JNK Contributes to Hif-1α Regulation in Hypoxic Neurons


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Differential susceptibility of young/immature compared to adult/mature neurons to hypoxic injury
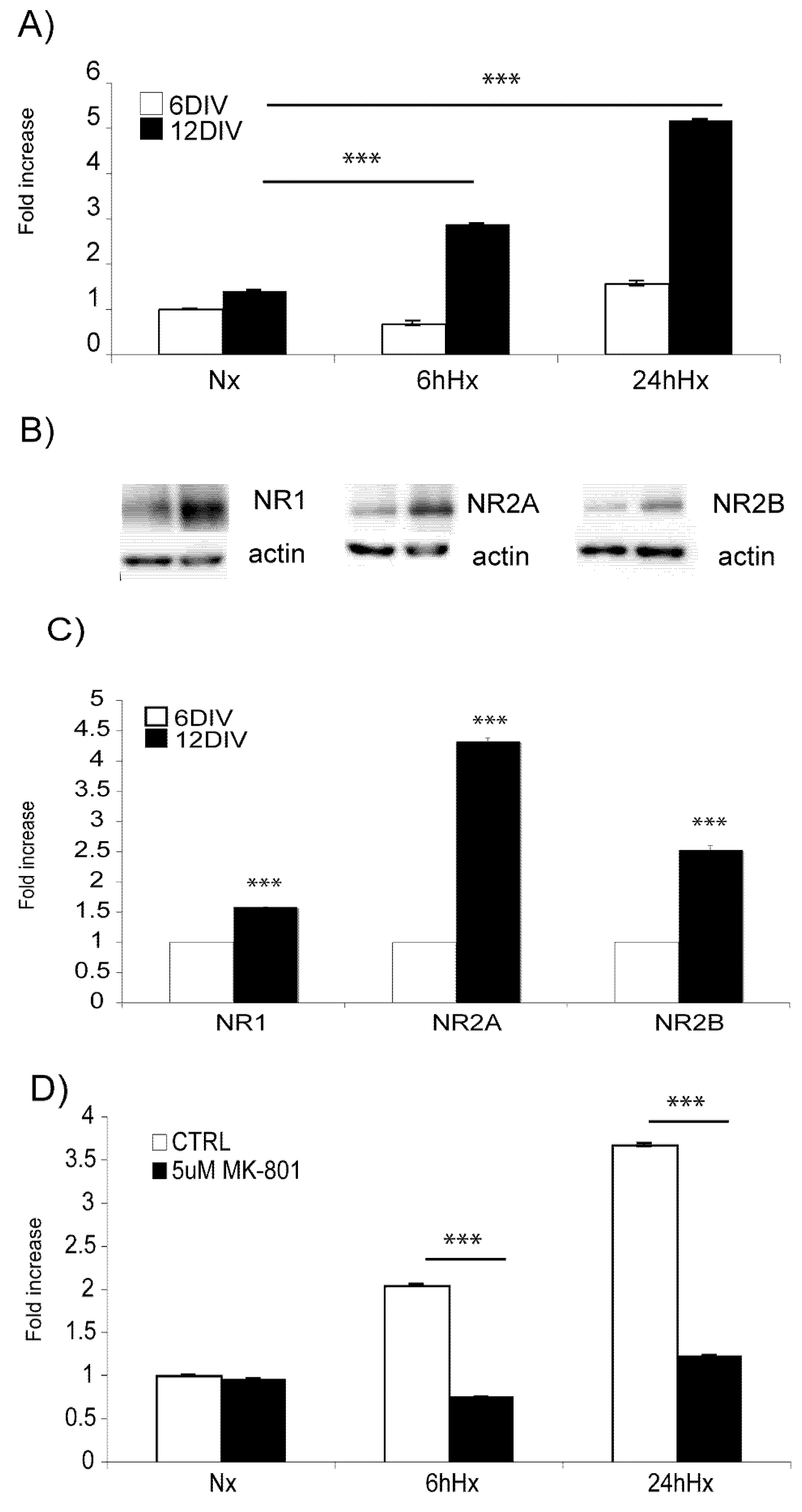
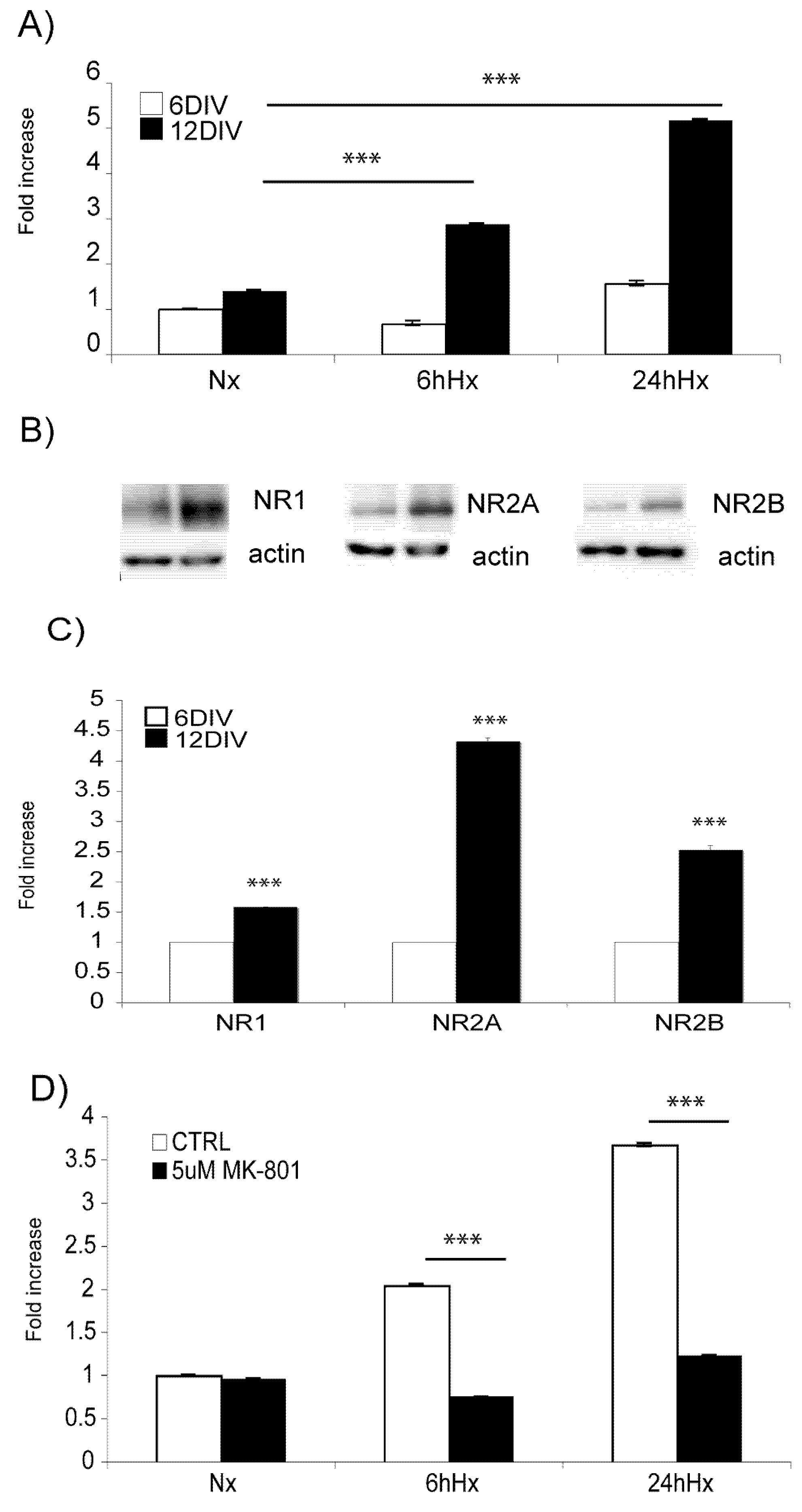
2.2. Basal levels of JNK signaling proteins increase with increasing age, while Hif-1α hypoxic induction decreases
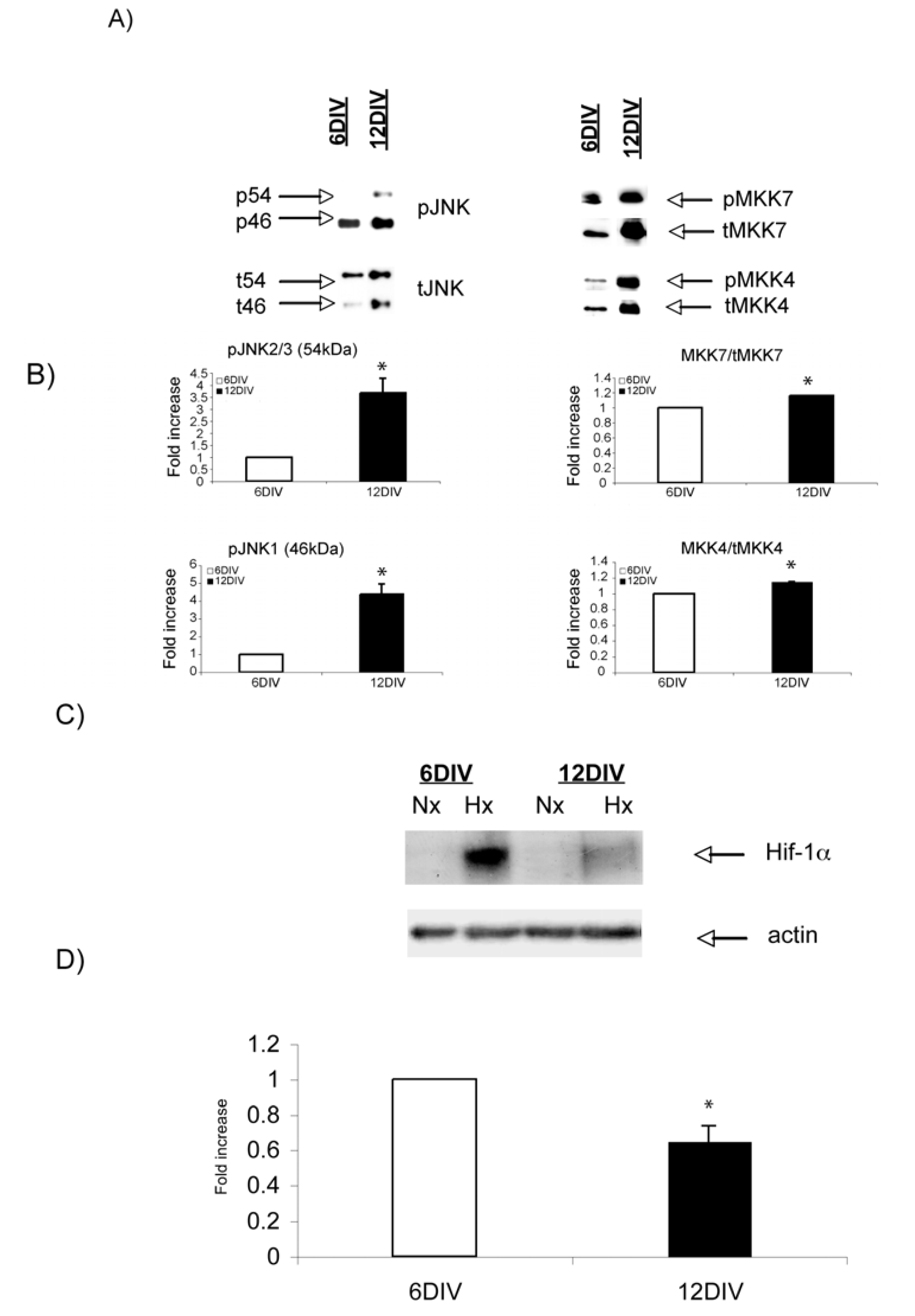
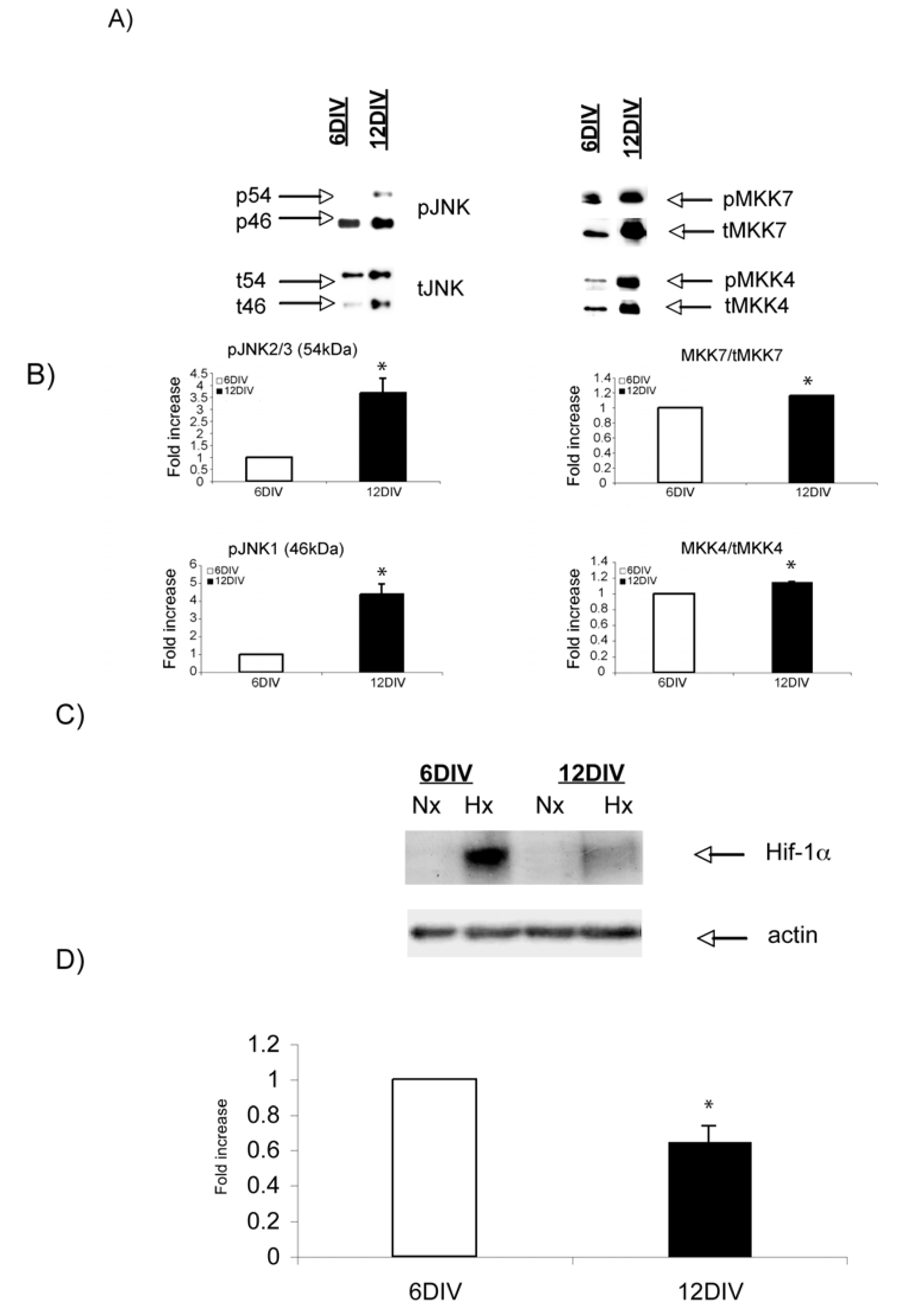
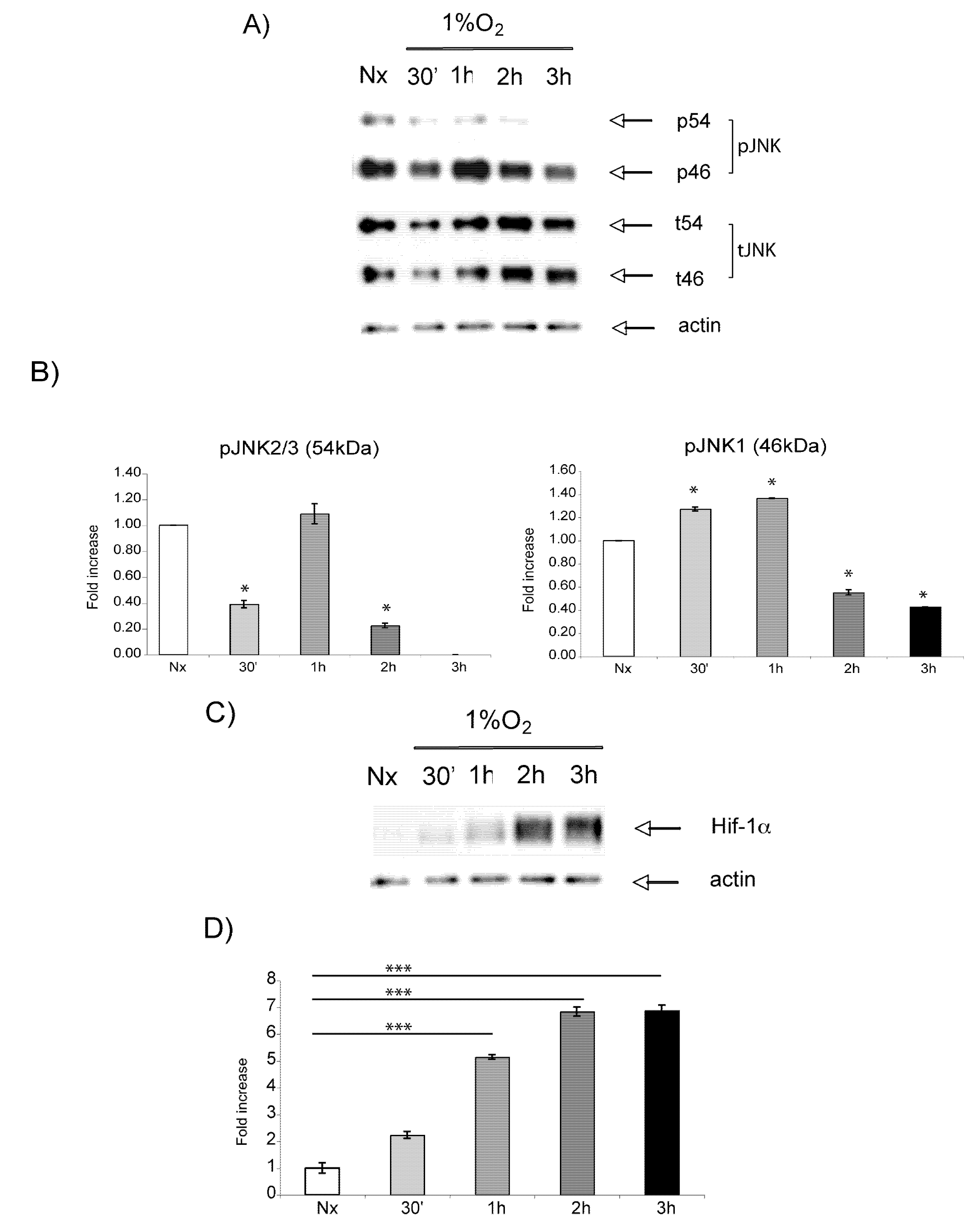
2.3. Hypoxic regulation of Hif-1α and JNK signaling pathways
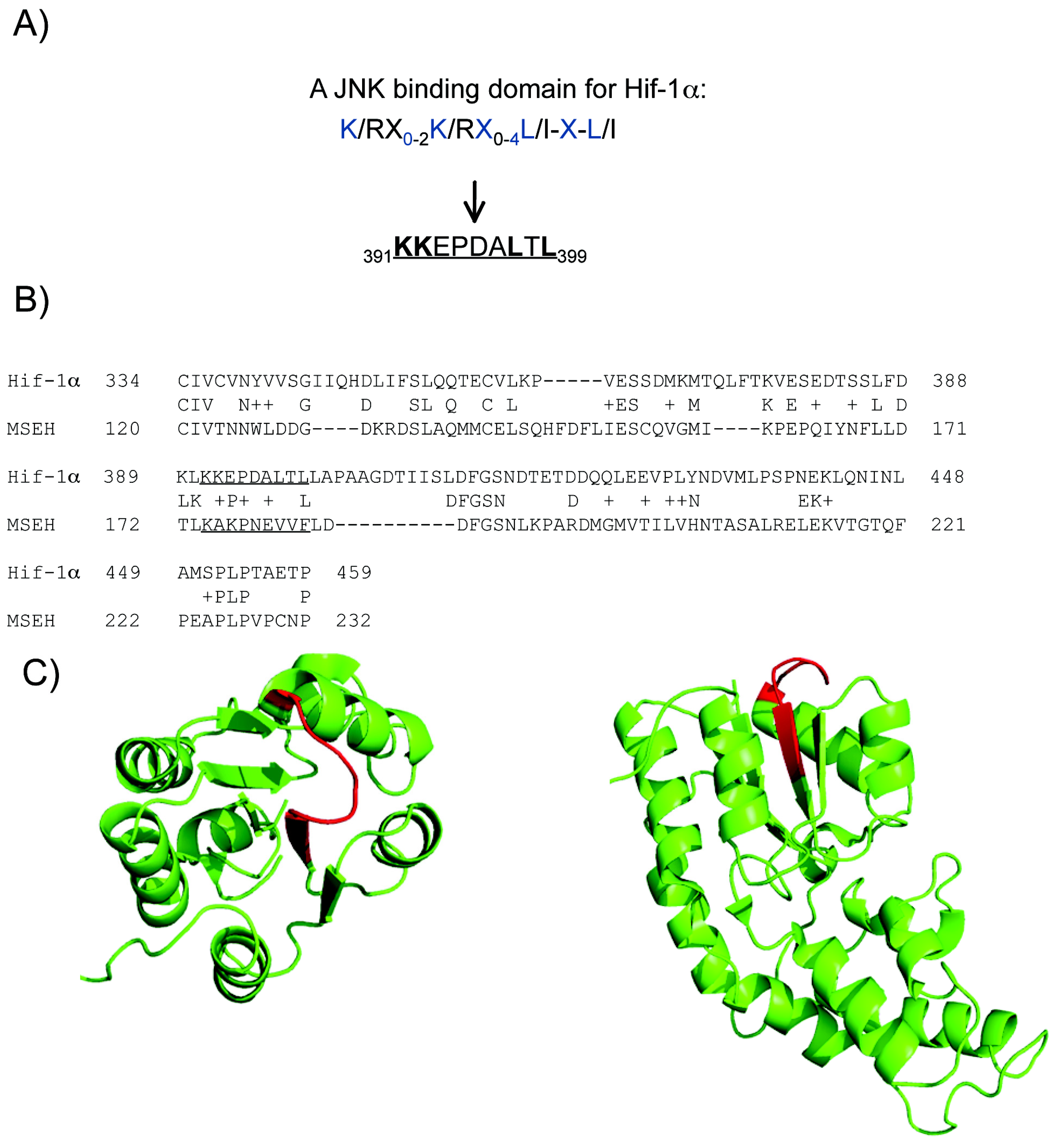
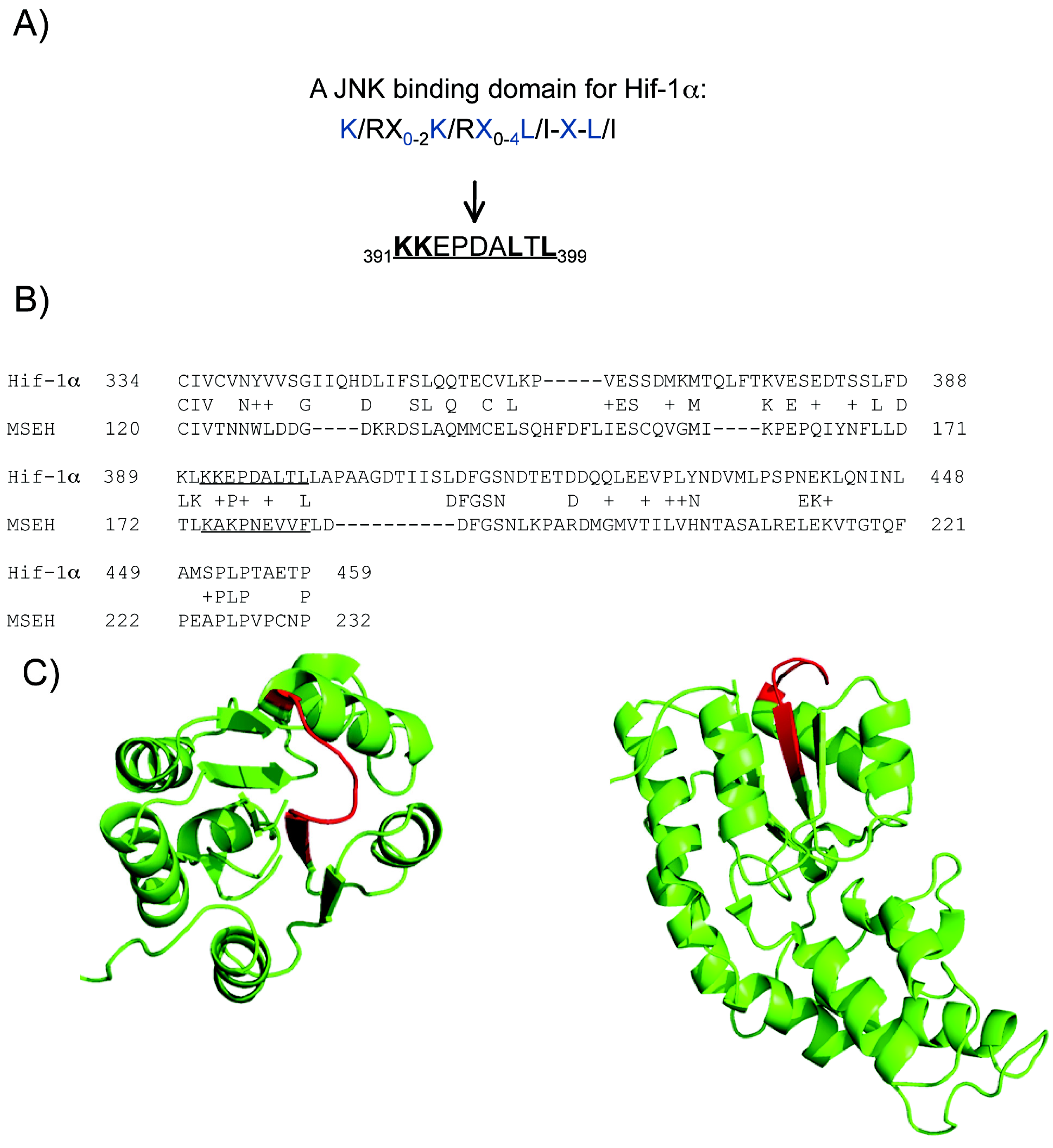
2.4. A putative JNK binding domain on Hif1-α

2.5. D-JNKI1 treatment rescues hypoxic adult neurons from death
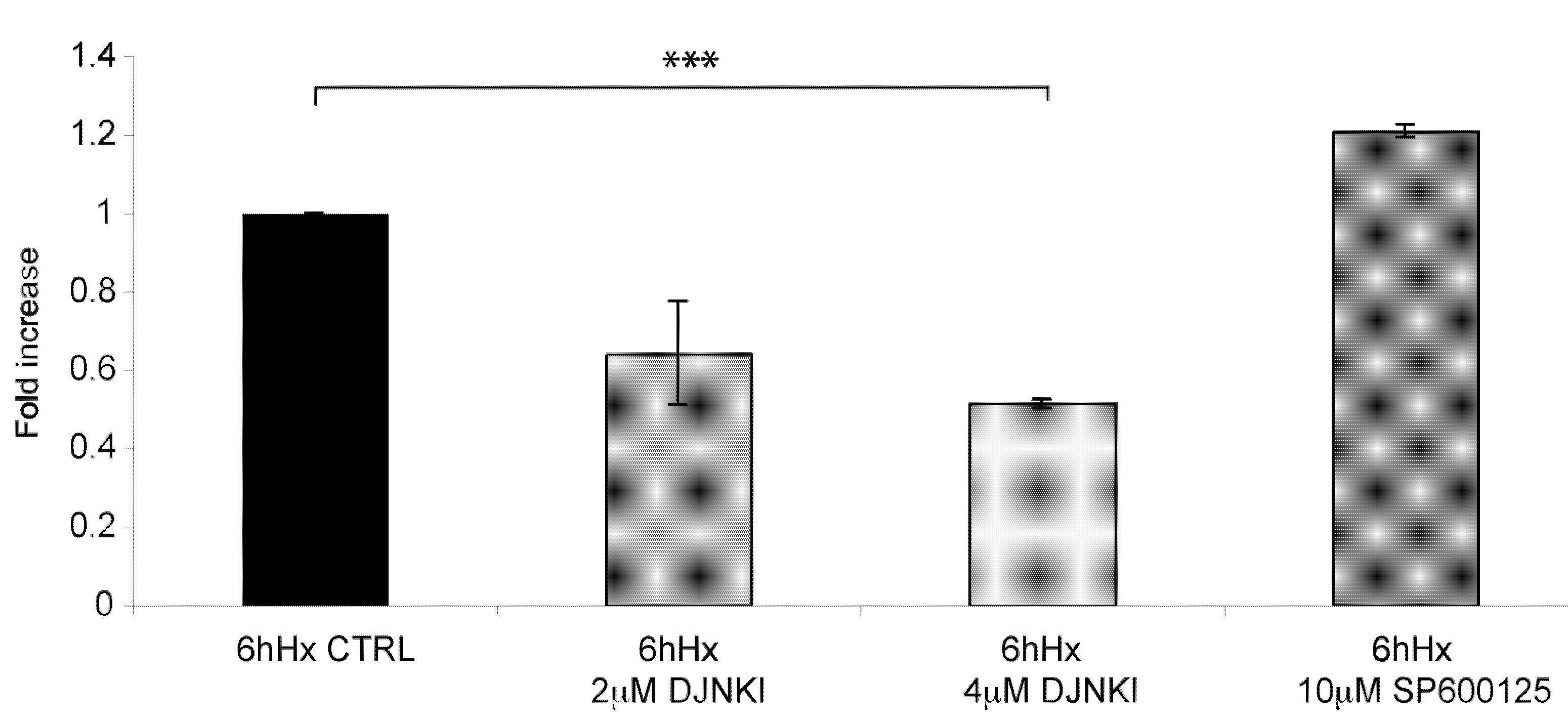
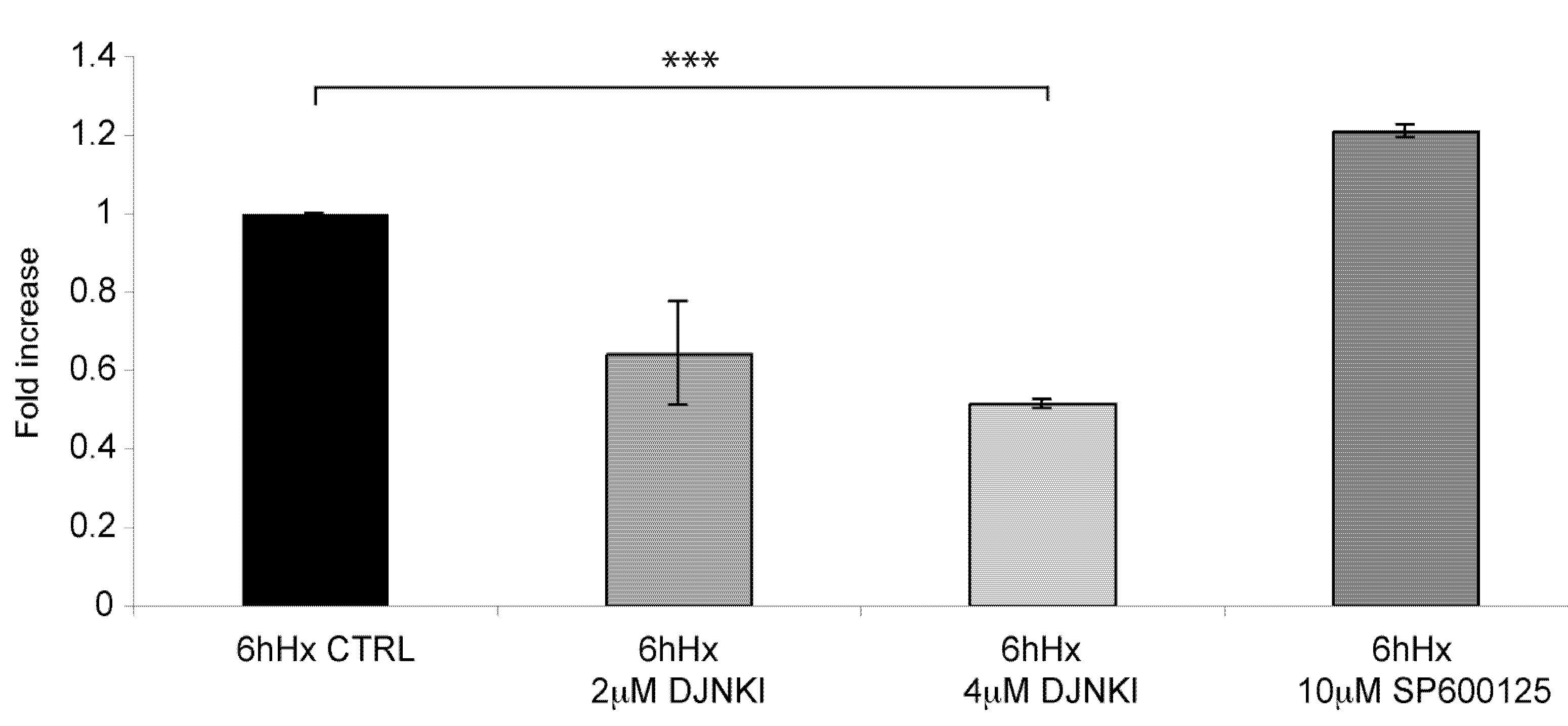
2.6. D-JNKI1 application increases hypoxia-induced Hif-1a levels

