholar] [CrossRef]
- Bernhard, D.; Tinhofer, I.; Tonko, M.; Hubl, H.; Ausserlechner, M.J.; Greil, R.; Kofler, R.; Csordas, A. Resveratrol causes arrest in the S-phase prior to Fas-independent apoptosis in CEM-C7H2 acute leukemia cells. Cell Death Differ. 2000, 7, 834–842. [Google Scholar] [CrossRef]
- Estrov, Z.; Shishodia, S.; Faderl, S.; Harris, D.; Van, Q.; Kantarjian, H.M.; Talpaz, M.; Aggarwal, B.B. Resveratrol blocks interleukin-1beta-induced activation of the nuclear transcription factor NF-kappaB, inhibits proliferation, causes S-phase arrest, and induces apoptosis of acute myeloid leukemia cells. Blood 2003, 102, 987–995. [Google Scholar]
- Zou, J.; Huang, Y.; Chen, Q.; Wang, N.; Cao, K.; Hsieh, T.C.; Wu, J.M. Suppression of mitogenesis and regulation of cell cycle traverse by resveratrol in cultured smooth muscle cells. Int. J. Oncol. 1999, 15, 647–651. [Google Scholar]
- Stivala, L.A.; Savio, M.; Carafoli, F.; Perucca, P.; Bianchi, L.; Maga, G.; Forti, L.; Pagnoni, U.M.; Albini, A.; Prosperi, E.; Vannini, V. Specific structural determinants are responsible for the antioxidant activity and the cell cycle effects of resveratrol. J. Biol. Chem. 2001, 276, 22586–22594. [Google Scholar]
- Locatelli, G.A.; Savio, M.; Forti, L.; Shevelev, I.; Ramadan, K.; Stivala, L.A.; Vannini, V.; Hubscher, U.; Spadari, S.; Maga, G. Inhibition of mammalian DNA polymerases by resveratrol: mechanism and structural determinants. Biochem. J. 2005, 389, 259–268. [Google Scholar] [CrossRef]
- Hayashibara, T.; Yamada, Y.; Nakayama, S.; Harasawa, H.; Tsuruda, K.; Sugahara, K.; Miyanishi, T.; Kamihira, S.; Tomonaga, M.; Maita, T. Resveratrol induces downregulation in survivin expression and apoptosis in HTLV-1-infected cell lines: a prospective agent for adult T cell leukemia chemotherapy. Nutr. Cancer 2002, 44, 193–201. [Google Scholar] [CrossRef]
- Billard, C.; Izard, J.C.; Roman, V.; Kern, C.; Mathiot, C.; Mentz, F.; Kolb, J.P. Comparative antiproliferative and apoptotic effects of resveratrol, epsilon-viniferin and vine-shots derived polyphenols (vineatrols) on chronic B lymphocytic leukemia cells and normal human lymphocytes. Leuk. Lymphoma 2002, 43, 1991–2002. [Google Scholar] [CrossRef]
- Roman, V.; Billard, C.; Kern, C.; Ferry-Dumazet, H.; Izard, J.C.; Mohammad, R.; Mossalayi, D. M.; Kolb, J.P. Analysis of resveratrol-induced apoptosis in human B-cell chronic leukaemia. Br. J. Haematol. 2002, 117, 842–851. [Google Scholar] [CrossRef]
- Kolb, J.P.; Roman, V.; Mentz, F.; Zhao, H.; Rouillard, D.; Dugas, N.; Dugas, B.; Sigaux, F. Contribution of nitric oxide to the apoptotic process in human B cell chronic lymphocytic leukaemia. Leuk. Lymphoma 2001, 40, 243–257. [Google Scholar]
- Bhardwaj, A.; Sethi, G.; Vadhan-Raj, S.; Bueso-Ramos, C.; Takada, Y.; Gaur, U.; Nair, A.S.; Shishodia, S.; Aggarwal, B.B. Resveratrol inhibits proliferation, induces apoptosis, and overcomes chemoresistance through down-regulation of STAT3 and nuclear factor-kappaB-regulated antiapoptotic and cell survival gene products in human multiple myeloma cells. Blood 2007, 109, 2293–2302. [Google Scholar]
- Youn, J.; Lee, J.S.; Na, H.K.; Kundu, J.K.; Surh, Y.J. Resveratrol and piceatannol inhibit iNOS expression and NF-kappaB activation in dextran sulfate sodium-induced mouse colitis. Nutr. Cancer 2009, 61, 847–854. [Google Scholar] [CrossRef]
- Kundu, J.K.; Shin, Y.K.; Surh, Y.J. Resveratrol modulates phorbol ester-induced pro-inflammatory signal transduction pathways in mouse skin in vivo: NF-kappaB and AP-1 as prime targets. Biochem Pharmacol 2006, 72, 1506–1515. [Google Scholar] [CrossRef]
- Kundu, J.K.; Surh, Y.J. Molecular basis of chemoprevention by resveratrol: NF-kappaB and AP-1 as potential targets. Mutat. Res. 2004, 555, 65–80. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.J.; Adair, B.; Alder, H.; Limagne, E.; Taccioli, C.; Ferracin, M.; Delmas, D.; Latruffe, N.; Croce, C.M. Resveratrol decreases the levels of miR-155 by upregulating miR-663, a microRNA targeting JunB and JunD. Carcinogenesis 2010. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.J.; Alder, H.; Volinia, S.; Delmas, D.; Latruffe, N.; Croce, C.M. Resveratrol modulates the levels of microRNAs targeting genes encoding tumor-suppressors and effectors of TGFbeta signaling pathway in SW480 cells. Biochem. Pharmacol. 2010.
