Results and Discussion
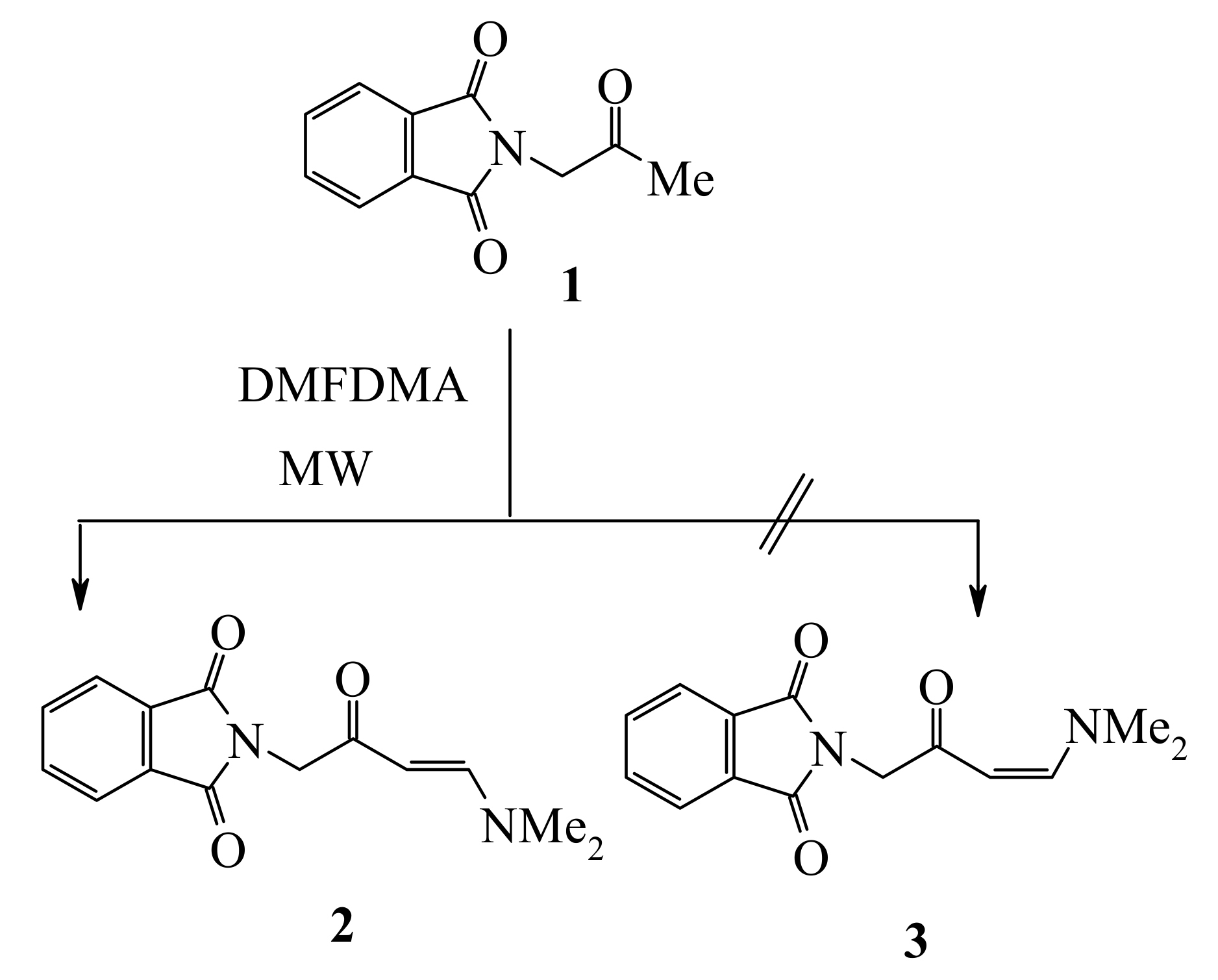
Condensation of phthalimidoacetone (
1) with dimethylformamide dimethyl acetal (DMFDMA) has afforded enaminone
2 in 76% and 77 % yields, respectively, either by refluxing in xylene for 8 h or by heating in MW without solvent at 180 °C for 20 min. The same condensation product had been obtained earlier by Al-Mousawi
et al. [
10,
11]. Fischer
et al. [
12] have used coupling of carbonyl carbons with protons to establish relative geometry extensively, but using
13C-NMR to identify the long range coupling of C-H as an indication of the stereochemistry from the CO group is very difficult because of the appearance of carbonyl carbons as broad signals. This is expected theoretically because this carbonyl carbon has three different 2
J, 2
J and 3
J couplings with nearby protons. We have run NOE difference experimentx to establish the stereo-orientation of either
E or
Z-forms for enaminone
2. The NOE showed that irradiating the alkene CH at δ 5.04 ppm enhanced the dimethylamino protons at δ 2.79 and 3.08 ppm, confirming that they are proximal in space as required by the
E-form (
cf. Scheme 1).
