Synthesis and in Vitro Cytotoxic Activity of Compounds with Pro-Apoptotic Potential

Abstract
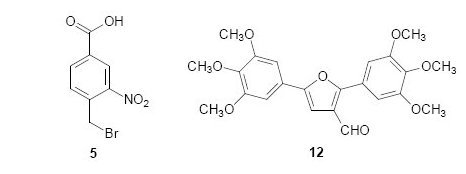
:
Introduction
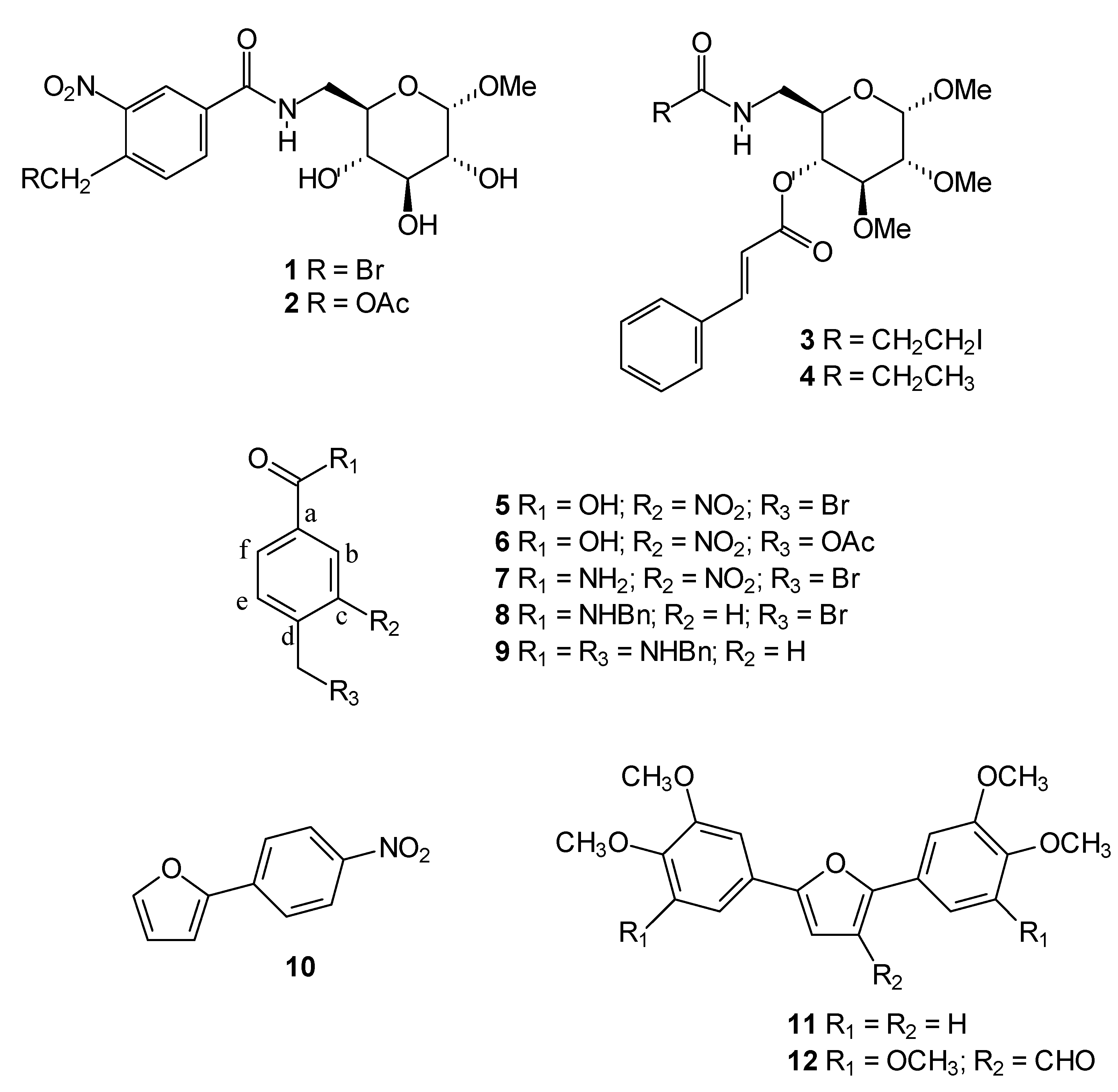
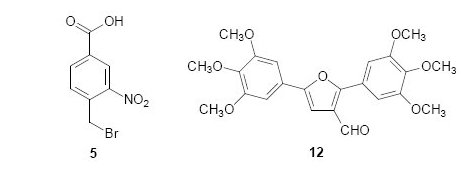
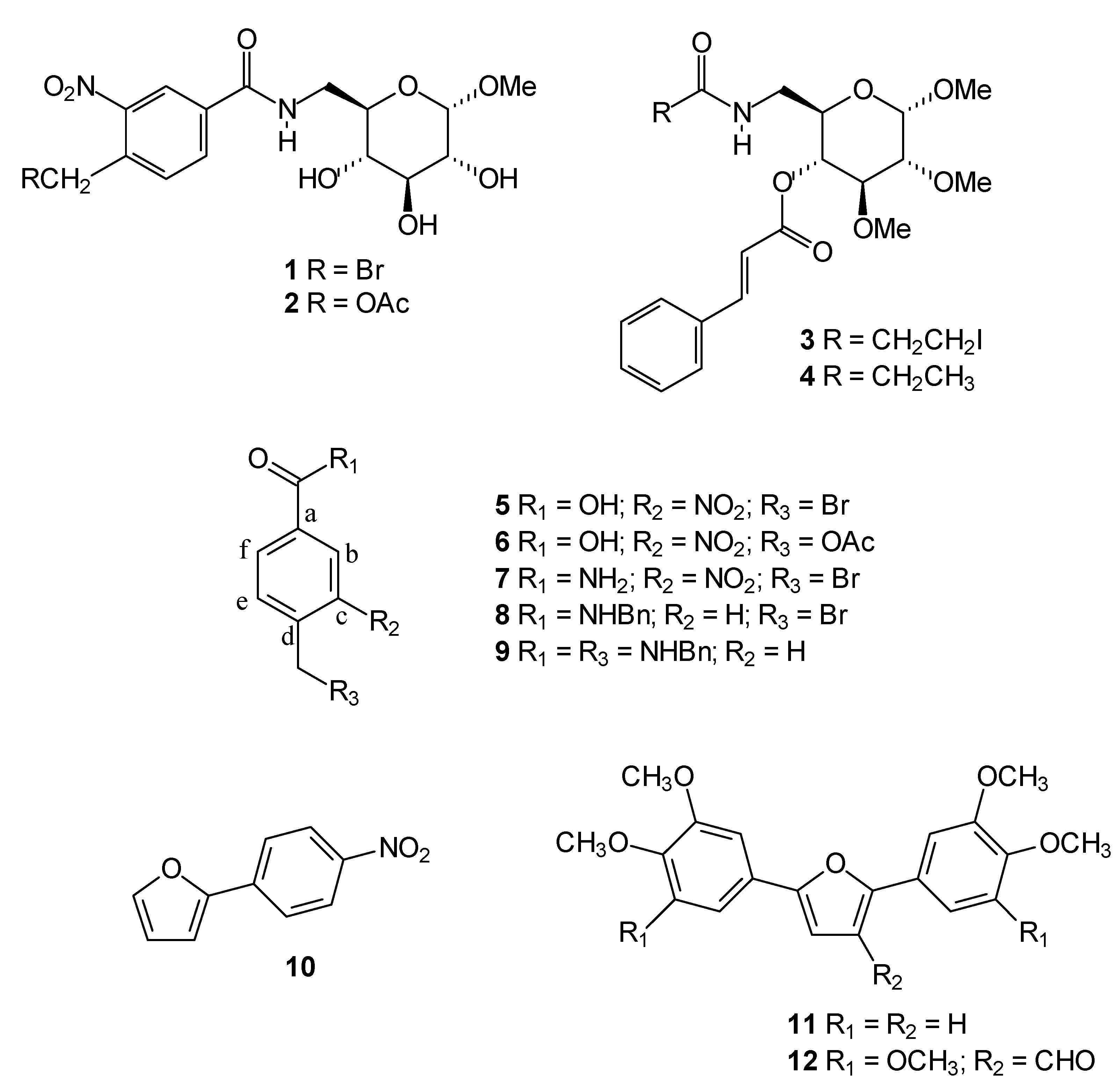
Chemistry
Antiproliferative activity
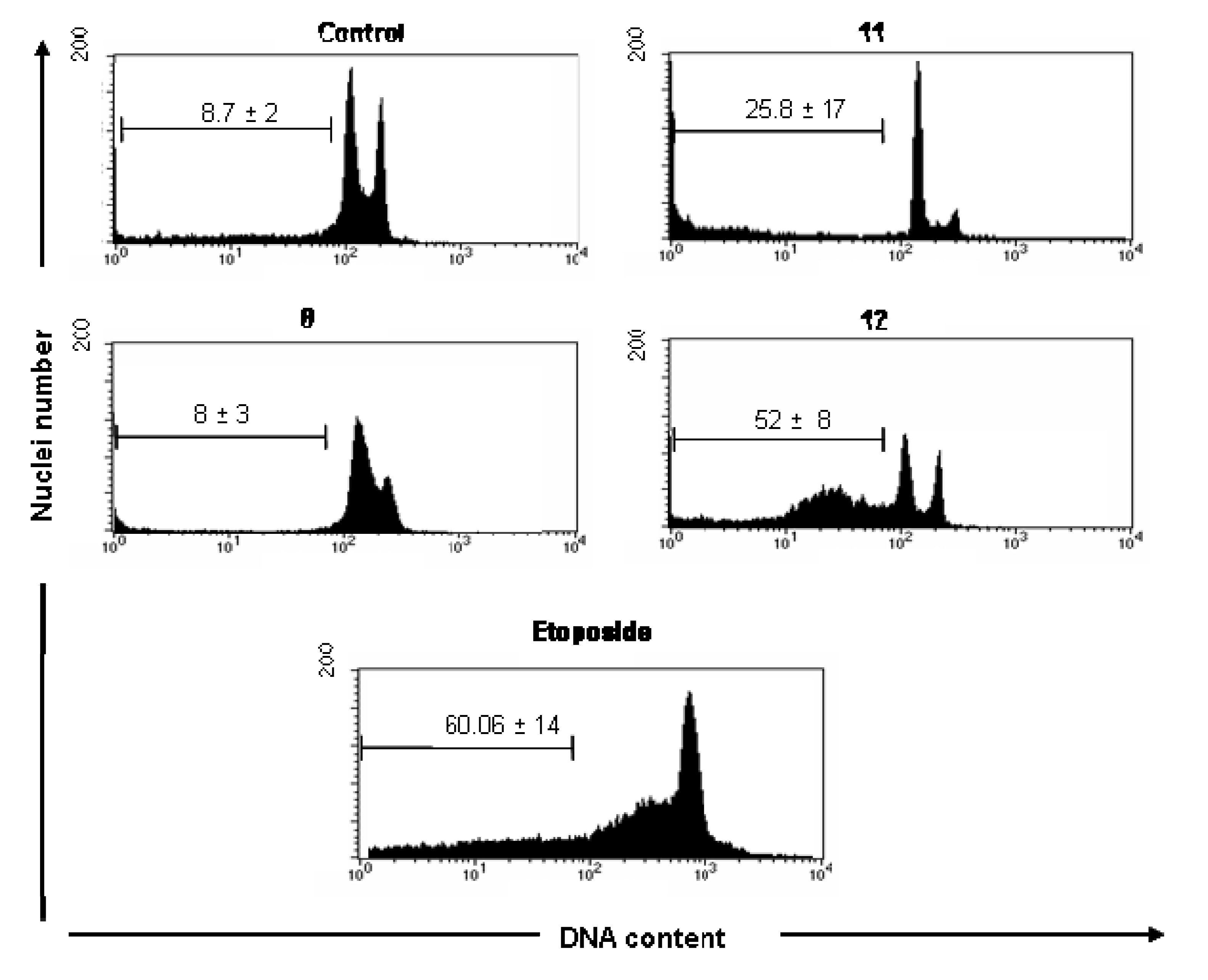
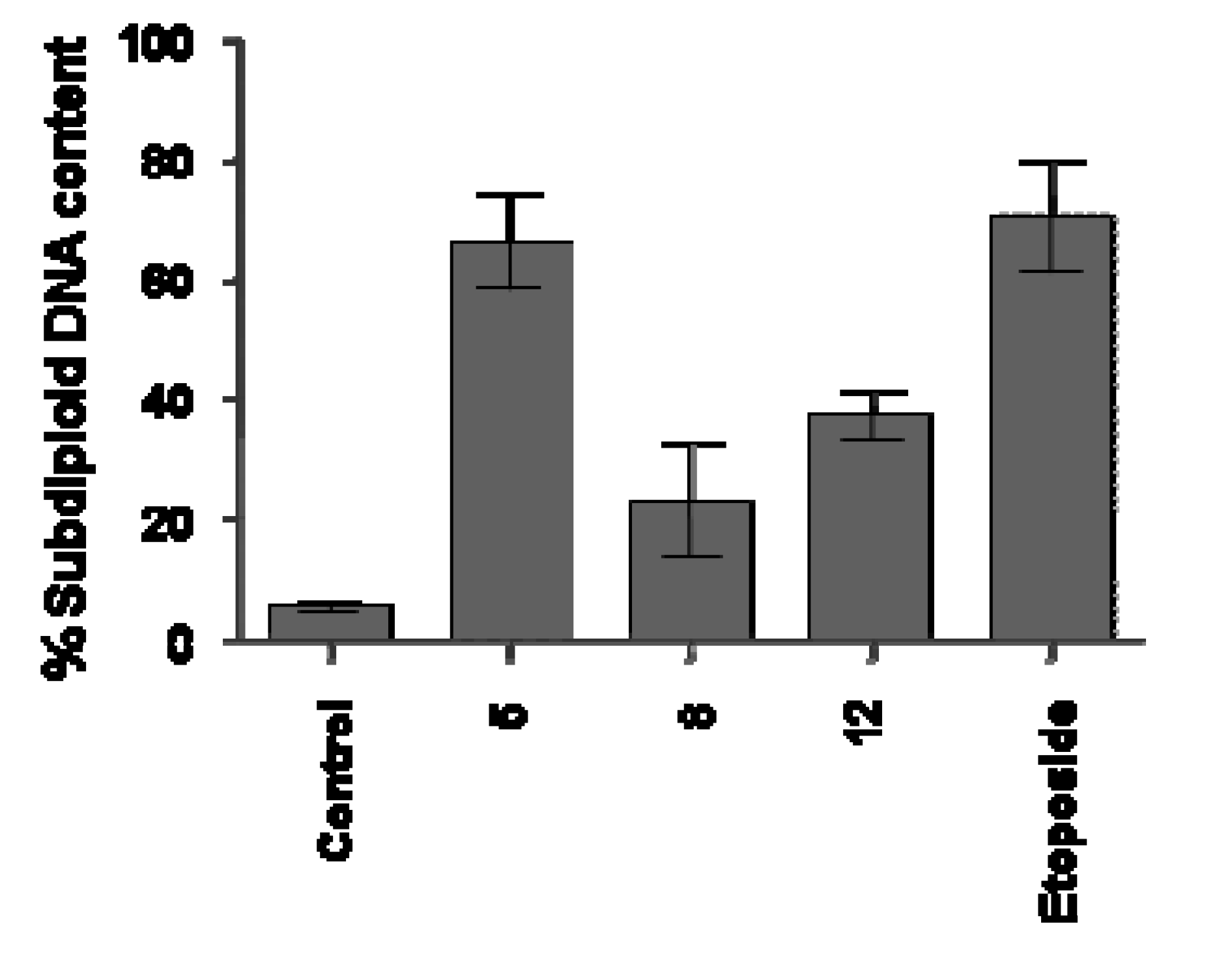
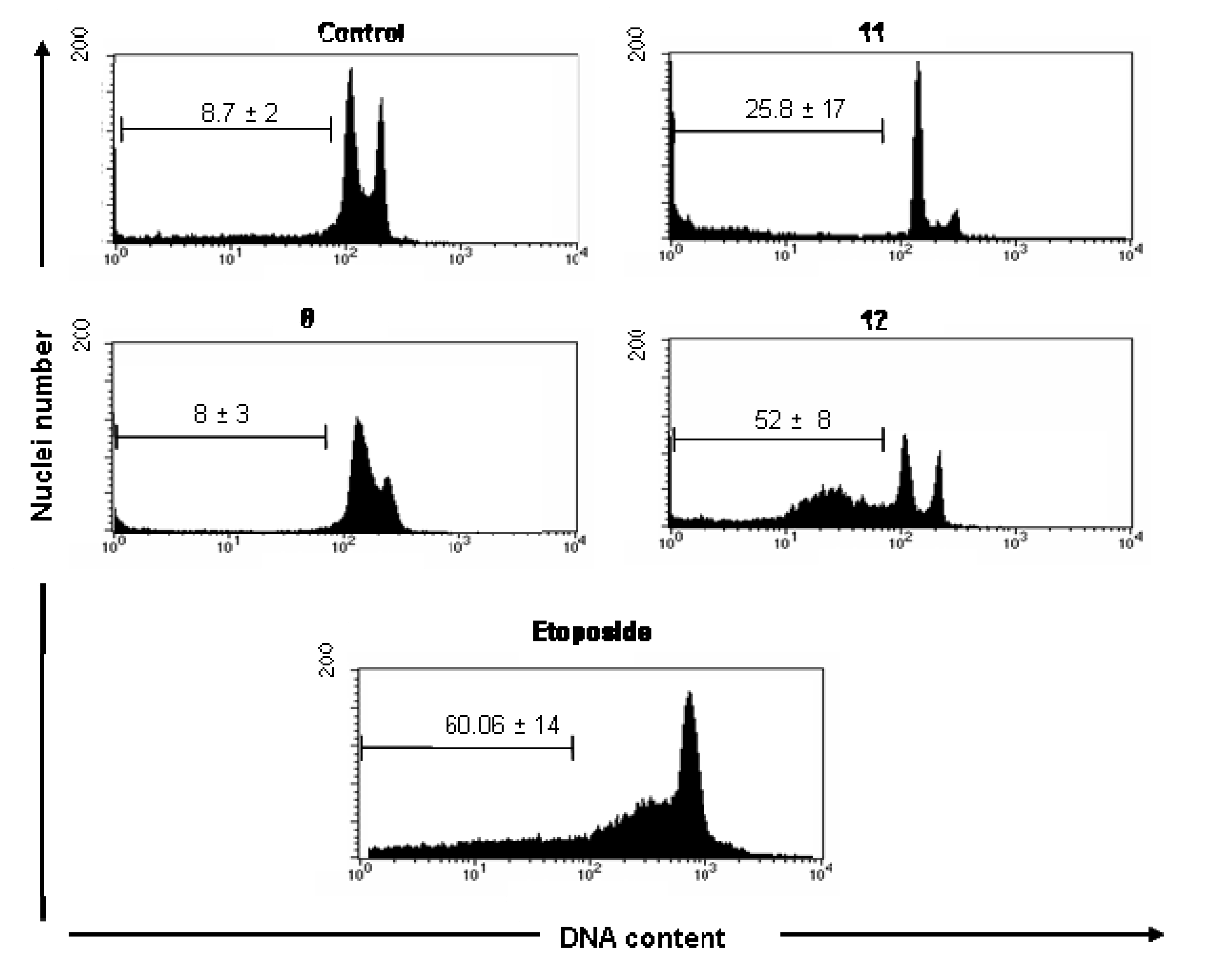
Analysis of DNA Fragmentation
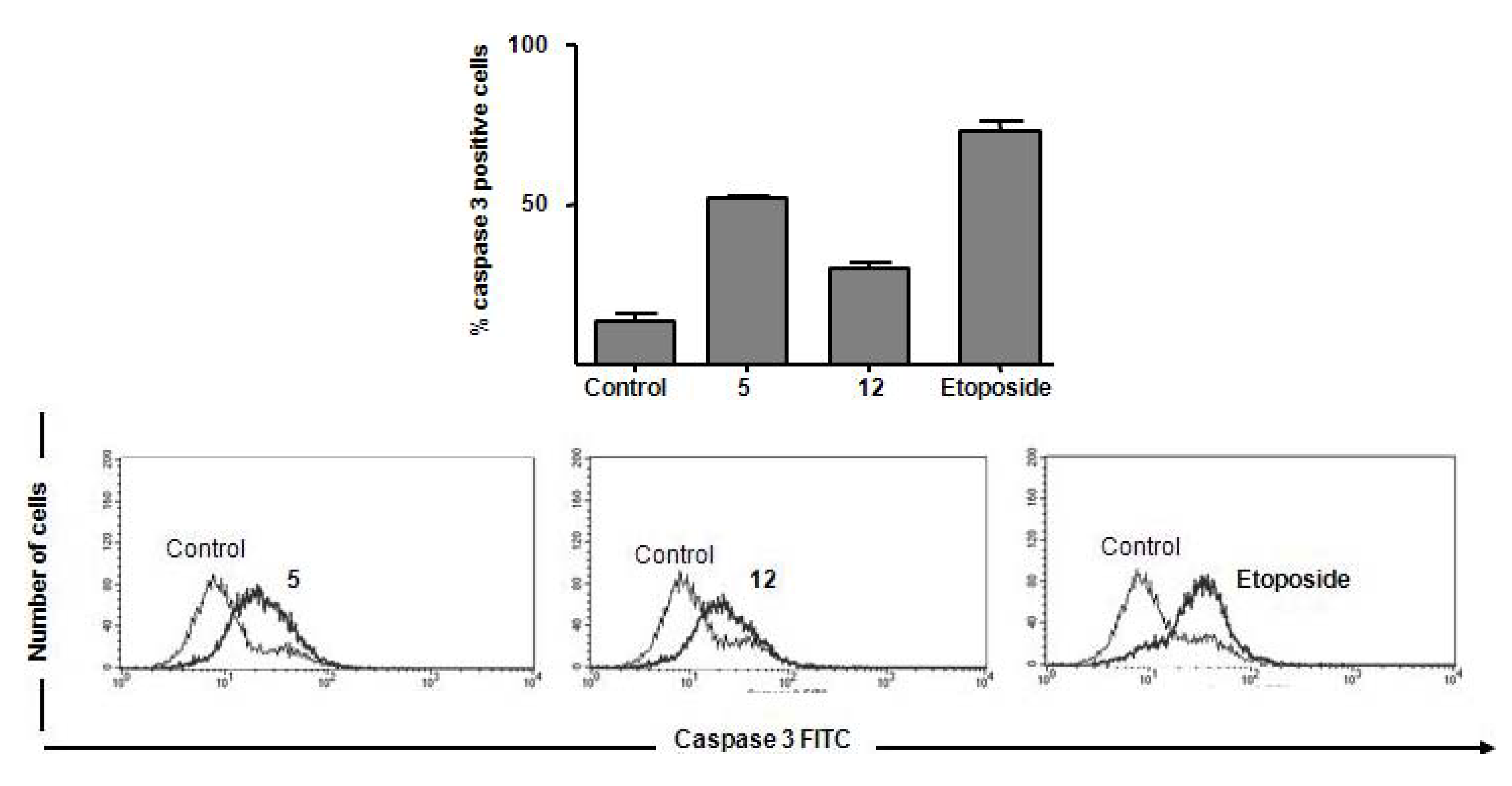
Caspase 3 Activation
Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound a | % Proliferation versus control | |
|---|---|---|
| UACC-62 (melanoma) | Jurkat (lymphoma) | |
| 1 | 105 ± 16 | 88 ± 9 |
| 2 | 117 ± 28 | 107 ± 9 |
| 3 | 104 ± 32 | 92 ± 9 |
| 4 | 99 ± 14 | 128 ± 22 |
| 5 | 84 ± 17 | 41 ± 18 |
| 6 | 106 ± 16 | 110 ± 10 |
| 7 | 106 ± 25 | 93 ± 5 |
| 8 | 39 ± 14 | 32 ± 5 |
| 9 | 110 ± 15 | 99 ± 6 |
| 10 | 87 ± 13 | 113 ± 19 |
| 11 | 25 ± 6 | 120 ± 14 |
| 12 | 60 ± 4 | 54 ± 12 |
| Cell control | 100 | 100 |
| Etoposide | 70 ± 3 | 60 ± 15 |