- Kundu, J.K.; Shin, Y.K.; Kim, S.H.; Surh, Y.J. Resveratrol inhibits phorbol ester-induced expression of COX-2 and activation of NF-kappaB in mouse skin by blocking IkappaB kinase activity. Carcinogenesis 2006, 27, 1465–1474. [Google Scholar] [CrossRef]
- Harikumar, K.B.; Kunnumakkara, A.B.; Sethi, G.; Diagaradjane, P.; Anand, P.; Pandey, M.K.; Gelovani, J.; Krishnan, S.; Guha, S.; Aggarwal, B.B. Resveratrol, a multitargeted agent, can enhance antitumor activity of gemcitabine in vitro and in orthotopic mouse model of human pancreatic cancer. Int. J. Cancer 2010, 127, 257–268. [Google Scholar]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; Scherer, B.; Sinclair, D.A. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar]
- Chung, S.; Yao, H.; Caito, S.; Hwang, J.W.; Arunachalam, G.; Rahman, I. Regulation of SIRT1 in cellular functions: role of polyphenols. Arch. Biochem. Biophys. 2010, 501, 79–90. [Google Scholar] [CrossRef]
- Knutson, M.D.; Leeuwenburgh, C. Resveratrol and novel potent activators of SIRT1: effects on aging and age-related diseases. Nutr. Rev. 2008, 66, 591–596. [Google Scholar] [CrossRef]
- Kaeberlein, M.; McDonagh, T.; Heltweg, B.; Hixon, J.; Westman, E.A.; Caldwell, S.D.; Napper, A.; Curtis, R.; DiStefano, P.S.; Fields, S.; Bedalov, A.; Kennedy, B.K. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005, 280, 17038–17045. [Google Scholar]
- Beher, D.; Wu, J.; Cumine, S.; Kim, K.W.; Lu, S.C.; Atangan, L.; Wang, M. Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem. Biol. Drug Des. 2009, 74, 619–624. [Google Scholar] [CrossRef]
- Pacholec, M.; Bleasdale, J.E.; Chrunyk, B.; Cunningham, D.; Flynn, D.; Garofalo, R.S.; Griffith, D.; Griffor, M.; Loulakis, P.; Pabst, B.; Qiu, X.; Stockman, B.; Thanabal, V.; Varghese, A.; Ward, J.; Withka, J.; Ahn, K. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 2010, 285, 8340–8351. [Google Scholar]
- Boily, G.; He, X.H.; Pearce, B.; Jardine, K.; McBurney, M.W. SirT1-null mice develop tumors at normal rates but are poorly protected by resveratrol. Oncogene 2009, 28, 2882–2893. [Google Scholar] [CrossRef]
- Zee, R.S.; Yoo, C.B.; Pimentel, D.R.; Perlman, D.H.; Burgoyne, J.R.; Hou, X.; McComb, M.E.; Costello, C.E.; Cohen, R.A.; Bachschmid, M.M. Redox regulation of sirtuin-1 by S-glutathiolation. Antioxid. Redox Signal. 2010, 13, 1023–1032. [Google Scholar] [CrossRef]
- Kao, C.L.; Chen, L.K.; Chang, Y.L.; Yung, M.C.; Hsu, C.C.; Chen, Y.C.; Lo, W.L.; Chen, S.J.; Ku, H.H.; Hwang, S.J. Resveratrol Protects Human Endothelium from H(2)O(2)-Induced Oxidative Stress and Senescence via SirT1 Activation. J. Atheroscler. Thromb. 2010, 17, 970–979. [Google Scholar] [CrossRef]
- Caito, S.; Rajendrasozhan, S.; Cook, S.; Chung, S.; Yao, H.; Friedman, A.E.; Brookes, P.S.; Rahman, I. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 2010, 24, 3145–3159. [Google Scholar] [CrossRef]
- Clement, M.V.; Hirpara, J.L.; Chawdhury, S.H.; Pervaiz, S. Chemopreventive agent resveratrol, a natural product derived from grapes, triggers CD95 signaling-dependent apoptosis in human tumor cells. Blood 1998, 92, 996–1002. [Google Scholar]
- Surh, Y.J.; Hurh, Y.J.; Kang, J.Y.; Lee, E.; Kong, G.; Lee, S.J. Resveratrol, an antioxidant present in red wine, induces apoptosis in human promyelocytic leukemia (HL-60) cells. Cancer Lett. 1999, 140, 1–10. [Google Scholar]
- Dorrie, J.; Gerauer, H.; Wachter, Y.; Zunino, S.J. Resveratrol induces extensive apoptosis by depolarizing mitochondrial membranes and activating caspase-9 in acute lymphoblastic leukemia cells. Cancer Res. 2001, 61, 4731–4739. [Google Scholar]
- Ovesna, Z.; Horvathova-Kozics, K. Structure-activity relationship of trans-resveratrol and its analogues. Neoplasma 2005, 52, 450–455. [Google Scholar]
- Fang, J.G.; Lu, M.; Chen, Z.H.; Zhu, H.H.; Li, Y.; Yang, L.; Wu, L.M.; Liu, Z.L. Antioxidant effects of resveratrol and its analogues against the free-radical-induced peroxidation of linoleic acid in micelles. Chemistry 2002, 8, 4191–4198. [Google Scholar] [CrossRef]
- Shang, Y.J.; Qian, Y.P.; Liu, X.D.; Dai, F.; Shang, X.L.; Jia, W.Q.; Liu, Q.; Fang, J.G.; Zhou, B. Radical-scavenging activity and mechanism of resveratrol-oriented analogues: influence of the solvent, radical, and substitution. J. Org. Chem. 2009, 74, 5025–5031. [Google Scholar] [CrossRef]
- Fan, G.J.; Liu, X.D.; Qian, Y.P.; Shang, Y.J.; Li, X.Z.; Dai, F.; Fang, J.G.; Jin, X.L.; Zhou, B. 4,4'-Dihydroxy-trans-stilbene, a resveratrol analogue, exhibited enhanced antioxidant activity and cytotoxicity. Bioorg. Med. Chem. 2009, 17, 2360–2365. [Google Scholar]
- Qian, Y.P.; Cai, Y.J.; Fan, G.J.; Wei, Q.Y.; Yang, J.; Zheng, L.F.; Li, X.Z.; Fang, J.G.; Zhou, B. Antioxidant-based lead discovery for cancer chemoprevention: the case of resveratrol. J. Med. Chem. 2009, 52, 1963–1974. [Google Scholar]
- Murias, M.; Jager, W.; Handler, N.; Erker, T.; Horvath, Z.; Szekeres, T.; Nohl, H.; Gille, L. Antioxidant, prooxidant and cytotoxic activity of hydroxylated resveratrol analogues: structure-activity relationship. Biochem. Pharmacol. 2005, 69, 903–912. [Google Scholar]
- Horvath, Z.; Murias, M.; Saiko, P.; Erker, T.; Handler, N.; Madlener, S.; Jaeger, W.; Grusch, M.; Fritzer-Szekeres, M.; Krupitza, G.; Szekeres, T. Cytotoxic and biochemical effects of 3,3',4,4',5,5'-hexahydroxystilbene, a novel resveratrol analog in HL-60 human promyelocytic leukemia cells. Exp. Hematol. 2006, 34, 1377–1384. [Google Scholar] [CrossRef]