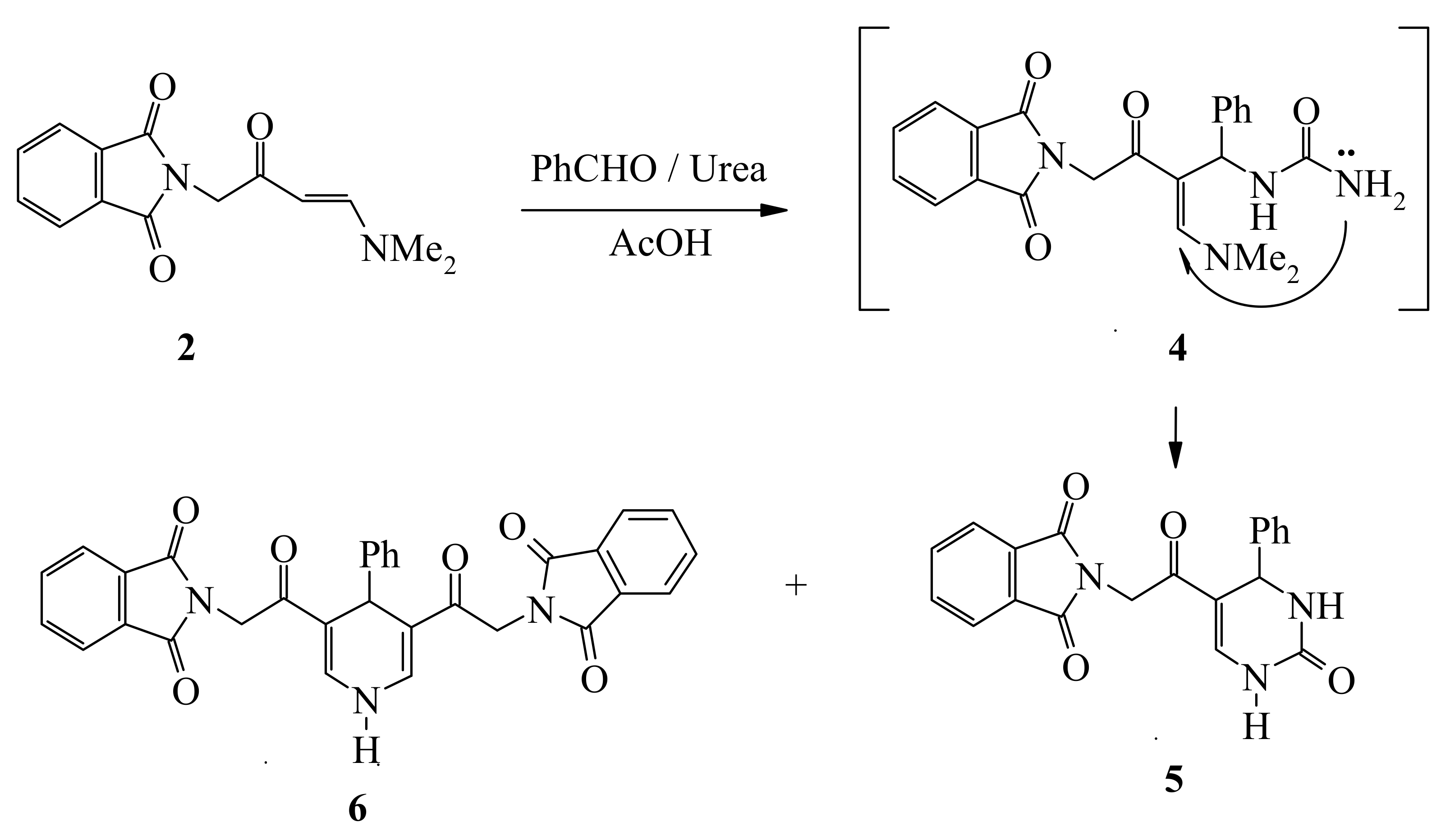
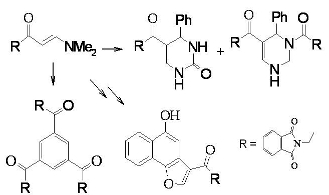
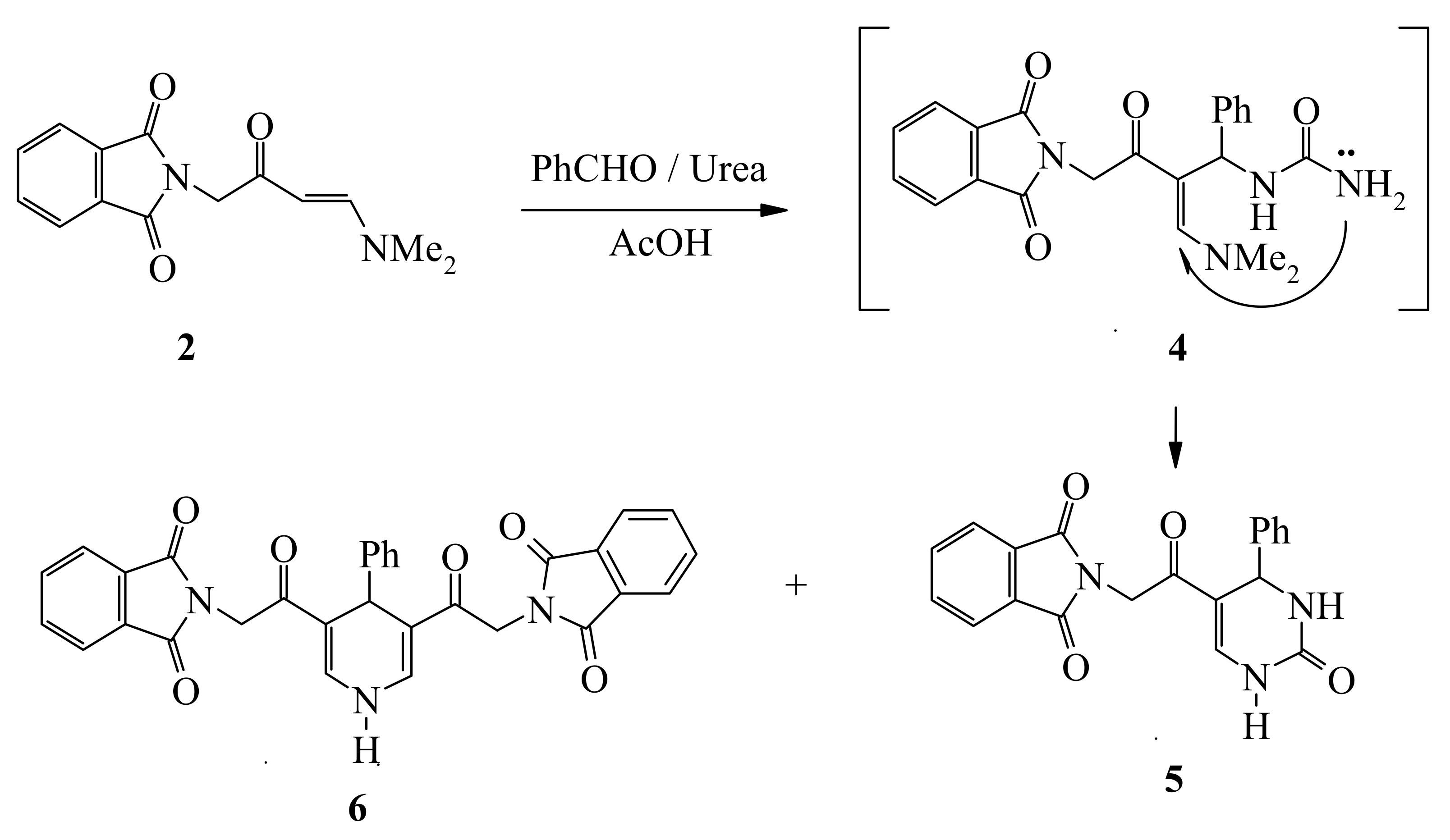
Compound
2, thus formed, has proven to be a versatile starting material for a variety of otherwise not readily obtainable functionally substituted aromatics and heteroaromatics. Thus, refluxing
2 with equimolecular amounts of benzaldehyde and urea in acetic acid afforded a 3:1 mixture of tetrahydropyrimidine
5 and the dihydropyridine
6, which was readily separated by fractional crystallization (
cf. Scheme 2).