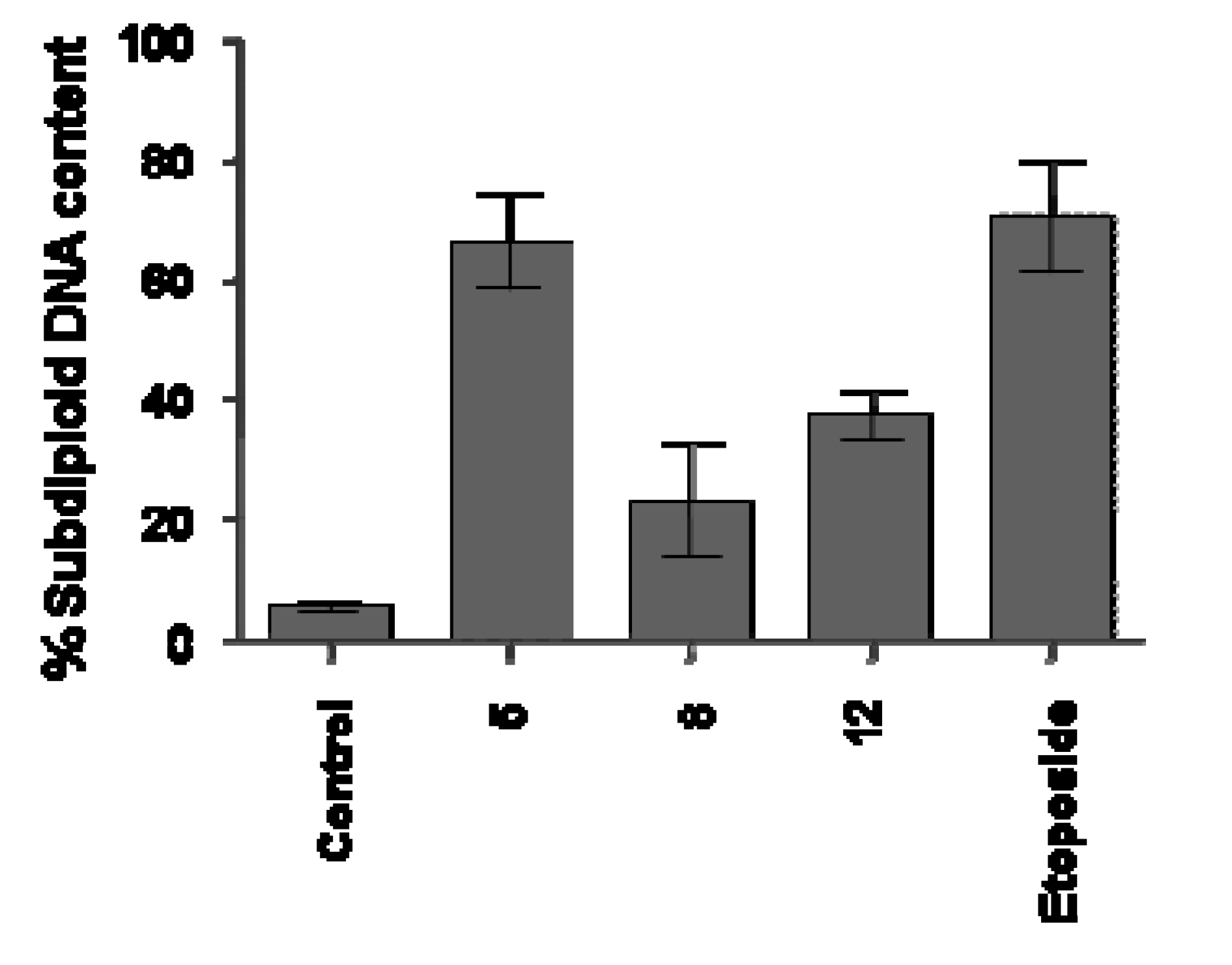
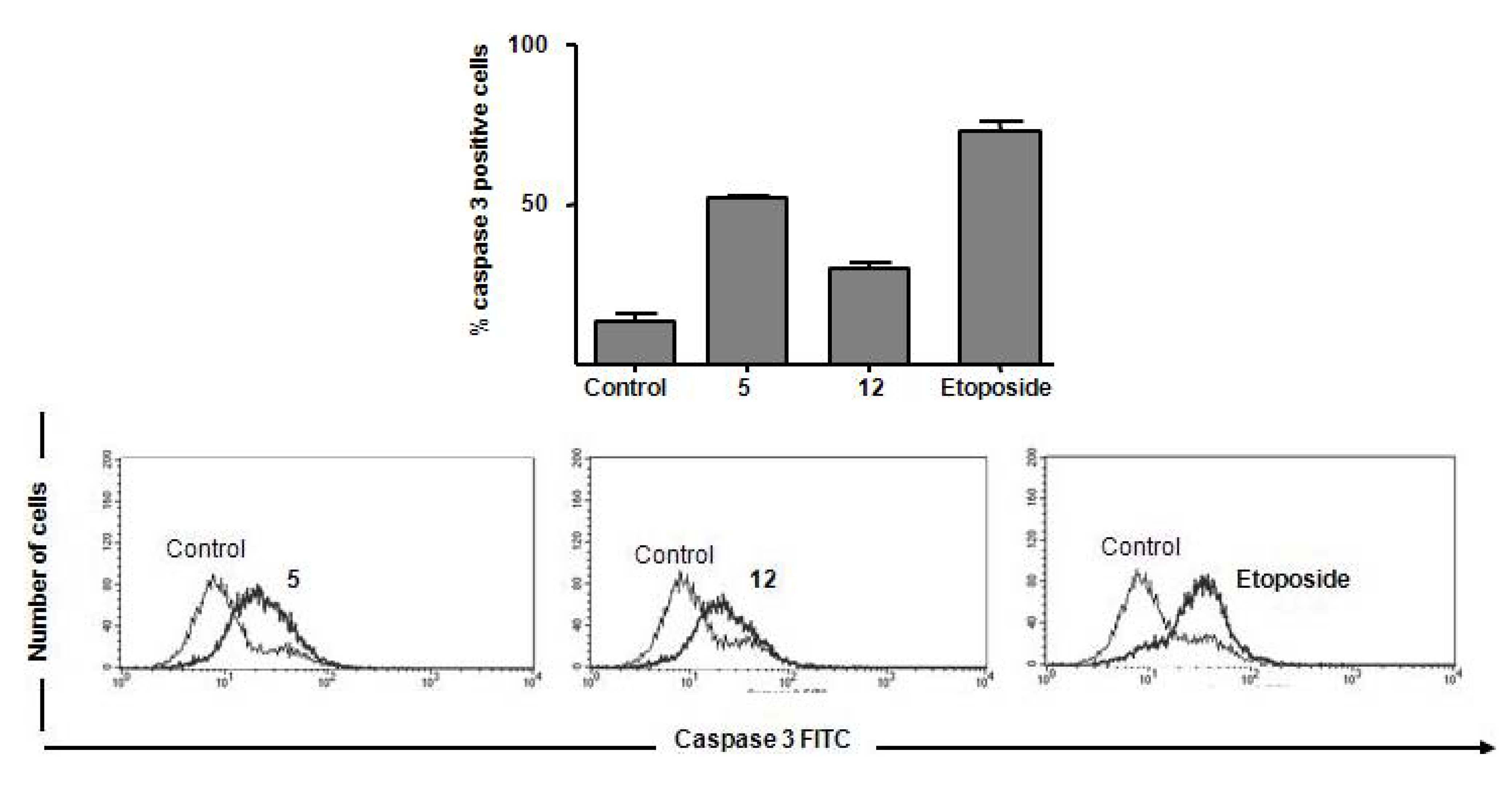
Experimental
General
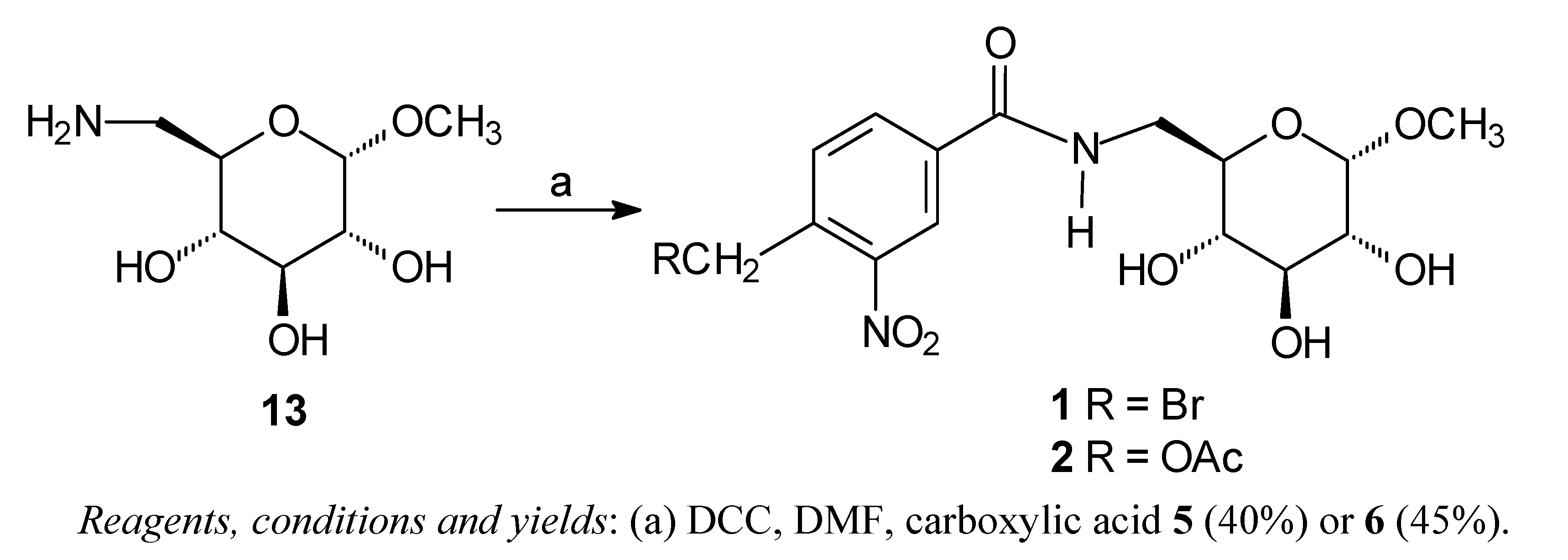
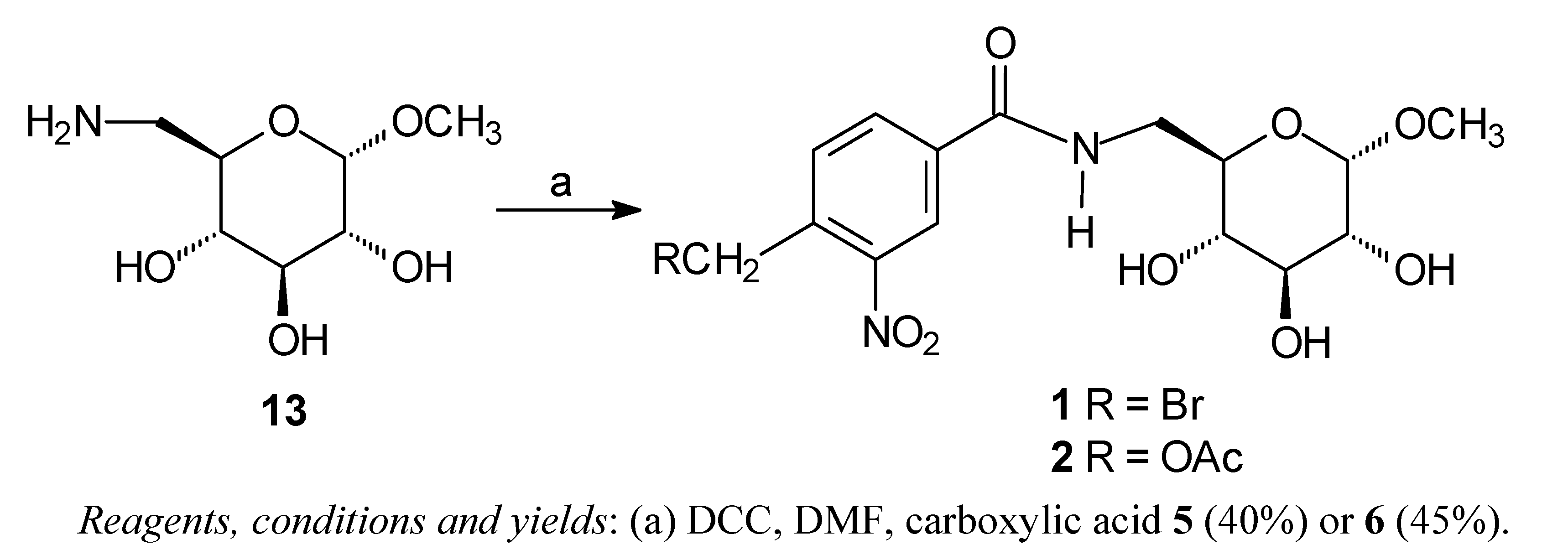
General procedure for synthesis of compounds 1 and 2 [13]
4-Bromomethyl-3-nitrobenzamide (7) [13]
N-Benzyl-4-bromomethylbenzamide (8) [13]
N-Benzyl-4-benzylaminomethylbenzamide (9) [13]
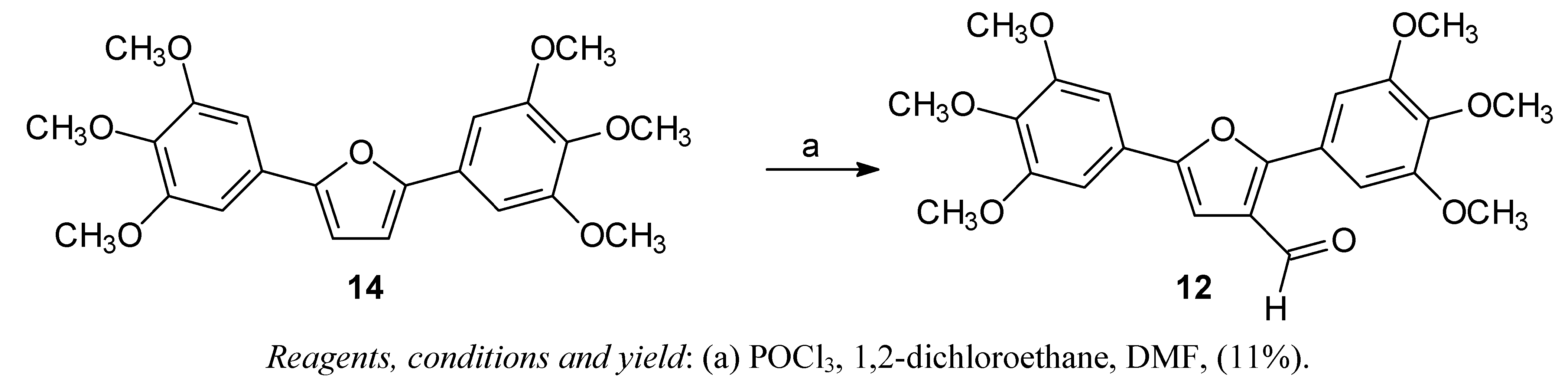
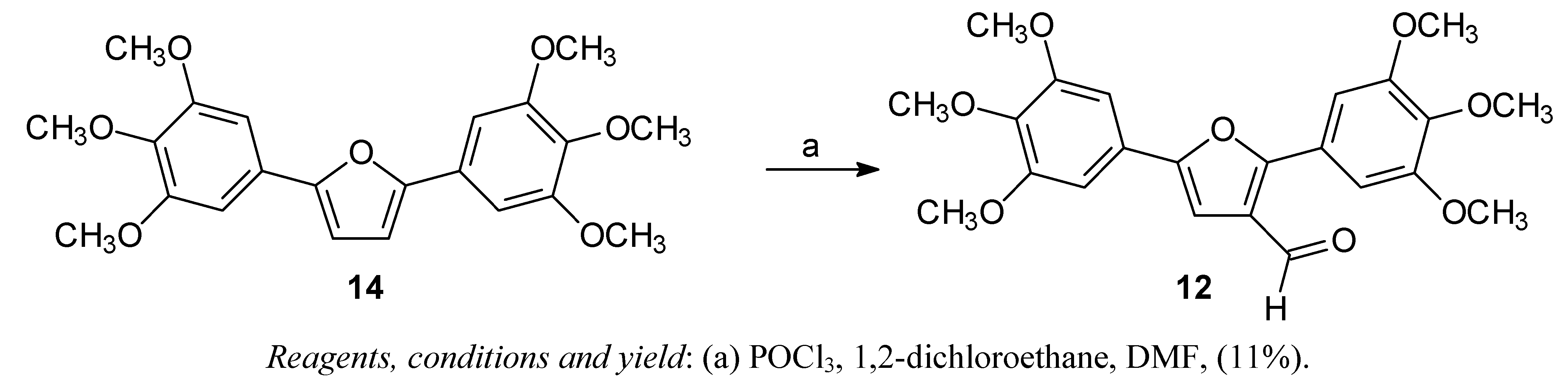
2,5-Bis-(3,4,5-trimethoxyphenyl)-3-formylfuran (12)
Biological evaluation
Antitumor activity
Analysis of DNA Fragmentation - DNA labeling and flow cytometry analysis
Flow cytometric analysis of caspase 3 activation
4. Conclusions
Acknowledgements
- Samples Availability: Contact the authors.
References
- Sanmartín, C.; Echeverría, M.; Mendívil, B.; Cordeu, L.; Cubedo, E.; García-Foncilla,s , J.; Font, M.; Palop, J.A. Synthesis and biological evaluation of new symmetrical derivatives as cytotoxic agents and apoptosis inducers. Bioorg. Med. Chem. 2005, 13, 2031–2044. [Google Scholar]