- Ovesna, Z.; Kozics, K.; Bader, Y.; Saiko, P.; Handler, N.; Erker, T.; Szekeres, T. Antioxidant activity of resveratrol, piceatannol and 3,3',4,4',5,5'-hexahydroxy-trans-stilbene in three leukemia cell lines. Oncol. Rep. 2006, 16, 617–624. [Google Scholar]
- Fulda, S. Resveratrol and derivatives for the prevention and treatment of cancer. Drug Discov. Today 2010. [Google Scholar] [CrossRef]
- Saiko, P.; Ozsvar-Kozma, M.; Bernhaus, A.; Jaschke, M.; Graser, G.; Lackner, A.; Grusch, M.; Horvath, Z.; Madlener, S.; Krupitza, G.; Handler, N.; Erker, T.; Jaeger, W.; Fritzer-Szekeres, M.; Szekeres, T. N-hydroxy-N'-(3,4,5-trimethoxyphenyl)-3,4,5-trimethoxy-benzamidine, a novel resveratrol analog, inhibits ribonucleotide reductase in HL-60 human promyelocytic leukemia cells: synergistic antitumor activity with arabinofuranosylcytosine. Int. J. Oncol. 2007, 31, 1261–1266. [Google Scholar]
- Roberti, M.; Pizzirani, D.; Simoni, D.; Rondanin, R.; Baruchello, R.; Bonora, C.; Buscemi, F.; Grimaudo, S.; Tolomeo, M. Synthesis and biological evaluation of resveratrol and analogues as apoptosis-inducing agents. J. Med. Chem. 2003, 46, 3546–3554. [Google Scholar] [CrossRef]
- Colin, D.; Gimazane, A.; Lizard, G.; Izard, J.C.; Solary, E.; Latruffe, N.; Delmas, D. Effects of resveratrol analogs on cell cycle progression, cell cycle associated proteins and 5fluoro-uracil sensitivity in human derived colon cancer cells. Int. J. Cancer 2009, 124, 2780–2788. [Google Scholar]
- Mazue, F.; Colin, D.; Gobbo, J.; Wegner, M.; Rescifina, A.; Spatafora, C.; Fasseur, D.; Delmas, D.; Meunier, P.; Tringali, C.; Latruffe, N. Structural determinants of resveratrol for cell proliferation inhibition potency: experimental and docking studies of new analogs. Eur. J. Med. Chem. 2010, 45, 2972–2980. [Google Scholar]
- Schneider, Y.; Chabert, P.; Stutzmann, J.; Coelho, D.; Fougerousse, A.; Gosse, F.; Launay, J.F.; Brouillard, R.; Raul, F. Resveratrol analog (Z)-3,5,4'-trimethoxystilbene is a potent anti-mitotic drug inhibiting tubulin polymerization. Int. J. Cancer 2003, 107, 189–196. [Google Scholar] [CrossRef]
- Rimando, A.M.; Suh, N. Biological/chemopreventive activity of stilbenes and their effect on colon cancer. Planta Med. 2008, 74, 1635–1643. [Google Scholar] [CrossRef]
- Nigro, P.; Bloise, E.; Turco, M.C.; Skhirtladze, A.; Montoro, P.; Pizza, C.; Piacente, S.; Belisario, M.A. Antiproliferative and pro-apoptotic activity of novel phenolic derivatives of resveratrol. Life Sci. 2007, 81, 873–883. [Google Scholar] [CrossRef]
- Tolomeo, M.; Grimaudo, S.; Di Cristina, A.; Roberti, M.; Pizzirani, D.; Meli, M.; Dusonchet, L.; Gebbia, N.; Abbadessa, V.; Crosta, L.; Barucchello, R.; Grisolia, G.; Invidiata, F.; Simoni, D. Pterostilbene and 3'-hydroxypterostilbene are effective apoptosis-inducing agents in MDR and BCR-ABL-expressing leukemia cells. Int. J. Biochem. Cell. Biol. 2005, 37, 1709–1726. [Google Scholar] [CrossRef]
- Gao, X.; Xu, Y.X.; Divine, G.; Janakiraman, N.; Chapman, R.A.; Gautam, S.C. Disparate in vitro and in vivo antileukemic effects of resveratrol, a natural polyphenolic compound found in grapes. J. Nutr. 2002, 132, 2076–2081. [Google Scholar]
- Aggarwal, S.; Ichikawa, H.; Takada, Y.; Sandur, S.K.; Shishodia, S.; Aggarwal, B.B. Curcumin (diferuloylmethane) down-regulates expression of cell proliferation and antiapoptotic and metastatic gene products through suppression of IkappaBalpha kinase and Akt activation. Mol. Pharmacol. 2006, 69, 195–206. [Google Scholar]
- Chainani-Wu, N. Safety and anti-inflammatory activity of curcumin: a component of tumeric (Curcuma longa). J. Altern. Complement. Med. 2003, 9, 161–168. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Sundaram, C.; Malani, N.; Ichikawa, H. Curcumin: the Indian solid gold. Adv. Exp. Med. Biol. 2007, 595, 1–75. [Google Scholar] [CrossRef]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef]
- Iqbal, M.; Okazaki, Y.; Okada, S. Curcumin attenuates oxidative damage in animals treated with a renal carcinogen, ferric nitrilotriacetate (Fe-NTA): implications for cancer prevention. Mol. Cell. Biochem. 2009, 324, 157–164. [Google Scholar] [CrossRef]
- Duvoix, A.; Blasius, R.; Delhalle, S.; Schnekenburger, M.; Morceau, F.; Henry, E.; Dicato, M.; Diederich, M. Chemopreventive and therapeutic effects of curcumin. Cancer Lett. 2005, 223, 181–190. [Google Scholar] [CrossRef]
- Reuter, S.; Eifes, S.; Dicato, M.; Aggarwal, B.B.; Diederich, M. Modulation of anti-apoptotic and survival pathways by curcumin as a strategy to induce apoptosis in cancer cells. Biochem. Pharmacol. 2008, 76, 1340–1351. [Google Scholar] [CrossRef]
- Peeyush, K.T.; Gireesh, G.; Jobin, M.; Paulose, C.S. Neuroprotective role of curcumin in the cerebellum of streptozotocin-induced diabetic rats. Life Sci. 2009, 85, 704–710. [Google Scholar] [CrossRef]
- Mazumder, A.; Raghavan, K.; Weinstein, J.; Kohn, K.W.; Pommier, Y. Inhibition of human immunodeficiency virus type-1 integrase by curcumin. Biochem. Pharmacol. 1995, 49, 1165–1170. [Google Scholar] [CrossRef]
- Conney, A.H.; Lysz, T.; Ferraro, T.; Abidi, T.F.; Manchand, P.S.; Laskin, J.D.; Huang, M.T. Inhibitory effect of curcumin and some related dietary compounds on tumor promotion and arachidonic acid metabolism in mouse skin. Adv. Enzyme Regul. 1991, 31, 385–396. [Google Scholar] [CrossRef]
- Goel, A.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin as "Curecumin": from kitchen to clinic. Biochem. Pharmacol. 2008, 75, 787–809. [Google Scholar] [CrossRef]