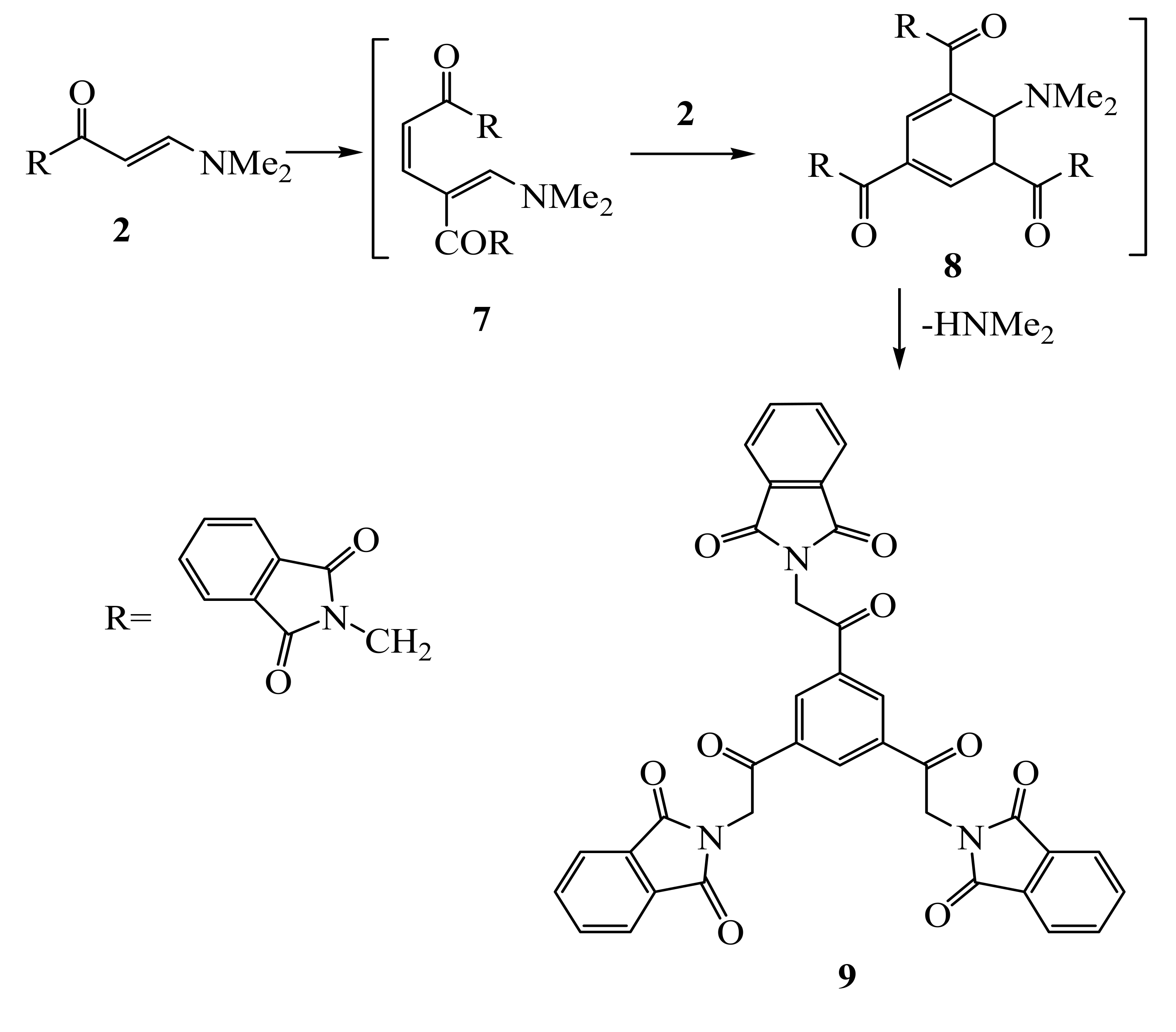
Compound
2 undergoes self-condensation upon heating in acetic acid for 10 hours or upon irradiation at 150 ºC for 1 hour in a focused microwave oven in the presence of an acidic zeolite (Montmorillonite K-10) to give 1,3,5-triacylbenzene
9. We believe that compound
2 is initially converted to the open chain intermediate
7 that further adds to one molecule of enaminone
2 to yield the intermediate
8, which aromatize under these reaction conditions to give the final isolable product
9 (
cf. Scheme 3).
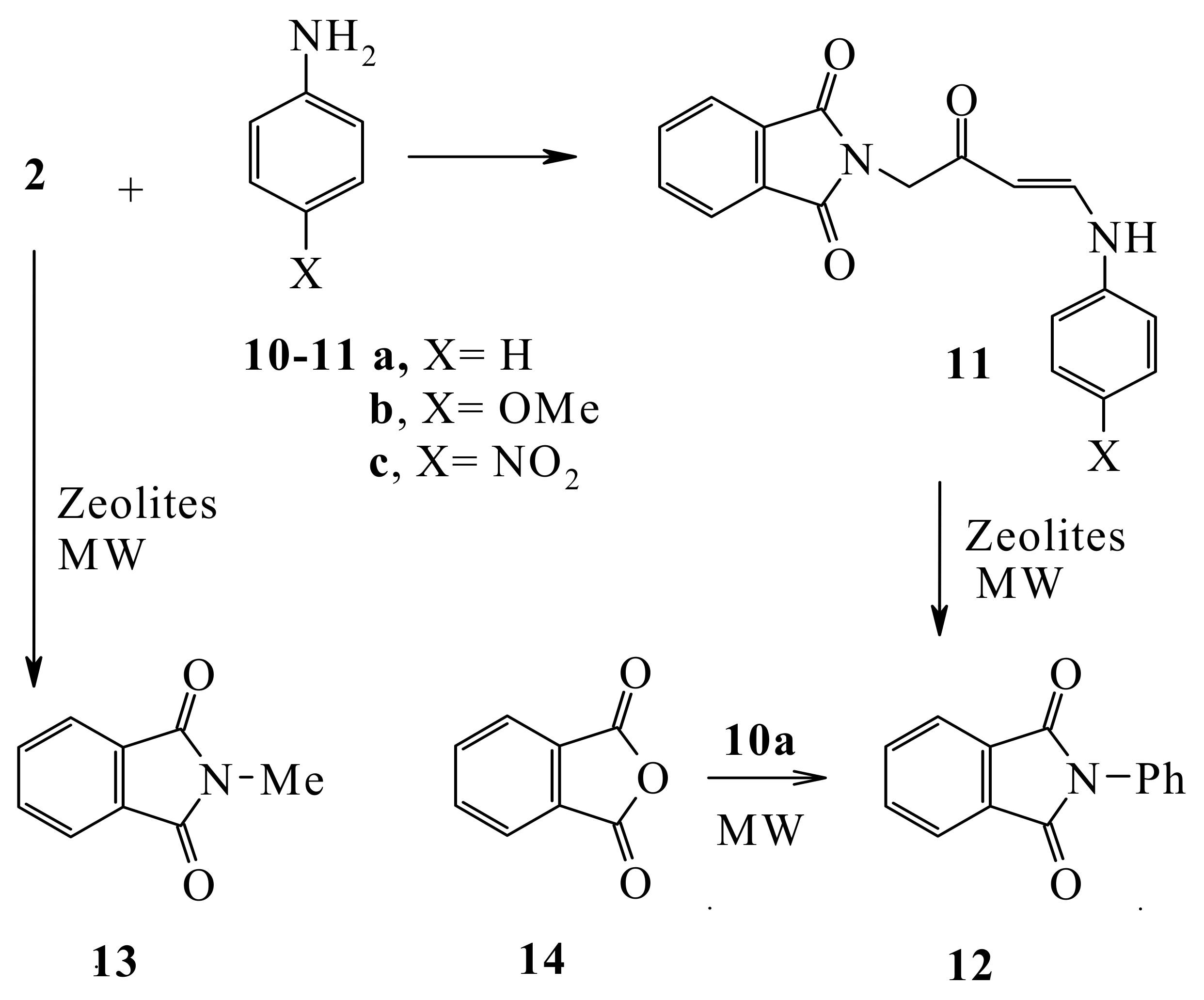
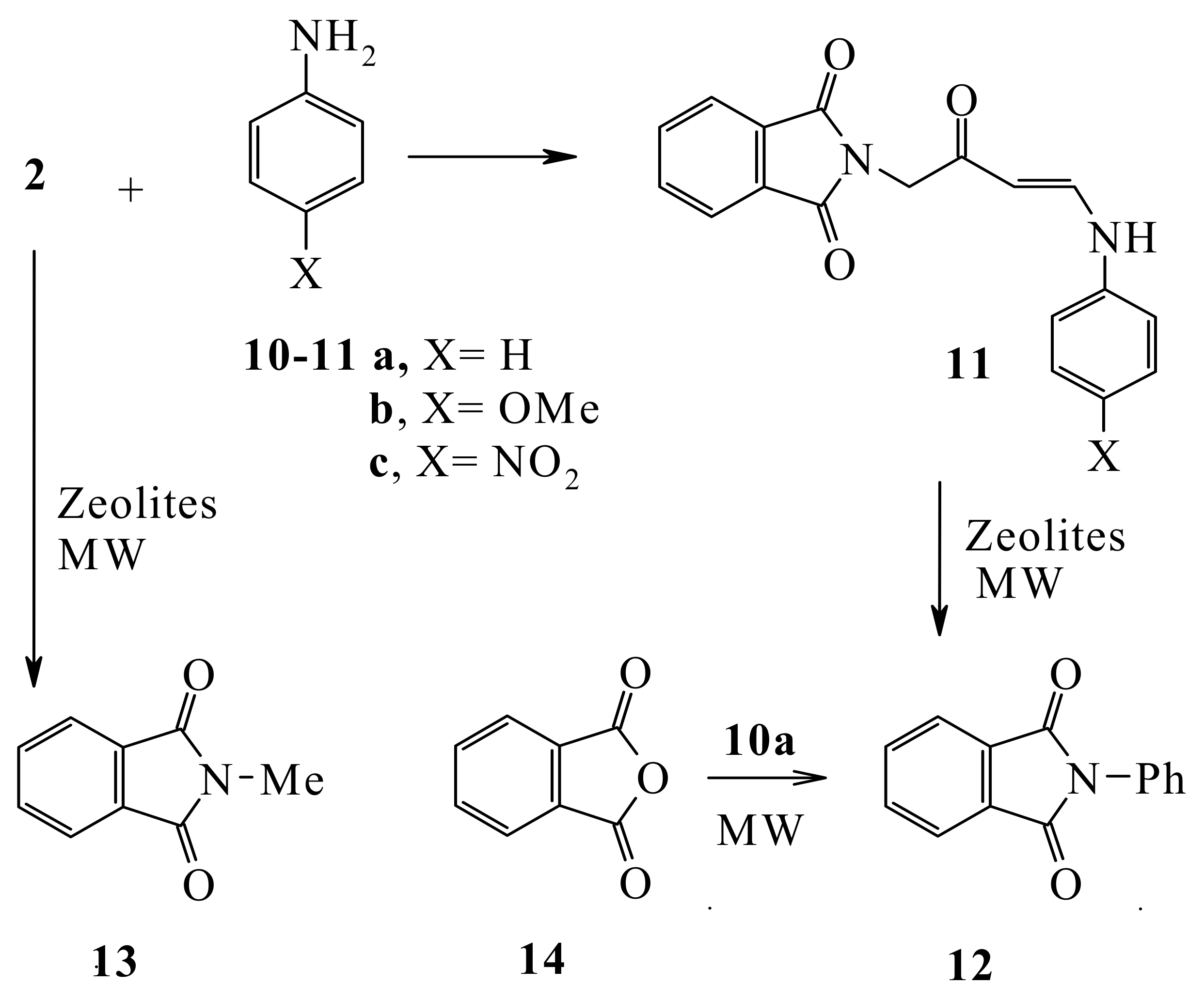
Compound
2 reacted with anilines
10a-c to yield the aniline derivatives
11a-c. Compound
11a when heated with zeolite in an attempt to effect an intramolecular Friedel Craft’s cyclization reaction under MW in the absence of solvent at 160 °C for 30 min afforded
N-phenylphthalimide (
12) that was readily obtained from the MW assisted reaction of phthalic anhydride (
14) with aniline without solvent at 180 °C for 60 min. It is most likely that in the initial step in this transformation the nitrogen atom lone pair attacks the phthalimidocarbonyl moiety. Similarly, heating (
2) by MW in Zeolite without solvent at 160 °C for 30 min. afforded
N-methylphthalimide (
13) (
cf. Scheme 4).