- Li, W.; Lam, M.S.; Birkeland, A.; Riffel, A.; Montana, L.; Sullivan, M.E.; Post, J.M. Cell-based assays for profiling activity and safety properties of cancer drugs. J. Pharmacol. Toxicol. Methods 2006, 54, 313–319. [Google Scholar] [CrossRef]
- Kamb, A.; Lassota, P. Disease models of cancer: Apoptosis. Drug Discov. Today Dis. Models 2004, 1, 31–36. [Google Scholar] [CrossRef]
- Garber, K.; Arbor, A. New apoptosis drug face critical test. Nature Biotech. 2005, 23, 409–411. [Google Scholar] [CrossRef]
- Wyllie, A.H. Apoptosis: An overview. Br. Med. Bull. 1997, 53, 451–465. [Google Scholar]
- Fleischer, A.; Ghadiri, A.; Dessauge, A.F.; Duhamela, M.; Rebollo, M.P.; Alvarez-Franco, F.; Rebollo, A. Modulating apoptosis as a target for effective therapy. Mol. Immunol. 2006, 43, 1065–1079. [Google Scholar]
- Reed, J.C.; Tomaselli, K.J. Drug discovery opportunities from apoptosis research. Curr. Opin. Biotechnol. 2000, 11, 586–592. [Google Scholar] [CrossRef]
- Fesik, S.W. Promoting apoptosis as a strategy for cancer drug discovery. Nature Rev. Cancer 2005, 5, 876–885. [Google Scholar] [CrossRef]