- Huang, M.T.; Lou, Y.R.; Xie, J.G.; Ma, W.; Lu, Y.P.; Yen, P.; Zhu, B.T.; Newmark, H.; Ho, C.T. Effect of dietary curcumin and dibenzoylmethane on formation of 7,12-dimethylbenz[a]anthracene-induced mammary tumors and lymphomas/leukemias in Sencar mice. Carcinogenesis 1998, 19, 1697–1700. [Google Scholar] [CrossRef]
- Huang, M.T.; Smart, R.C.; Wong, C.Q.; Conney, A.H. Inhibitory effect of curcumin, chlorogenic acid, caffeic acid, and ferulic acid on tumor promotion in mouse skin by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1988, 48, 5941–5946. [Google Scholar]
- Huang, M.T.; Wang, Z.Y.; Georgiadis, C.A.; Laskin, J.D.; Conney, A.H. Inhibitory effects of curcumin on tumor initiation by benzo[a]pyrene and 7,12-dimethylbenz[a]anthracene. Carcinogenesis 1992, 13, 2183–2186. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Anand, P.; Aggarwal, B.B. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008, 269, 199–225. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Diagaradjane, P.; Anand, P.; Harikumar, K.B.; Deorukhkar, A.; Gelovani, J.; Guha, S.; Krishnan, S.; Aggarwal, B.B. Curcumin sensitizes human colorectal cancer to capecitabine by modulation of cyclin D1, COX-2, MMP-9, VEGF and CXCR4 expression in an orthotopic mouse model. Int. J. Cancer 2009, 125, 2187–2197. [Google Scholar] [CrossRef]
- Ralhan, R.; Pandey, M.K.; Aggarwal, B.B. Nuclear factor-kappa B links carcinogenic and chemopreventive agents. Front Biosci (Schol Ed) 2009, 1, 45–60. [Google Scholar]
- Tharakan, S.T.; Inamoto, T.; Sung, B.; Aggarwal, B.B.; Kamat, A.M. Curcumin potentiates the antitumor effects of gemcitabine in an orthotopic model of human bladder cancer through suppression of proliferative and angiogenic biomarkers. Biochem. Pharmacol. 2010, 79, 218–228. [Google Scholar] [CrossRef]
- Sandur, S.K.; Pandey, M.K.; Sung, B.; Ahn, K.S.; Murakami, A.; Sethi, G.; Limtrakul, P.; Badmaev, V.; Aggarwal, B.B. Curcumin, demethoxycurcumin, bisdemethoxycurcumin, tetrahydrocurcumin and turmerones differentially regulate anti-inflammatory and anti-proliferative responses through a ROS-independent mechanism. Carcinogenesis 2007, 28, 1765–1773. [Google Scholar] [CrossRef]
- Somparn, P.; Phisalaphong, C.; Nakornchai, S.; Unchern, S.; Morales, N.P. Comparative antioxidant activities of curcumin and its demethoxy and hydrogenated derivatives. Biol. Pharm. Bull. 2007, 30, 74–78. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Kawakishi, S.; Osawa, T. Involvement of the beta-diketone moiety in the antioxidative mechanism of tetrahydrocurcumin. Biochem. Pharmacol. 1996, 52, 519–525. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Kumar, A.; Bharti, A.C. Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res. 2003, 23, 363–398. [Google Scholar]
- Naidu, K.A.; Thippeswamy, N.B. Inhibition of human low density lipoprotein oxidation by active principles from spices. Mol. Cell. Biochem. 2002, 229, 19–23. [Google Scholar] [CrossRef]
- Chen, W.F.; Deng, S.L.; Zhou, B.; Yang, L.; Liu, Z L. Curcumin and its analogues as potent inhibitors of low density lipoprotein oxidation: H-atom abstraction from the phenolic groups and possible involvement of the 4-hydroxy-3-methoxyphenyl groups. Free Radic. Biol. Med. 2006, 40, 526–535. [Google Scholar] [CrossRef]
- Wei, Q.Y.; Chen, W.F.; Zhou, B.; Yang, L.; Liu, Z.L. Inhibition of lipid peroxidation and protein oxidation in rat liver mitochondria by curcumin and its analogues. Biochim. Biophys. Acta. 2006, 1760, 70–77. [Google Scholar]
- Dai, F.; Chen, W.F.; Zhou, B.; Yang, L.; Liu, Z.L. Antioxidative effects of curcumin and its analogues against the free-radical-induced peroxidation of linoleic acid in micelles. Phytother Res 2009, 23, 1220–1228. [Google Scholar] [CrossRef]
- Ak, T.; Gulcin, I. Antioxidant and radical scavenging properties of curcumin. Chem. Biol. Interact. 2008, 174, 27–37. [Google Scholar] [CrossRef]
- Biswas, J.; Sinha, D.; Mukherjee, S.; Roy, S.; Siddiqi, M.; Roy, M. Curcumin protects DNA damage in a chronically arsenic-exposed population of West Bengal. Hum. Exp. Toxicol. 2010, 29, 513–524. [Google Scholar] [CrossRef]
- Iqbal, M.; Okazaki, Y.; Okada, S. In vitro curcumin modulates ferric nitrilotriacetate (Fe-NTA) and hydrogen peroxide (H2O2)-induced peroxidation of microsomal membrane lipids and DNA damage. Teratog Carcinog Mutagen 2003, 23 (Suppl. 1), 151–160. [Google Scholar]
- Iqbal, M.; Sharma, S.D.; Okazaki, Y.; Fujisawa, M.; Okada, S. Dietary supplementation of curcumin enhances antioxidant and phase II metabolizing enzymes in ddY male mice: possible role in protection against chemical carcinogenesis and toxicity. Pharmacol. Toxicol. 2003, 92, 33–38. [Google Scholar] [CrossRef]
- Bhaumik, S.; Anjum, R.; Rangaraj, N.; Pardhasaradhi, B.V.; Khar, A. Curcumin mediated apoptosis in AK-5 tumor cells involves the production of reactive oxygen intermediates. FEBS Lett. 1999, 456, 311–314. [Google Scholar] [CrossRef]
- Chen, J.; Wanming, D.; Zhang, D.; Liu, Q.; Kang, J. Water-soluble antioxidants improve the antioxidant and anticancer activity of low concentrations of curcumin in human leukemia cells. Pharmazie 2005, 60, 57–61. [Google Scholar]
- Galati, G.; Sabzevari, O.; Wilson, J.X.; O'Brien, P.J. Prooxidant activity and cellular effects of the phenoxyl radicals of dietary flavonoids and other polyphenolics. Toxicology 2002, 177, 91–104. [Google Scholar] [CrossRef]