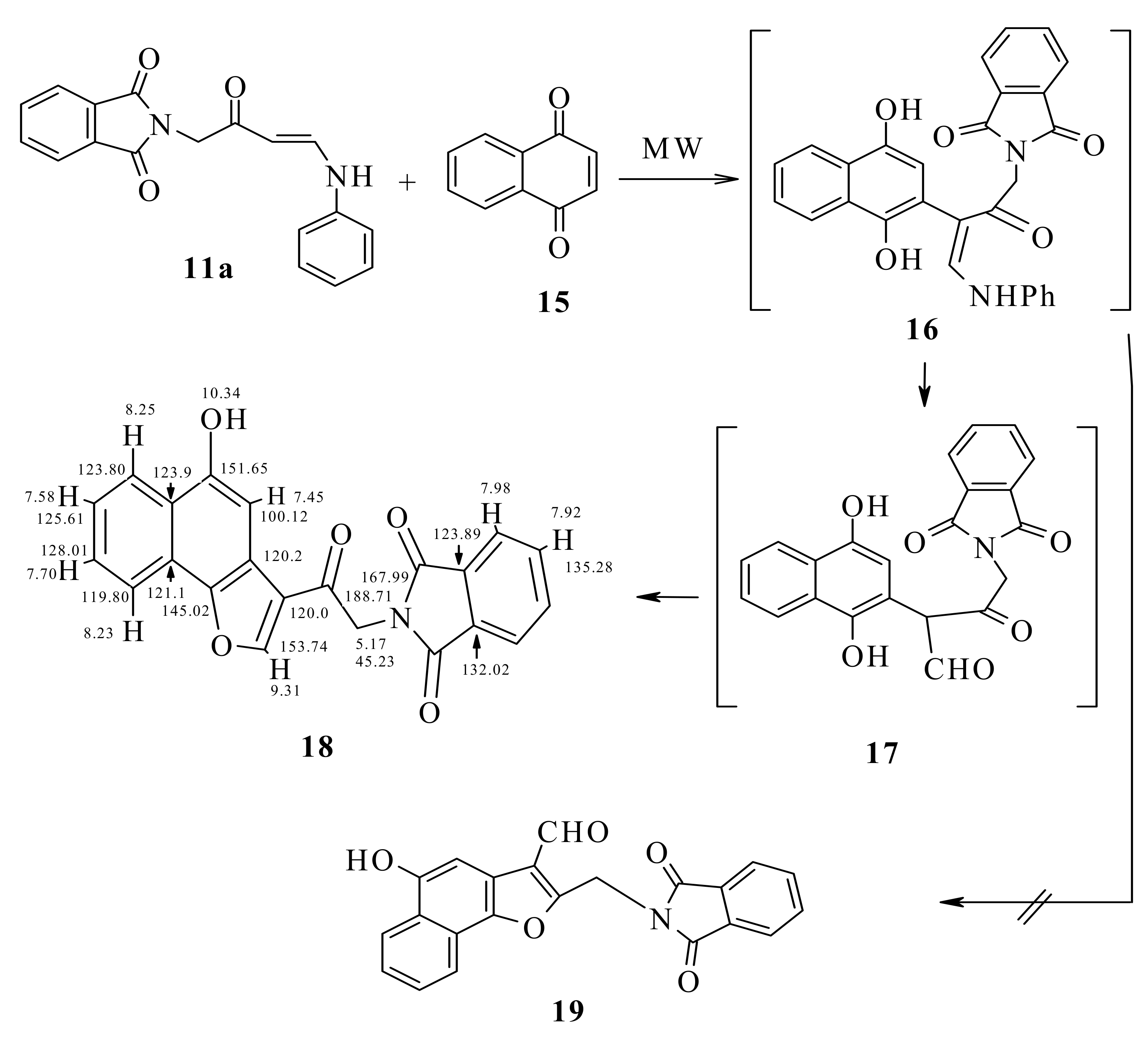
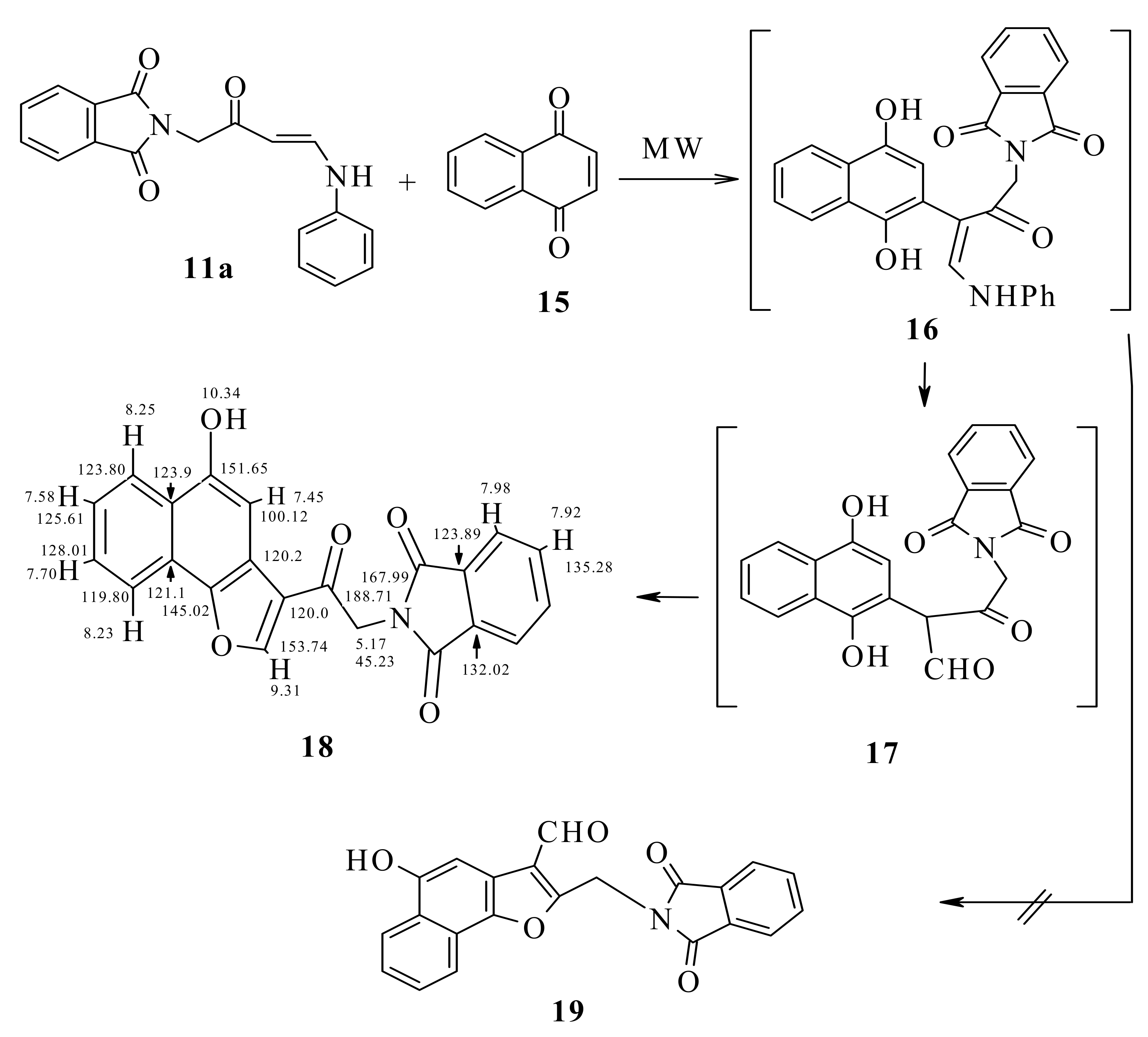
We decided to investigate the reaction utilizing microwave irradiation, while keeping the temperature at 30 °C. This should also allow maintaining the maximum power input of 80 W during the full run of the irradiation. To ensure a correct temperature measurement, a fiber optic sensor was used. Using the same reagent ratios and solvent as those for the reaction run at room temperature (~25 °C), we performed the reaction of enaminone 11a with naphthoquinone 15 under microwave irradiation with simultaneous cooling at 30 °C.