- Fischer, U.; Janssen, K.; Schulze-Osthoff, K. Cutting-edge apoptosis-based therapeutics: A panacea for cancer? BioDrugs 2007, 21, 273–297. [Google Scholar]
- Fischer, U.; Schulze-Osthoff, K. New approaches and therapeutics targeting apoptosis in disease. Pharmacol. Rev. 2005, 57, 187–215. [Google Scholar] [CrossRef]
- Sampathkumar, S.G.; Jones, M.B.; Meledeo, M.A.; Campbell, C.T.; Choi, S.S.; Hida, K.; Gomutputra, P.; Sheh, A.; Gilmartin, T.; Head, S.R.; Yarema, K.J. Targeting glycosylation pathways and the cell cycle: Sugar-dependent activity of butyrate-carbohydrate cancer prodrugs. Chem. Biol. 2006, 13, 1265–1275. [Google Scholar]
- Bartzatt, R. Synthesis and Activity of an anti-neoplastic aromatic nitrogen mustard agent that alkylates nucleophiles at 37 °C and pH 7.4. Preclinica 2003, 1, 127–132. [Google Scholar]
- Oliveira, R.B.; Passos, A.P.F.; Alves, R.O.; Romanha, A.J.; Prado, M.A.F.; Souza Filho, J.D.; Alves, R.J. In vitro evaluation of the activity of aromatic nitrocompounds against Trypanosoma cruzi. Mem. Inst. Oswaldo Cruz 2003, 98, 141–144. [Google Scholar]