- Kong, Y.; Ma, W.; Liu, X.; Zu, Y.; Fu, Y.; Wu, N.; Liang, L.; Yao, L.; Efferth, T. Cytotoxic activity of curcumin towards CCRF-CEM leukemia cells and its effect on DNA damage. Molecules 2009, 14, 5328–5338. [Google Scholar] [CrossRef]
- Kang, J.; Chen, J.; Shi, Y.; Jia, J.; Zhang, Y. Curcumin-induced histone hypoacetylation: the role of reactive oxygen species. Biochem. Pharmacol. 2005, 69, 1205–1213. [Google Scholar] [CrossRef]
- Fry, C.J.; Peterson, C.L. Transcription. Unlocking the gates to gene expression. Science 2002, 295, 1847–1848. [Google Scholar] [CrossRef]
- Fang, J.; Lu, J.; Holmgren, A. Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. J. Biol. Chem. 2005, 280, 25284–25290. [Google Scholar] [CrossRef]
- Syng-Ai, C.; Kumari, A.L.; Khar, A. Effect of curcumin on normal and tumor cells: role of glutathione and bcl-2. Mol. Cancer Ther. 2004, 3, 1101–1108. [Google Scholar]
- Awasthi, S.; Pandya, U.; Singhal, S.S.; Lin, J.T.; Thiviyanathan, V.; Seifert, W.E.Jr.; Awasthi, Y.C.; Ansari, G.A. Curcumin-glutathione interactions and the role of human glutathione S-transferase P1-1. Chem. Biol. Interact. 2000, 128, 19–38. [Google Scholar] [CrossRef]
- Ghibelli, L.; Coppola, S.; Rotilio, G.; Lafavia, E.; Maresca, V.; Ciriolo, M.R. Non-oxidative loss of glutathione in apoptosis via GSH extrusion. Biochem. Biophys. Res. Commun. 1995, 216, 313–320. [Google Scholar] [CrossRef]
- Coppola, S.; Ghibelli, L. GSH extrusion and and the mitochondrial pathway of apoptotic signalling. Biochem. Soc. Trans. 2000, 28, 56–61. [Google Scholar]
- Armstrong, J.S.; Steinauer, K.K.; Hornung, B.; Irish, J.M.; Lecane, P.; Birrell, G.W.; Peehl, D.M.; Knox, S.J. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 2002, 9, 252–263. [Google Scholar] [CrossRef]
- Franco, R.; Panayiotidis, M.I.; Cidlowski, J.A. Glutathione depletion is necessary for apoptosis in lymphoid cells independent of reactive oxygen species formation. J. Biol. Chem. 2007, 282, 30452–30465. [Google Scholar] [CrossRef]
- Voehringer, D.W.; McConkey, D.J.; McDonnell, T.J.; Brisbay, S.; Meyn, R.E. Bcl-2 expression causes redistribution of glutathione to the nucleus. Proc. Natl. Acad. Sci. USA 1998, 95, 2956–2960. [Google Scholar] [CrossRef]
- Meredith, M.J.; Cusick, C.L.; Soltaninassab, S.; Sekhar, K.S.; Lu, S.; Freeman, M.L. Expression of Bcl-2 increases intracellular glutathione by inhibiting methionine-dependent GSH efflux. Biochem. Biophys. Res. Commun. 1998, 248, 458–463. [Google Scholar] [CrossRef]
- Chendil, D.; Ranga, R.S.; Meigooni, D.; Sathishkumar, S.; Ahmed, M.M. Curcumin confers radiosensitizing effect in prostate cancer cell line PC-3. Oncogene 2004, 23, 1599–1607. [Google Scholar] [CrossRef]
- Piwocka, K.; Jaruga, E.; Skierski, J.; Gradzka, I.; Sikora, E. Effect of glutathione depletion on caspase-3 independent apoptosis pathway induced by curcumin in Jurkat cells. Free Radic. Biol. Med. 2001, 31, 670–678. [Google Scholar] [CrossRef]
- Na, H.K.; Surh, Y.J. Transcriptional regulation via cysteine thiol modification: a novel molecular strategy for chemoprevention and cytoprotection. Mol. Carcinog. 2006, 45, 368–380. [Google Scholar] [CrossRef]
- Lin, J.K. Molecular targets of curcumin. Adv. Exp. Med. Biol. 2007, 595, 227–243. [Google Scholar] [CrossRef]
- Ravindran, J.; Prasad, S.; Aggarwal, B.B. Curcumin and cancer cells: how many ways can curry kill tumor cells selectively? AAPS J. 2009, 11, 495–510. [Google Scholar] [CrossRef]
- Dang, C.V. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar]
- Basile, V.; Ferrari, E.; Lazzari, S.; Belluti, S.; Pignedoli, F.; Imbriano, C. Curcumin derivatives: molecular basis of their anti-cancer activity. Biochem. Pharmacol. 2009, 78, 1305–1315. [Google Scholar] [CrossRef]
- Sun, C.; Liu, X.; Chen, Y.; Liu, F. Anticancer effect of curcumin on human B cell non-Hodgkin's lymphoma. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2005, 25, 404–407. [Google Scholar] [CrossRef]
- Duvoix, A.; Morceau, F.; Schnekenburger, M.; Delhalle, S.; Galteau, M.M.; Dicato, M.; Diederich, M. Curcumin-induced cell death in two leukemia cell lines: K562 and Jurkat. Ann. NY Acad. Sci. 2003, 1010, 389–392. [Google Scholar] [CrossRef]
- Skommer, J.; Wlodkowic, D.; Pelkonen, J. Cellular foundation of curcumin-induced apoptosis in follicular lymphoma cell lines. Exp. Hematol. 2006, 34, 463–474. [Google Scholar] [CrossRef]
- Hussain, A.R.; Al-Rasheed, M.; Manogaran, P.S.; Al-Hussein, K.A.; Platanias, L.C.; Al Kuraya, K.; Uddin, S. Curcumin induces apoptosis via inhibition of PI3'-kinase/AKT pathway in acute T cell leukemias. Apoptosis 2006, 11, 245–254. [Google Scholar] [CrossRef]
- William, B.M.; Goodrich, A.; Peng, C.; Li, S. Curcumin inhibits proliferation and induces apoptosis of leukemic cells expressing wild-type or T315I-BCR-ABL and prolongs survival of mice with acute lymphoblastic leukemia. Hematology 2008, 13, 333–343. [Google Scholar] [CrossRef]
- Anto, R.J.; Mukhopadhyay, A.; Shishodia, S.; Gairola, C.G.; Aggarwal, B.B. Cigarette smoke condensate activates nuclear transcription factor-kappaB through phosphorylation and degradation of IkappaB(alpha): correlation with induction of cyclooxygenase-2. Carcinogenesis 2002, 23, 1511–1518. [Google Scholar] [CrossRef]
- Blasius, R.; Dicato, M.; Diederich, M. Effect of curcumin treatment on protein phosphorylation in K562 cells. Ann. NY Acad. Sci. 2007, 1095, 377–387. [Google Scholar] [CrossRef]