The reaction proceeded smoothly, furnishing the corresponding naphthofuran 18. The yield of the product was increased to 76%; The conventional method reaction at room temperature yielded only 68%. It is noteworthy that the reactions could be performed using an irradiation power of 80 W continuously during the whole run, as the high power level was needed to maintain the temperature at 30 °C because of the efficient external cooling. The differences in yield could be attributed to a lower rate of decomposition of the compounds when using simultaneous cooling.
The possibility of the formation of the aldehyde 19 was excluded based on the HMQC data which revealed that the carbonyl carbon is not linked to any hydrogen.
From H,H-COSY, H-8 at δ 7.70 correlates with H-9 at δ 8.23 and H-7 at δ 7.58 correlates with H-8 and with H-6 at δ 8.25 ppm. From HMBC, H-9 was detected at δ 8.23 ppm because it correlates with C-9b at δ 145.0, which also correlates with H-2 at δ 9.31 ppm. CO-12 correlated at δ 167.99 with CH
2-11 at δ 5.70 ppm and CH-13 at δ 7.98 ppm (
cf. Scheme 5).
Experimental
General
Melting points are uncorrected. All the reactions were conducted under microwave irradiation in heavy-walled Pyrex tubes (capacity 10 mL) fitted with PCS caps. Microwave heating was carried out with a single mode cavity Explorer Microwave Synthesizer (CEM Corporation, NC, USA), producing continuous irradiation and equipped with simultaneous external air-cooling system. Compound 17 was prepared in a microwave irradiation experiment which was carried out in a dedicated CEM-Discover-Coolmate monomode microwave apparatus operating at a frequency of 2.45 GHz with continuous irradiation power from 0 to 300 W. The reaction was carried out in an open 10 mL double walled glass vial, which was cooled to 10 °C using a microwave transparent cooling liquid. The temperature was measured with a fiber-optic device inserted into the reaction vessel. IR spectra were recorded in KBr disks using a Perkin-Elmer System 2000 FT-IR spectrophotometer. 1H-NMR (400 MHz) and 13C-NMR (100 MHz) spectra were recorded on a Bruker DPX 400, super-conducting NMR spectrometer in CDCl3 or DMSO-d6 as solvent and TMS as internal standard; chemical shifts were reported in δ units (ppm). Mass spectra were measured on a VG Autospec-Q (high resolution, high performance, tri-sector GC/MS/MS). Microanalyses were performed on a LECO CHNS-932 Elemental Analyzer.
2-(4-Dimethylamino-2-oxo-but-3-enyl)isoindole-1,3-dione (2)
Thermal method: A mixture of phthalimidoacetone (2.03 g, 0.01 mol) and DMFDMA (1.19 g, 0.01 mol) in xylene (5 mL) was heated under reflux for 8 h. The solid product was collected by filtration and crystallized from EtOH. The reaction gave yellow crystal, yield (76%); mp 162 °C (lit. [
11] mp 159–162 °C ); IR (KBr) ν 1,769, 1,714, 1,660 (CO) (cm
-1); MS (EI)
m/z (%): 258 [M
+, 20%], 160(20), 76(10);
1H-NMR (DMSO-
d6): δ(ppm) 2.72 (s, 3H, CH
3), 3.05 (s, 3H, CH
3), 4.40 (s, 2H, CH
2), 5.04 (d,1H,
J = 12 Hz, CH), 7.61 (d,1H,
J = 12 Hz, CH), 7.85-7.91 (m, 4H, phthalimidyl-H); Anal. Calcd. for C
14H
14N
2O
3: C, 65.11; H, 5.46; N, 10.85; Found: C, 65.18; H, 5.44; N, 10.76.
Microwave method: A mixture of phthalimidoacetone (2.03 g, 0.01 mol) and DMFDMA (1.19 g, 0.01 mol) was irradiated by focused microwave at 180 °C for 20 min. The build-up of pressure in the closed reaction vessel was carefully monitored. After the irradiation, the reaction tube was cooled with high-pressure air through an inbuilt system in the instrument until the temperature had fallen below 50 °C. The solid product, so formed, was collected by filtration and crystallized from EtOH to give compound 2 in 77% yield.
General procedure for the synthesis of compounds 5 and 6
A solution of compound 2 (2.58 g 0.01 mol), benzaldehyde (1.06 g 0.01 mol), urea (0.76 g 0.01 mol), and acetic acid (10 mL) was heated under reflux for 3 h (completion of reaction was monitored by TLC). The precipitate thus formed was then collected by filtration, washed with water and crystallized from acetic acid to give (61%) of tetrahydropyrimidine 5. The filtrate was then washed thoroughly with water the solid product, so formed, was collected by filtration and crystallized from ethanol to afford dihydropyridine 6.
2-[2-Oxo-2-(2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidin-5-yl)ethyl]isoindole-1,3-dione (5): Yellow crystals, yield 61%; mp 280–282 °C; IR (KBr, cm-1): 3,236, 3,101 (NH), 1,714, 1,655 (CO); MS (EI) m/z: (%) 361 [M+, 15%]; 342 (100), 316 (22), 200 (86), 172 (25). 157 (100), 131 (76), 103 (38), 77 (52); 1H-NMR (DMSO-d6): The diasterotopic protons NCH2 appear as two doublets 4.81, 4.91 (d, 1H, J = 13.6 Hz, d, 1H, J = 13.6 Hz, NCH2), 5.15 (s, 1H, CH), 7.23 (t, 3H, J = 8.0 Hz, arom-H), 7.31 (t, 2H, J = 7.2 Hz, arom-H), 7.83-7.90 (m, 6H, arom-H, NH), 9.74 (s, 1H, NH); 13C-NMR (DMSO-d6): δ = 187.41, 168.08, 152.09 (CO), 144.41, 140.92, 135.19, 132.80, 128.88, 127.84, 126.72, 123.70, 110.84, 53.20, 42.85 (CH2); Anal. Calcd. for C20H15N3O4 (361.35): C, 66.48; H, 4.18; N, 11.63. Found: C, 66.49; H, 4.40; N, 11.54.