- De Castro, S.L. The challenge of Chagas disease chemotherapy: An update of drugs assayed against Trypanosoma cruzi. Acta Trop. 1993, 53, 83–98. [Google Scholar] [CrossRef]
- Oliveira, R.B.; Souza-Fagundes, E.M.; Siqueira, H.A.J.; Leite, R.S.; Donnici, C.L.; Zani, C.L. Synthesis and evaluation of cytotoxic actity of arylfurans. Eur. J. Med. Chem. 2006, 41, 756–760. [Google Scholar] [CrossRef]
- Faraco, A.A.G.; Prado, M.A.F.; Alves, R.J.; Oliveira, A.B.; Souza-Filho, J.D. Sintese de Amino e Tioacucares: Ligantes para a obtencao d complexos com potencial atividade biológica. Rev. Farm. Bras. 1996, 77, 2–4. [Google Scholar]
- Oliveira, R.B.; Souza-Filho, J.D.; Prado, M.A.F.; Eberlin, M.N.; Meurer, E.C.; Santos, L.S.; Alves, R.J. Synthesis of unexpected six-membered imides by free-radical carbocyclisation on carbohydrate templates. Tetrahedron 2004, 60, 9901–9908. [Google Scholar] [CrossRef]
- Rich, D.H.; Gurwara, S.K. Preparation of a new o-nitrobenzyl resin for solid-phase synthesis of tert-butyloxycarbonyl protected peptide acids. J. Am. Chem. Soc. 1975, 97, 1575–1579. [Google Scholar] [CrossRef]