- Blasius, R.; Reuter, S.; Henry, E.; Dicato, M.; Diederich, M. Curcumin regulates signal transducer and activator of transcription (STAT) expression in K562 cells. Biochem. Pharmacol. 2006, 72, 1547–1554. [Google Scholar] [CrossRef]
- Rajasingh, J.; Raikwar, H.P.; Muthian, G.; Johnson, C.; Bright, J.J. Curcumin induces growth-arrest and apoptosis in association with the inhibition of constitutively active JAK-STAT pathway in T cell leukemia. Biochem. Biophys. Res. Commun. 2006, 340, 359–368. [Google Scholar] [CrossRef]
- Uddin, S.; Hussain, A.R.; Manogaran, P.S.; Al-Hussein, K.; Platanias, L.C.; Gutierrez, M.I.; Bhatia, K.G. Curcumin suppresses growth and induces apoptosis in primary effusion lymphoma. Oncogene 2005, 24, 7022–7030. [Google Scholar] [CrossRef]
- Shishodia, S.; Singh, T.; Chaturvedi, M.M. Modulation of transcription factors by curcumin. Adv. Exp. Med. Biol. 2007, 595, 127–148. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Kay, N.E.; Secreto, C.R.; Shanafelt, T.D. Curcumin inhibits prosurvival pathways in chronic lymphocytic leukemia B cells and may overcome their stromal protection in combination with EGCG. Clin. Cancer Res. 2009, 15, 1250–1258. [Google Scholar] [CrossRef]
- Zhang, C.; Li, B.; Zhang, X.; Hazarika, P.; Aggarwal, B.B.; Duvic, M. Curcumin selectively induces apoptosis in cutaneous T-cell lymphoma cell lines and patients' PBMCs: potential role for STAT-3 and NF-kappaB signaling. J. Invest. Dermatol. 2010, 130, 2110–2119. [Google Scholar] [CrossRef]
- Reuter, S.; Schnekenburger, M.; Cristofanon, S.; Buck, I.; Teiten, M.H.; Daubeuf, S.; Eifes, S.; Dicato, M.; Aggarwal, B.B.; Visvikis, A.; Diederich, M. Tumor necrosis factor alpha induces gamma-glutamyltransferase expression via nuclear factor-kappaB in cooperation with Sp1. Biochem. Pharmacol. 2009, 77, 397–411. [Google Scholar] [CrossRef]
- Reuter, S.; Charlet, J.; Juncker, T.; Teiten, M.H.; Dicato, M.; Diederich, M. Effect of curcumin on nuclear factor kappaB signaling pathways in human chronic myelogenous K562 leukemia cells. Ann. NY Acad. Sci. 2009, 1171, 436–447. [Google Scholar] [CrossRef]
- Teiten, M.H.; Eifes, S.; Reuter, S.; Duvoix, A.; Dicato, M.; Diederich, M. Gene expression profiling related to anti-inflammatory properties of curcumin in K562 leukemia cells. Ann. NY Acad. Sci. 2009, 1171, 391–398. [Google Scholar] [CrossRef]
- Teiten, M.H.; Reuter, S.; Schmucker, S.; Dicato, M.; Diederich, M. Induction of heat shock response by curcumin in human leukemia cells. Cancer Lett. 2009, 279, 145–154. [Google Scholar] [CrossRef]
- Knowlton, A.A. NFkappaB, heat shock proteins, HSF-1, and inflammation. Cardiovasc Res. 2006, 69, 7–8. [Google Scholar] [CrossRef]
- Bhagat, L.; Singh, V.P.; Dawra, R.K.; Saluja, A.K. Sodium arsenite induces heat shock protein 70 expression and protects against secretagogue-induced trypsinogen and NF-kappaB activation. J. Cell. Physiol. 2008, 215, 37–46. [Google Scholar] [CrossRef]
- Chen, Y.; Currie, R.W. Small interfering RNA knocks down heat shock factor-1 (HSF-1) and exacerbates pro-inflammatory activation of NF-kappaB and AP-1 in vascular smooth muscle cells. Cardiovasc Res. 2006, 69, 66–75. [Google Scholar] [CrossRef]
- Duvoix, A.; Morceau, F.; Delhalle, S.; Schmitz, M.; Schnekenburger, M.; Galteau, M.M.; Dicato, M.; Diederich, M. Induction of apoptosis by curcumin: mediation by glutathione S-transferase P1-1 inhibition. Biochem. Pharmacol. 2003, 66, 1475–1483. [Google Scholar] [CrossRef]
- Blasius, R.; Duvoix, A.; Morceau, F.; Schnekenburger, M.; Delhalle, S.; Henry, E.; Dicato, M.; Diederich, M. Curcumin stability and its effect on glutathione S-transferase P1-1 mRNA expression in K562 cells. Ann. NY Acad. Sci. 2004, 1030, 442–448. [Google Scholar] [CrossRef]
- Ravindranath, V.; Chandrasekhara, N. Absorption and tissue distribution of curcumin in rats. Toxicology 1980, 16, 259–265. [Google Scholar] [CrossRef]
- Tidefelt, U.; Elmhorn-Rosenborg, A.; Paul, C.; Hao, X.Y.; Mannervik, B.; Eriksson, L.C. Expression of glutathione transferase pi as a predictor for treatment results at different stages of acute nonlymphoblastic leukemia. Cancer Res. 1992, 52, 3281–3285. [Google Scholar]
- Bengmark, S. Curcumin, an atoxic antioxidant and natural NFkappaB, cyclooxygenase-2, lipooxygenase, and inducible nitric oxide synthase inhibitor: a shield against acute and chronic diseaes. JPEN J. Parenter. Enteral. Nutr. 2006, 30, 45–51. [Google Scholar] [CrossRef]
- Menon, V.P.; Sudheer, A.R. Antioxidant and anti-inflammatory properties of curcumin. Adv. Exp. Med. Biol. 2007, 595, 105–125. [Google Scholar] [CrossRef]
- Kay, N.E.; Shanafelt, T.D.; Strege, A.K.; Lee, Y.K.; Bone, N.D.; Raza, A. Bone biopsy derived marrow stromal elements rescue chronic lymphocytic leukemia B-cells from spontaneous and drug induced cell death and facilitates an "angiogenic switch". Leuk. Res. 2007, 31, 899–906. [Google Scholar] [CrossRef]
- Lee, Y.K.; Shanafelt, T.D.; Bone, N.D.; Strege, A.K.; Jelinek, D.F.; Kay, N.E. VEGF receptors on chronic lymphocytic leukemia (CLL) B cells interact with STAT 1 and 3: implication for apoptosis resistance. Leukemia 2005, 19, 513–523. [Google Scholar]
- Plate, J.M.; Long, B.W.; Kelkar, S.B. Role of beta2 integrins in the prevention of apoptosis induction in chronic lymphocytic leukemia B cells. Leukemia 2000, 14, 34–39. [Google Scholar] [CrossRef]