2-[2-(5-(2-(1,3-Dioxoisoindolin-2-yl)acetyl)-4-phenyl-1,4-dihydropyridin-3-yl)-2-oxoethyl]isoindoline-1,3-dione (6): Orange crystals, yield 20%; mp 274–276 °C; IR (KBr, cm-1): 3,236, 3,101 (NH), 1,714, 1,655 (CO); MS (EI) m/z: (%) 531 [M+, 14%]; 513 (100), 454 (32), 369 (55), 343 (22). 284 (18), 182 (26), 160 (100), 147 (72), 104 (74), 76 (61); 1H-NMR (DMSO-d6): 4.76-4.90 (m, 4H, 2NCH2), 5.16 (s, 1H, CH), 7.10 (t, 3H, J = 7.6 Hz, arom-H), 7.16 (d, 2H, J = 7.2 Hz, arom-H), 7.21-7.37 (m, 2H, arom-H), 7.87-7.97 (m, 8H, arom-H), 10.14 (s, 1H, NH); 13C-NMR (DMSO-d6): δ = 188.41, 167.62 (CO), 146.11, 138.00, 134.68, 131.58, 128.80, 128.44, 126.28, 126.09, 123.26, 114.01, 27.24, 42.80 (CH2); Anal. Calcd. for C31H21N3O6 (531.51): C, 70.05; H, 3.98; N, 7.91. Found: C, 70.21; H, 4.20; N, 8.13.
2-(2-3,5-Di[2-(1,3-dioxo-2,3-dihydro-1H-2-isoindolyl)acetyl]phenyl-2-oxoethyl)-1,3-isoindolinedione (9)
Thermal method: A mixture of compound 2 (2.58 g, 0.01 mol) and acetic acid (10 mL) was refluxed for 10 h, upon completion of the reaction, as checked by TLC, the product was extracted with EtOH. After evaporation of the solvent under reduced pressure, the product was recrystallized from toluene. This compound was obtained as light brown crystals, yield (84 %); mp. 300–301 °C; IR (KBr, cm-1): 1,776, 1,721, (CO); MS (EI) m/z (%) = 639 [M+]; 1H-NMR (DMSO-d6): δ (ppm) 5.57 (s, 6H, CH2), 7.93-7.99 (m-12H, phthalimidyl-H), 8.98 (s, 3H, benzyl-H); 13C-NMR (DMSO-d6): δ 192.23, 167.98 (2 CO), 135.33, 135.20, 133.39, 132.02, 123.93, 45.50 (CH2); Anal. Calcd. for C36H21N3O9 (639.56); C, 67.61; H, 3.31; N, 6.57. Found 67.46; H, 3.33; N, 6.81.
Microwave method: A mixture of compound 2 (2.58 g, 0.01 mol), zeolite (Montmorillonite k-10 clay, 1 g) and acetic acid (1 mL) was irradiated in microwave at 150 ºC for 60 min. The crude product was poured onto water, the solid product, so formed, was collected by filtration and crystallized from toluene. This gave compound 9 in 57% yield.
General procedure for the synthesis of compounds 11a-c
A solution of compound 2 (2.58 g 0.01 mol), compounds 10a-c (0.01 mol), and EtOH (20 mL) was heated under reflux for 5 h (completion of reaction was monitored by TLC). The reaction mixture was poured into cold water, filtered and crystallized from ethanol to afford compounds 11a-c.
2-(2-Oxo-4-phenylamino-but-3-enyl)isoindole-1,3-dione (11a): Yellow-orange crystals, yield 88%; mp 167–169 °C; IR (KBr, cm-1): 3,457 (NH), 1,773, 1,706, 1,640 (CO); MS (EI) m/z: (%) 306 [M+, 20%]; 160 (25), 146 (100), 117 (8), 104 (14), 77 (12); 1H-NMR (DMSO-d6): 4.50 (s, 2H, NCH2), 5.45 (d, 1H, J = 8.0 Hz, CH), 7.03 (t, 1H, J = 7.2 Hz, phenyl-H), 7.25 (d, 2H, J = 8.0 Hz, phenyl-H), 7.30 (t, 2H, J = 7.2 Hz, phenyl-H), 7.77 (dd, 1H, J = 8.0 Hz, J = 8.0 Hz CH), 7.87-7.90 (m, 2H, phthalimidyl-H), 7.91-7.95 (m, 2H, phthalimidyl-H), 11.15 (d, 1H, J = 12.6 Hz, NH); 13C-NMR (DMSO-d6): δ = 191.47, 168.22 (CO), 145.75, 140.38, 135.16, 132.14, 130.06, 123.96, 123.69, 116.66, 93.58, 45.79 (NCH2); Anal. Calcd. for C18H14N2O3 (306.31): C, 70.58; H, 4.61, N, 9.15. Found: 70.55; H, 4.61, N, 9.27.
2-[4-(4-Methoxyphenylamino)-2-oxo-but-3-enyl]isoindole-1,3-dione (11b): Yellow crystals, yield 83%; mp 204–206 °C; IR (KBr, cm-1): 3,455 (NH), 1,772, 1,706, 1,632 (CO); MS (EI) m/z: (%) 336 [M+, 82%]; 175 (100), 160 (42), 132 (65), 103 (22), 76 (21); 1H-NMR (DMSO-d6): 3.71 (s, 3H, OCH3), 4.46 (s, 2H, NCH2), 5.37 (d, 1H, J = 7.8 Hz, CH), 6.88 (d, 2H, J = 8.4 Hz, arom-H), 7.19 (d, 2H, J = 8.4 Hz, arom-H), 7.64 (dd, 1H, J = 7.8 Hz, J = 7.8 Hz CH), 7.86-7.89 (m, 2H, phthalimidyl-H), 7.91-7.93 (m, 2H, phthalimidyl-H), 11.18 (d, 1H, J = 12.6 Hz, NH); 13C-NMR (DMSO-d6): δ = 190.27, 167.70 (CO), 155.80, 145.99, 134.62, 133.30, 131.65, 123.18, 117.65, 114.77, 92.08, 55.99 (NCH2), 44.34 (OCH3); Anal. Calcd. for C19H16N2O4 (336.34): C, 67.85; H, 4.79, N, 8.33. Found: C, 67.73; H, 4.85, N, 8.30.