Discussion
3. Conclusions
4. Experimental
4.1. Neuronal cultures
4.2. Hypoxic experiments and inhibitor treatments
4.3. Lactate dehydrogenase assay
4.4. Cellular lysis
4.5. Western blot analysis
4.6. Sequence analysis of the Hif-1α protein
4.7. Statistical analysis
Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References and Notes
- Munns, S.E.; Meloni, B.P.; Knuckey, N.W.; Arthur, P.G. Primary cortical neuronal cultures reduce cellular energy utilization during anoxic energy deprivation. J. Neurochem. 2003, 87, 764–772. [Google Scholar] [CrossRef]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef]
- Loerch, P.M.; Lu, T.; Dakin, K.A.; Vann, J.M.; Isaacs, A.; Geula, C.; Wang, J.; Pan, Y.; Gabuzda, D.H.; Li, C.; Prolla, T.A.; Yankner, B.A. Evolution of the aging brain transcriptome and synaptic regulation. PLoS One 2008, 3, e3329. [Google Scholar]
- Lu, T.; Pan, Y.; Kao, S.Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar]
- Ogunshola, O.O.; Antoniou, X. Contribution of hypoxia to Alzheimer's disease: Is HIF-1alpha a mediator of neurodegeneration? Cell. Mol. Life Sci. 2009, 66, 22. [Google Scholar]
- Chavez, J.C.; LaManna, J.C. Hypoxia-inducible factor-1alpha accumulation in the rat brain in response to hypoxia and ischemia is attenuated during aging. Adv. Exp. Med. Biol. 2003, 510, 337–341. [Google Scholar] [CrossRef]
- Anderson, J.; Sandhir, R.; Hamilton, E.S.; Berman, N.E. Impaired Expression of neuroprotective molecules in the HIF-1-alpha Pathway following traumatic brain injury in aged mice. J. Neurotrauma 2009. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad Sci. USA 1995, 92, 5510–5514. [Google Scholar]
- Semenza, G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef]