- Lee, Y.K.; Bone, N.D.; Strege, A.K.; Shanafelt, T.D.; Jelinek, D.F.; Kay, N.E. VEGF receptor phosphorylation status and apoptosis is modulated by a green tea component, epigallocatechin-3-gallate (EGCG), in B-cell chronic lymphocytic leukemia. Blood 2004, 104, 788–794. [Google Scholar] [CrossRef]
- Somers-Edgar, T.J.; Scandlyn, M.J.; Stuart, E.C.; Le Nedelec, M.J.; Valentine, S.P.; Rosengren, R.J. The combination of epigallocatechin gallate and curcumin suppresses ER alpha-breast cancer cell growth in vitro and in vivo. Int. J. Cancer 2008, 122, 1966–1971. [Google Scholar]
- Sandur, S.K.; Deorukhkar, A.; Pandey, M.K.; Pabon, A.M.; Shentu, S.; Guha, S.; Aggarwal, B.B.; Krishnan, S. Curcumin modulates the radiosensitivity of colorectal cancer cells by suppressing constitutive and inducible NF-kappaB activity. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 534–542. [Google Scholar]
- Siddique, Y.H.; Ara, G.; Beg, T.; Gupta, J.; Afzal, M. Assessment of cell viability, lipid peroxidation and quantification of DNA fragmentation after the treatment of anticancerous drug mitomycin C and curcumin in cultured human blood lymphocytes. Exp. Toxicol. Pathol. 2009. [Google Scholar] [CrossRef]
- Kikuchi, H.; Kuribayashi, F.; Kiwaki, N.; Nakayama, T. Curcumin dramatically enhances retinoic acid-induced superoxide generating activity via accumulation of p47-phox and p67-phox proteins in U937 cells. Biochem. Biophys. Res. Commun. 2010, 395, 61–65. [Google Scholar] [CrossRef]
- Bisht, S.; Feldmann, G.; Soni, S.; Ravi, R.; Karikar, C.; Maitra, A. Polymeric nanoparticle-encapsulated curcumin ("nanocurcumin"): a novel strategy for human cancer therapy. J. Nanobiotechnology 2007, 5, 3. [Google Scholar] [CrossRef]
- Narayanan, N.K.; Nargi, D.; Randolph, C.; Narayanan, B.A. Liposome encapsulation of curcumin and resveratrol in combination reduces prostate cancer incidence in PTEN knockout mice. Int. J. Cancer 2009, 125, 1–8. [Google Scholar] [CrossRef]
- Lapenna, S.; Bilia, A.R.; Morris, G.A.; Nilsson, M. Novel artemisinin and curcumin micellar formulations: drug solubility studies by NMR spectroscopy. J. Pharm. Sci. 2009, 98, 3666–3675. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Anand, P.; Nair, H.B.; Sung, B.; Kunnumakkara, A.B.; Yadav, V.R.; Tekmal, R.R.; Aggarwal, B.B. Design of curcumin-loaded PLGA nanoparticles formulation with enhanced cellular uptake, and increased bioactivity in vitro and superior bioavailability in vivo. Biochem. Pharmacol. 2010, 79, 330–338. [Google Scholar] [CrossRef]
- Yadav, V.R.; Prasad, S.; Kannappan, R.; Ravindran, J.; Chaturvedi, M.M.; Vaahtera, L.; Parkkinen, J.; Aggarwal, B.B. Cyclodextrin-complexed curcumin exhibits anti-inflammatory and antiproliferative activities superior to those of curcumin through higher cellular uptake. Biochem. Pharmacol. 2010, 80, 1021–1032. [Google Scholar] [CrossRef]
- Teiten, M.H.; Gaascht, F.; Eifes, S.; Dicato, M.; Diederich, M. Chemopreventive potential of curcumin in prostate cancer. Genes. Nutr. 2010, 5, 61–74. [Google Scholar] [CrossRef]
- Ravindran, J.; Subbaraju, G.V.; Ramani, M.V.; Sung, B.; Aggarwal, B.B. Bisdemethylcurcumin and structurally related hispolon analogues of curcumin exhibit enhanced prooxidant, anti-proliferative and anti-inflammatory activities in vitro. Biochem. Pharmacol. 2010, 79, 1658–1666. [Google Scholar] [CrossRef]
- Anand, P.; Thomas, S.G.; Kunnumakkara, A.B.; Sundaram, C.; Harikumar, K.B.; Sung, B.; Tharakan, S.T.; Misra, K.; Priyadarsini, I.K.; Rajasekharan, K.N.; Aggarwal, B.B. Biological activities of curcumin and its analogues (Congeners) made by man and Mother Nature. Biochem. Pharmacol. 2008, 76, 1590–1611. [Google Scholar] [CrossRef]
- Ruby, A.J.; Kuttan, G.; Babu, K.D.; Rajasekharan, K.N.; Kuttan, R. Anti-tumour and antioxidant activity of natural curcuminoids. Cancer Lett. 1995, 94, 79–83. [Google Scholar] [CrossRef]
- Ahsan, H.; Parveen, N.; Khan, N.U.; Hadi, S.M. Pro-oxidant, anti-oxidant and cleavage activities on DNA of curcumin and its derivatives demethoxycurcumin and bisdemethoxycurcumin. Chem. Biol. Interact. 1999, 121, 161–175. [Google Scholar] [CrossRef]
- Anuchapreeda, S.; Tima, S.; Duangrat, C.; Limtrakul, P. Effect of pure curcumin, demethoxycurcumin, and bisdemethoxycurcumin on WT1 gene expression in leukemic cell lines. Cancer Chemother. Pharmacol. 2008, 62, 585–594. [Google Scholar] [CrossRef]
- Inoue, K.; Ogawa, H.; Sonoda, Y.; Kimura, T.; Sakabe, H.; Oka, Y.; Miyake, S.; Tamaki, H.; Oji, Y.; Yamagami, T.; Tatekawa, T.; Soma, T.; Kishimoto, T.; Sugiyama, H. Aberrant overexpression of the Wilms tumor gene (WT1) in human leukemia. Blood 1997, 89, 1405–1412. [Google Scholar]
- Wu, Y.; Fraizer, G.C.; Saunders, G.F. GATA-1 transactivates the WT1 hematopoietic specific enhancer. J. Biol. Chem. 1995, 270, 5944–5949. [Google Scholar] [CrossRef]
- Ireson, C.R.; Jones, D.J.; Orr, S.; Coughtrie, M.W.; Boocock, D.J.; Williams, M.L.; Farmer, P.B.; Steward, W.P.; Gescher, A.J. Metabolism of the cancer chemopreventive agent curcumin in human and rat intestine. Cancer Epidemiol. Biomarkers. Prev. 2002, 11, 105–111. [Google Scholar]
- Fuchs, J.R.; Pandit, B.; Bhasin, D.; Etter, J.P.; Regan, N.; Abdelhamid, D.; Li, C.; Lin, J.; Li, P.K. Structure-activity relationship studies of curcumin analogues. Bioorg. Med. Chem. Lett. 2009, 19, 2065–2069. [Google Scholar] [CrossRef]
- Youssef, K.M.; El-Sherbeny, M.A. Synthesis and antitumor activity of some curcumin analogs. Arch. Pharm. (Weinheim) 2005, 338, 181–189. [Google Scholar] [CrossRef]