- Barany, G.; Alberico, F. A three-dimensional orthogonal protection scheme for solid-phase peptide synthesis under mild conditions. J. Am. Chem. Soc. 1985, 107, 4936–4942. [Google Scholar] [CrossRef]
- Lloyd-Wills, P.; Gairí, M.; Albericio, F.; Giralt, E. Convergent solid-phase peptide synthesis. X. Synthesis and purification of protected peptide fragments using the photolabile Nbb-resin. Tetrahedron 1991, 47, 9867–9880. [Google Scholar]
- Khalafi-Nezhad, A.; Parhami, A.; Rad, M.N.S.; Zarea, A. Efficient method for the direct preparation of amides from carboxylic acids using tosyl chloride under solvent-free conditions. Tetrahedron Lett. 2005, 46, 6879–6882. [Google Scholar]
- Oliveira, R.B.; Vaz, A.B.M.; Alves, R.O.; Liarte, D.B.; Donnici, C.L.; Romanha, A.J.; Zani, C.L. Arylfurans as potential Trypanossoma cruzi trypanothione redutase inhibitors. Mem. Inst. Oswaldo Cruz 2006, 101, 169–173. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Meth. 1991, 139, 271–279. [Google Scholar]
- Day, T.W.; Wu, C.H.; Safa, A.R. Etoposide induces protein kinase C {delta}- and caspase-3-dependent apoptosis in neuroblastoma cancer cells. Mol. Pharmacol. 2009, 76, 632–640. [Google Scholar] [CrossRef]
- Walsh, J.G.; Cullen, S.P.; Sheridan, C.; Lüthi, A.U.; Gerner, C.; Martin, S.J. Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci. 2008, 105, 12815–12819. [Google Scholar]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell. Biol. 2008, 9, 231–241. [Google Scholar]
- Iguchi, K.; Usui, S.; Ishida, R.; Hirano, K. Imidazole-induced cell death, associated with intracellular acidification, caspase-3 activation, DFF-45 cleavage, but not oligonucleosomal DNA fragmentation. Apoptosis 2002, 7, 519–525. [Google Scholar] [CrossRef]
- Langer, T.; Hoffman, R.; Bryant, S.; Lesur, B. Hit finding: towards ‘smater’ approaches. Curr. Opin. Pharmacol. 2009, 8, 1–5. [Google Scholar]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Apicela Soares, G.; Barbosa de Oliveira, R.; Fernandes de Andrade, S.; Alves, R.J.; Leomar Zani, C.; De