- Bae, S.H.; Jeong, J.W.; Park, J.A.; Kim, S.H.; Bae, M.K.; Choi, S.J.; Kim, K.W. Sumoylation increases HIF-1alpha stability and its transcriptional activity. Biochem. Biophys. Res. Commun. 2004, 324, 394–400. [Google Scholar] [CrossRef]
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)alpha: its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar]
- Comerford, K.M.; Cummins, E.P.; Taylor, C.T. c-Jun NH2-terminal kinase activation contributes to hypoxia-inducible factor 1alpha-dependent P-glycoprotein expression in hypoxia. Cancer Res. 2004, 64, 9057–9061. [Google Scholar] [CrossRef]
- Yu, B.; Miao, Z.H.; Jiang, Y.; Li, M.H.; Yang, N.; Li, T.; Ding, J. c-Jun protects hypoxia-inducible factor-1alpha from degradation via its oxygen-dependent degradation domain in a nontranscriptional manner. Cancer Res. 2009, 69, 7704–7712. [Google Scholar] [CrossRef]
- Borsello, T.; Clarke, P.G.; Hirt, L.; Vercelli, A.; Repici, M.; Schorderet, D.F.; Bogousslavsky, J.; Bonny, C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 2003, 9, 1180–1186. [Google Scholar] [CrossRef]
- Repici, M.; Centeno, C.; Tomasi, S.; Forloni, G.; Bonny, C.; Vercelli, A.; Borsello, T. Time-course of c-Jun N-terminal kinase activation after cerebral ischemia and effect of D-JNKI1 on c-Jun and caspase-3 activation. Neuroscience 2007, 150, 40–49. [Google Scholar] [CrossRef]
- Lesuisse, C.; Martin, L.J. Immature and mature cortical neurons engage different apoptotic mechanisms involving caspase-3 and the mitogen-activated protein kinase pathway. J. Cereb. Blood Flow Metab. 2002, 22, 935–950. [Google Scholar] [CrossRef]
- Li, J.H.; Wang, Y.H.; Wolfe, B.B.; Krueger, K.E.; Corsi, L.; Stocca, G.; Vicini, S. Developmental changes in localization of NMDA receptor subunits in primary cultures of cortical neurons. Eur. J. Neurosci. 1998, 10, 1704–1715. [Google Scholar] [CrossRef]