- Ohori, H.; Yamakoshi, H.; Tomizawa, M.; Shibuya, M.; Kakudo, Y.; Takahashi, A.; Takahashi, S.; Kato, S.; Suzuki, T.; Ishioka, C.; Iwabuchi, Y.; Shibata, H. Synthesis and biological analysis of new curcumin analogues bearing an enhanced potential for the medicinal treatment of cancer. Mol. Cancer Ther. 2006, 5, 2563–2571. [Google Scholar] [CrossRef]
- Venkatesan, P.; Rao, M.N. Structure-activity relationships for the inhibition of lipid peroxidation and the scavenging of free radicals by synthetic symmetrical curcumin analogues. J. Pharm. Pharmacol. 2000, 52, 1123–1128. [Google Scholar] [CrossRef]
- Youssef, K.M.; El-Sherbeny, M.A.; El-Shafie, F.S.; Farag, H.; Al-Deeb, O.A.; Awadalla, S.A. Synthesis of curcumin analogues as potential antioxidant, cancer chemopreventive agents. Arch. Pharm. (Weinheim) 2004, 337, 42–54. [Google Scholar] [CrossRef]
- Lin, L.; Shi, Q.; Nyarko, A.K.; Bastow, K.F.; Wu, C.C.; Su, C.Y.; Shih, C.C.; Lee, K.H. Antitumor agents. 250. Design and synthesis of new curcumin analogues as potential anti-prostate cancer agents. J. Med. Chem. 2006, 49, 3963–3972. [Google Scholar] [CrossRef]
- Adams, B.K.; Cai, J.; Armstrong, J.; Herold, M.; Lu, Y.J.; Sun, A.; Snyder, J.P.; Liotta, D.C.; Jones, D.P.; Shoji, M. EF24, a novel synthetic curcumin analog, induces apoptosis in cancer cells via a redox-dependent mechanism. Anticancer Drugs 2005, 16, 263–275. [Google Scholar] [CrossRef]
- Mosley, C.A.; Liotta, D.C.; Snyder, J.P. Highly active anticancer curcumin analogues. Adv. Exp. Med. Biol. 2007, 595, 77–103. [Google Scholar] [CrossRef]
- Lin, L.; Shi, Q.; Su, C.Y.; Shih, C.C.; Lee, K.H. Antitumor agents 247. New 4-ethoxycarbonylethyl curcumin analogs as potential antiandrogenic agents. Bioorg. Med. Chem. 2006, 14, 2527–2534. [Google Scholar] [CrossRef]
- Somers-Edgar, T.J.; Taurin, S.; Larsen, L.; Chandramouli, A.; Nelson, M.A.; Rosengren, R.J. Mechanisms for the activity of heterocyclic cyclohexanone curcumin derivatives in estrogen receptor negative human breast cancer cell lines. Invest. New Drugs 2009.
- Yadav, B.; Taurin, S.; Rosengren, R.J.; Schumacher, M.; Diederich, M.; Somers-Edgar, T.J.; Larsen, L. Synthesis and cytotoxic potential of heterocyclic cyclohexanone analogues of curcumin. Bioorg. Med. Chem. 2010. [Google Scholar] [CrossRef]
- Ferrari, E.; Lazzari, S.; Marverti, G.; Pignedoli, F.; Spagnolo, F.; Saladini, M. Synthesis, cytotoxic and combined cDDP activity of new stable curcumin derivatives. Bioorg. Med. Chem. 2009, 17, 3043–3052. [Google Scholar] [CrossRef]
- Everett, P.C.; Meyers, J.A.; Makkinje, A.; Rabbi, M.; Lerner, A. Preclinical assessment of curcumin as a potential therapy for B-CLL. Am. J. Hematol. 2007, 82, 23–30. [Google Scholar] [CrossRef]
- Bar-Sela, G.; Epelbaum, R.; Schaffer, M. Curcumin as an Anti-Cancer Agent: Review of the Gap between Basic and Clinical Applications. Curr. Med. Chem. 2010, 17, 190–197. [Google Scholar] [CrossRef]
- Shoba, G.; Joy, D.; Joseph, T.; Majeed, M.; Rajendran, R.; Srinivas, P. S. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998, 64, 353–356. [Google Scholar] [CrossRef]
- Cheng, A.L.; Hsu, C.H.; Lin, J.K.; Hsu, M.M.; Ho, Y.F.; Shen, T.S.; Ko, J.Y.; Lin, J.T.; Lin, B.R.; Ming-Shiang, W.; Yu, H.S.; Jee, S.H.; Chen, G.S.; Chen, T.M.; Chen, C.A.; Lai, M.K.; Pu, Y.S.; Pan, M.H.; Wang, Y.J.; Tsai, C.C.; Hsieh, C.Y. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001, 21, 2895–2900. [Google Scholar]
- Dance-Barnes, S.T.; Kock, N.D.; Moore, J.E.; Lin, E.Y.; Mosley, L.J.; D'Agostino, R.B.Jr.; McCoy, T.P.; Townsend, A.J.; Miller, M.S. Lung tumor promotion by curcumin. Carcinogenesis 2009, 30, 1016–1023. [Google Scholar] [CrossRef]
- Balaji, S.; Chempakam, B. Toxicity prediction of compounds from turmeric (Curcuma longa L.). Food Chem. Toxicol. 2010. [Google Scholar]
- Horvath, Z.; Saiko, P.; Illmer, C.; Madlener, S.; Hoechtl, T.; Bauer, W.; Erker, T.; Jaeger, W.; Fritzer-Szekeres, M.; Szekeres, T. Resveratrol, an ingredient of wine, acts synergistically with Ara-C and tiazofurin in HL-60 human promyelocytic leukemia cells. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1019–1024. [Google Scholar] [CrossRef]
- Mertens-Talcott, S.U.; Percival, S.S. Ellagic acid and quercetin interact synergistically with resveratrol in the induction of apoptosis and cause transient cell cycle arrest in human leukemia cells. Cancer Lett. 2005, 218, 141–151. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kelkel, M.; Jacob, C.; Dicato, M.; Diederich, M. Potential of the Dietary Antioxidants Resveratrol and Curcumin in Prevention and Treatment of Hematologic Malignancies. Molecules 2010, 15, 7035-7074. https://doi.org/10.3390/molecules15107035
Kelkel M, Jacob C, Dicato M, Diederich M. Potential of the Dietary Antioxidants Resveratrol and Curcumin in Prevention and Treatment of Hematologic Malignancies. Molecules. 2010; 15(10):7035-7074. https://doi.org/10.3390/molecules15107035
Chicago/Turabian StyleKelkel, Mareike, Claus Jacob, Mario Dicato, and Marc Diederich. 2010. "Potential of the Dietary Antioxidants Resveratrol and Curcumin in Prevention and Treatment of Hematologic Malignancies" Molecules 15, no. 10: 7035-7074. https://doi.org/10.3390/molecules15107035
APA StyleKelkel, M., Jacob, C., Dicato, M., & Diederich, M. (2010). Potential of the Dietary Antioxidants Resveratrol and Curcumin in Prevention and Treatment of Hematologic Malignancies. Molecules, 15(10), 7035-7074. https://doi.org/10.3390/molecules15107035