2-[4-(4-Nitrophenylamino)-2-oxo-but-3-enyl]isoindole-1,3-dione (11c): Yellow crystals, yield 79%; mp 234–236 °C; IR (KBr, cm-1): 3,460 (NH), 1,768, 1,715, 1,653 (CO); MS (EI) m/z: (%) 351 [M+, 84%]; 306 (53), 261 (28), 191 (60), 159 (41), 145 (43), 116 (45), 104 (70). 76 (100); 1H-NMR (DMSO-d6): 4.57 (s, 2H, NCH2), 5.65 (d, 1H, J = 8.4 Hz, CH), 7.49 (d, 2H, J = 9.0 Hz, arom-H), 7.82 (dd, 1H, J = 8.4 Hz, J = 8.4 Hz CH), 7.85-7.89 (m, 2H, phthalimidyl-H), 7.90-7.94 (m, 2H, phthalimidyl-H), 8.15 (d, 2H, J = 9.0 Hz, arom-H), 11.18 (d, 1H, J = 12.6 Hz, NH); 13C-NMR (DMSO-d6): δ = 190.26, 167.32 (CO), 155.72, 145.42, 141.53, 134.57, 131.69, 125.63, 123.32, 116.14, 108.33, 36.55 (NCH2); Anal. Calcd. for C18H13N3O5 (351.31): C, 61.54; H, 3.73, N, 11.96. Found: C, 61.53; H, 3.82, N, 11.96.
General procedure for the synthesis of 12 and 13
Equimolar quantities (0.01 mol) of either compound 2 or 11a and corresponding zeolite were heated under solvent-free conditions for 30 min in a focused microwave oven at 160 °C. Upon completion of the reaction, as checked by TLC, the product was extracted with EtOH (3 × 10 mL). After evaporation of the solvent under reduced pressure, the product was recrystallized from toluene.
2-Phenylisoindole-1,3-dione (
12): Buff crystals, yield 96%; mp 206-208 °C (lit. [
13] mp 205–207 °C yield 95%); MS (EI) m/z: (%) 223 [M+, 100%]; 179 (78), 152 (10), 104 (16), 76 (40).
1H-NMR (DMSO-
d6): 7.44 (d, 3H,
J = 7.2 Hz, phenyl-H), 7.53 (t, 2H,
J = 7.2 Hz, phenyl-H), 7.91-7.94 (m, 2H, phthalimidyl-H), 7.96-7.99 (m, 2H, phthalimidyl-H);
13C-NMR (DMSO-
d6): δ = 167.49 (CO), 135.17, 132.37, 132.02, 129.31, 128.53, 127.89, 123.89; Anal. Calcd. for C
14H
9NO
2 (223.23): C, 75.33; H, 4.06, N, 6.27. Found: C, 75.57; H, 4.12, N, 6.36.
2-Methyl-isoindole-1,3-dione (
13): Buff crystals, yield 87%, mp 131–133 °C (lit. [
14] mp 134 °C and yield 68%); MS (EI) m/z: (%) 161 [M+, 100%]; 132 (50), 117 (74), 104 (82), 76 (81), 66 (44);
1H-NMR (DMSO-
d6): 3.36 (s, 3H, CH
3), 7.81-7.83 (m, 2H, phthalimidyl-H), 7.84-7.88 (m, 2H, phthalimidyl-H);
13C-NMR (DMSO-
d6): δ = 168.53 (CO), 134.72, 132.30, 123.36, 24.20 (CH
3); Anal. Calcd. for C
9H
7NO
2 (161.16): C, 67.07; H, 4.38, N, 8.69. Found: C, 67.01; H, 4.39, N, 9.19.
Reaction of phthalic anhydride with aniline
Equimolar quantities (1.48 g, 0.01 mol) of phathalic anhydride and aniline (0.93 g, 0.01 mol) were heated for 60 min in a focused microwave oven at 180 °C under solvent-free conditions. Upon completion of the reaction, as checked by TLC, the product was extracted with EtOH. After evaporation of the solvent under reduced pressure, the product was recrystallized from toluene to afford compound 12, yield 94%.
2-[2-(5-Hydroxynaphtho[1,2-b]furan-3-yl)-2-oxoethyl]isoindole-1,3-dione (18)
Thermal method: A mixture of compound
11a (3.06 g, 0.01 mol) and naphthoquinone (1.54 g, 0.01 mol) was dissolved in glacial acetic acid (10 mL), then stirred overnight at room temperature. The so formed crystals were collected by filtration and crystallized from dioxane. This compound was obtained in 68 % yield, mp 303 °C (lit. [
15] mp 301–303 °C); IR (KBr, cm
-1): 3,309 (OH), 1,776, 1,722, 1,666 (CO); MS (EI) m/z (%) = 371 [M
+];
1H-NMR (DMSO-
d6): δ 5.17 (s, 2H, CH
2), 7.45 (s, 1H, H-4), 7.58 (dt, 1H,
J = 7.8, 1.2 Hz, H-7), 7.70 (dt, 1H,
J = 7.8, 1.2 Hz, H-8), 7.91-7.92 (m, 1H, H-14), 7.97-7.98 (m, 1H, H-13), 8.23 (d, 1H,
J = 8.4 Hz, H-9), 8.25 (d, 1H,
J = 8.4 Hz, H-6), 9.31 (s, 1H, H-2), 10.34 (s, 1H, OH);
13C-NMR (DMSO-
d6): δ 188.71 (C-10), 167.99 (C-12), 153.74 (C-2), 151.65 (C-5), 145.02 (C-9b), 135.28 (C-14), 132.02 (C-12a), 128.01 (C-8), 125.61 (C-7), 123.97 (C-5a), 123.89 (C-13), 123.80 (C-6), 121.19 (C-9a), 120.28 (C-3a), 120.08 (C-3), 119.85 (C-9), 100.12 (C-4), 45.23 (CH
2, C-11); Anal. Calcd. for C
22H
13NO
5 (371.35); C, 71.16; H, 3.53; N, 3.77. Found C, 71.14; H, 3.65; N, 3.95.
Microwave method: A mixture of compound 11a (3.06 g, 0.01 mol), naphthoquinone (1.54 g, 0.01 mol), and glacial acetic acid (1 mL) was irradiated at 30 °C for 30 min, continuously at the maximum power of 80 W. After completion of the reaction the solvent was evaporated. The crude product was then collected by filtration and crystallized from dioxane to give the title compound in 76% yield.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}