Souza-Fagundes, E.M. Synthesis and in Vitro Cytotoxic Activity of Compounds with Pro-Apoptotic Potential. Molecules 2010, 15, 12-26. https://doi.org/10.3390/molecules15010012
Apicela Soares G, Barbosa de Oliveira R, Fernandes de Andrade S, Alves RJ, Leomar Zani C, De Souza-Fagundes EM. Synthesis and in Vitro Cytotoxic Activity of Compounds with Pro-Apoptotic Potential. Molecules. 2010; 15(1):12-26. https://doi.org/10.3390/molecules15010012
Chicago/Turabian StyleApicela Soares, Gisell, Renata Barbosa de Oliveira, Saulo Fernandes de Andrade, Ricardo José Alves, Carlos Leomar Zani, and Elaine Maria De Souza-Fagundes. 2010. "Synthesis and in Vitro Cytotoxic Activity of Compounds with Pro-Apoptotic Potential" Molecules 15, no. 1: 12-26. https://doi.org/10.3390/molecules15010012
APA StyleApicela Soares, G., Barbosa de Oliveira, R., Fernandes de Andrade, S., Alves, R. J., Leomar Zani, C., & De Souza-Fagundes, E. M. (2010). Synthesis and in Vitro Cytotoxic Activity of Compounds with Pro-Apoptotic Potential. Molecules, 15(1), 12-26. https://doi.org/10.3390/molecules15010012