- McDonald, J.W.; Behrens, M.I.; Chung, C.; Bhattacharyya, T.; Choi, D.W. Susceptibility to apoptosis is enhanced in immature cortical neurons. Brain Res. 1997, 759, 228–232. [Google Scholar] [CrossRef]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vockler, J.; Dikranian, K.; Tenkova, T.I.; Stefovska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar]
- Ginet, V.; Puyal, J.; Magnin, G.; Clarke, P.G.; Truttmann, A.C. Limited role of the c-Jun N-terminal kinase pathway in a neonatal rat model of cerebral hypoxia-ischemia. J. Neurochem 2009, 108, 552–562. [Google Scholar] [CrossRef]
- Vannucci, S.J.; Hagberg, H. Hypoxia-ischemia in the immature brain. J. Exp. Biol. 2004, 207, 3149–3154. [Google Scholar] [CrossRef]
- Jin, Y.; Yan, E.Z.; Li, X.M.; Fan, Y.; Zhao, Y.J.; Liu, Z.; Liu, W.Z. Neuroprotective effect of sodium ferulate and signal transduction mechanisms in the aged rat hippocampus. Acta Pharmacol. Sin. 2008, 29, 1399–1408. [Google Scholar] [CrossRef]
- Rivard, A.; Berthou-Soulie, L.; Principe, N.; Kearney, M.; Curry, C.; Branellec, D.; Semenza, G.L.; Isner, J.M. Age-dependent defect in vascular endothelial growth factor expression is associated with reduced hypoxia-inducible factor 1 activity. J. Biol. Chem. 2000, 275, 29643–29647. [Google Scholar]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Acker, T.; Acker, H. Cellular oxygen sensing need in CNS function: physiological and pathological implications. J. Exp. Biol. 2004, 207, 3171–3188. [Google Scholar] [CrossRef]
- Bonny, C.; Oberson, A.; Negri, S.; Sauser, C.; Schorderet, D.F. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 2001, 50, 77–82. [Google Scholar] [CrossRef]
- The Universal Protein Resource (UniProt) 2009. Nucleic Acids Res. 2009, 37, D169–D174.
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Antoniou, X.; Sclip, A.; Ploia, C.; Colombo, A.; Moroy, G.; Borsello, T. JNK Contributes to Hif-1α Regulation in Hypoxic Neurons. Molecules 2010, 15, 114-127. https://doi.org/10.3390/molecules15010114
Antoniou X, Sclip A, Ploia C, Colombo A, Moroy G, Borsello T. JNK Contributes to Hif-1α Regulation in Hypoxic Neurons. Molecules. 2010; 15(1):114-127. https://doi.org/10.3390/molecules15010114
Chicago/Turabian StyleAntoniou, Xanthi, Alessandra Sclip, Cristina Ploia, Alessio Colombo, Gautier Moroy, and Tiziana Borsello. 2010. "JNK Contributes to Hif-1α Regulation in Hypoxic Neurons" Molecules 15, no. 1: 114-127. https://doi.org/10.3390/molecules15010114
APA StyleAntoniou, X., Sclip, A., Ploia, C., Colombo, A., Moroy, G., & Borsello, T. (2010). JNK Contributes to Hif-1α Regulation in Hypoxic Neurons. Molecules, 15(1), 114-127. https://doi.org/10.3390/molecules15010114