3-Nitropropionic Acid as a Tool to Study the Mechanisms Involved in Huntington’s Disease: Past, Present and Future

Abstract
:1. Introduction
2. The Past
2.1. The 3-nitropropionic acid (3-NP) model
2.1.1. Treatment with 3-NP and behavioral changes

{kind=link}
{kind=link}
{kind=link}
| Rats | Acute | Sub-chronic and Chronic |
|---|---|---|
| Fischer | 10 mg/kg/day (2–3 days, i.p.) | |
| 10–30 mg/kg/day (1 day, i.p.) | ||
| Lewis | 38 mg/kg/day (2 days, s.c.) | 50–60mg/kg/day (3–4 days, i.p.) |
| 15 mg/kg/day (1 day, i.p.) | 38 mg/kg/day (5 days, i.p.) | |
| 10–15 mg/kg/day (28 days, i.p.) | ||
| Sprague-Dawley | 30 mg/kg (0.5–4 hours, s.c.) | 20 mg/kg/day (3–4 days, i.p.) |
| 15 mg/kg/day (5 days, i.p.) | ||
| Wistar | 10–20 mg/kg/day (1–4 days, i.p.) | 10–20 mg/kg/day (7 days, i.p.) |
| 20 mg/kg/day (2 days, s.c.) | 10 mg/kg/day (twice dose by week/4 weeks, i.p.) | |
| 20, 40, 60 mg/kg/day (9 days, i.p.) |
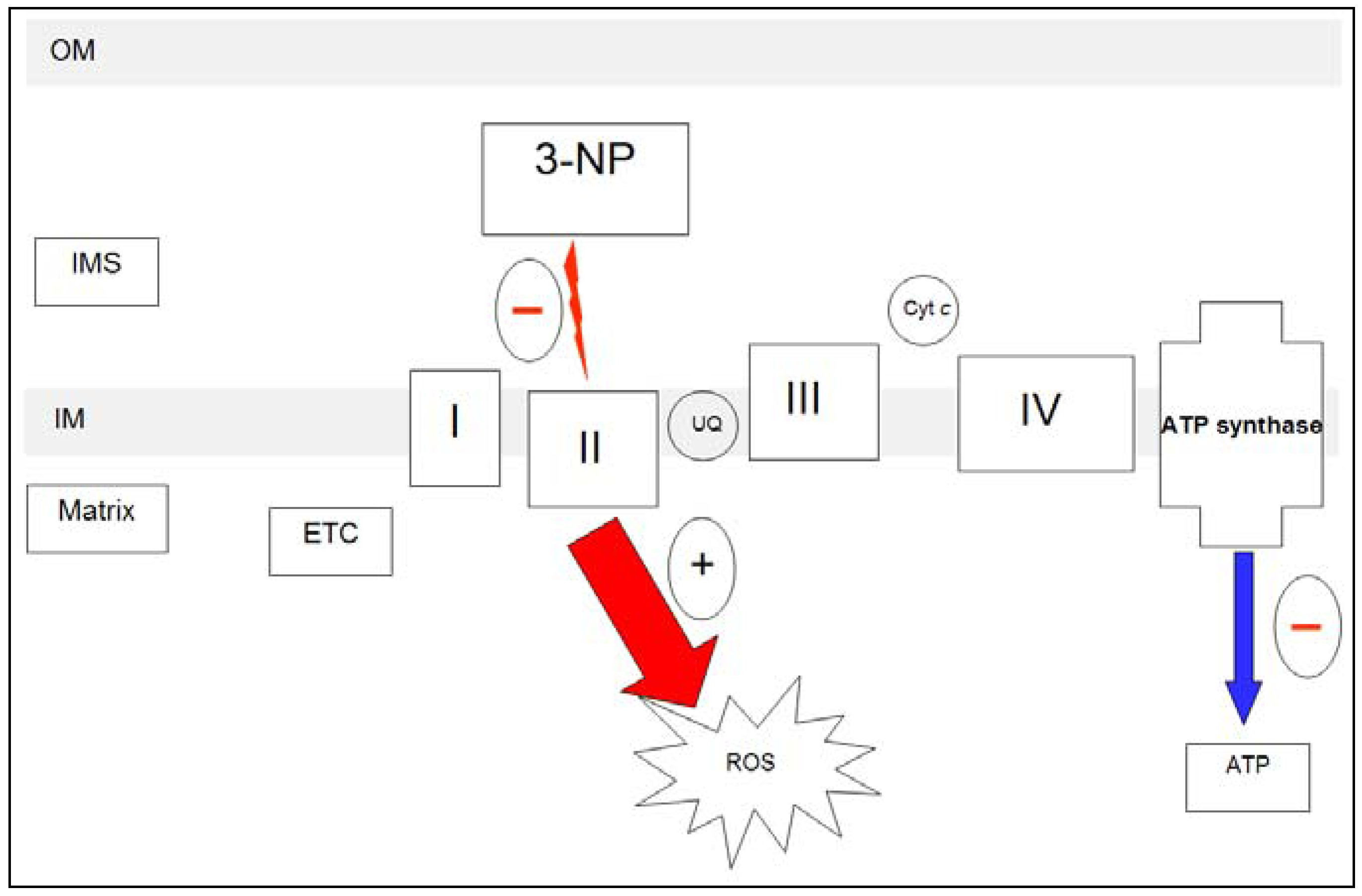
2.1.2. 3-NP and mitochondria
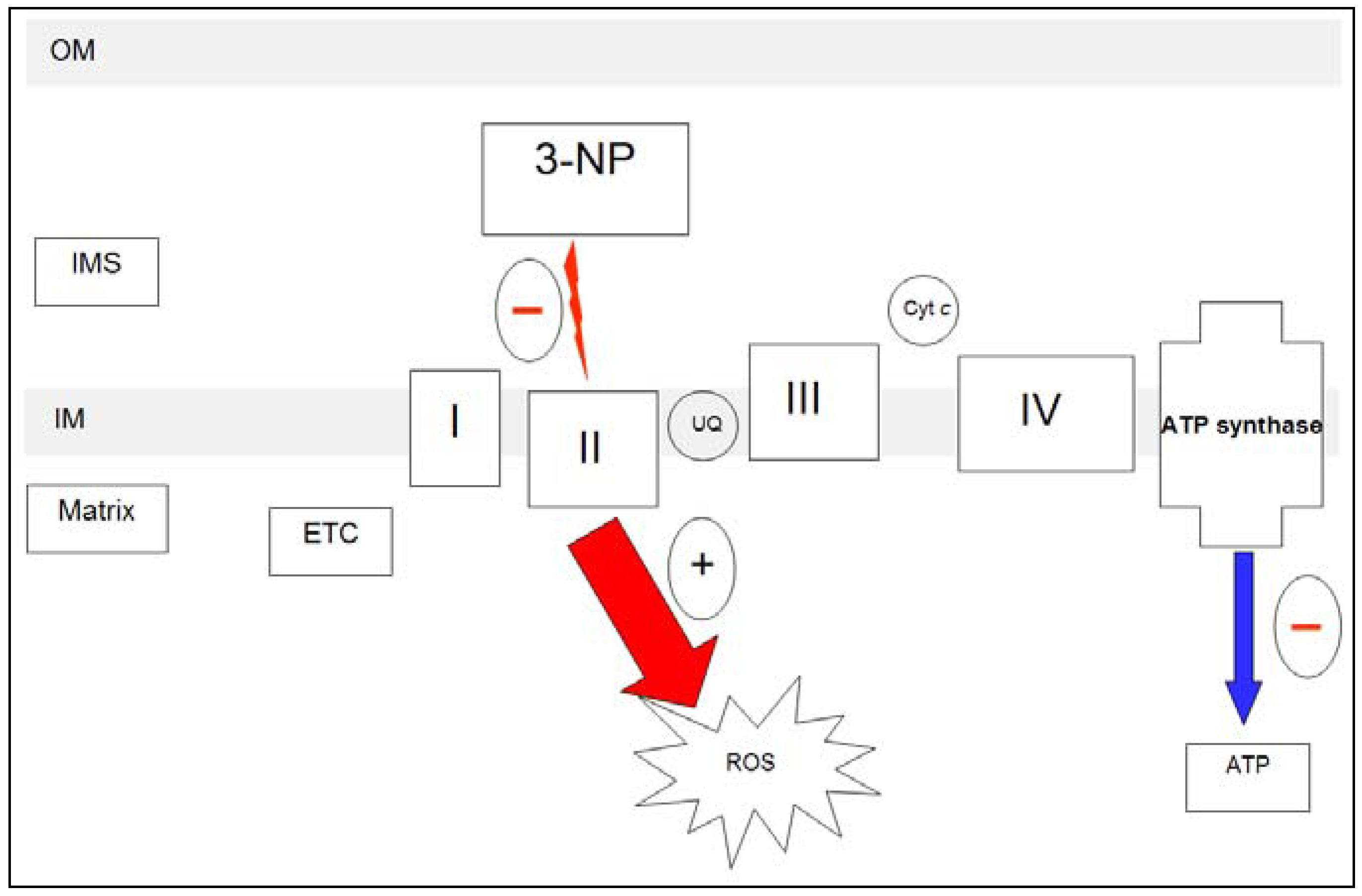
2.1.3. 3-NP and neurotoxicity
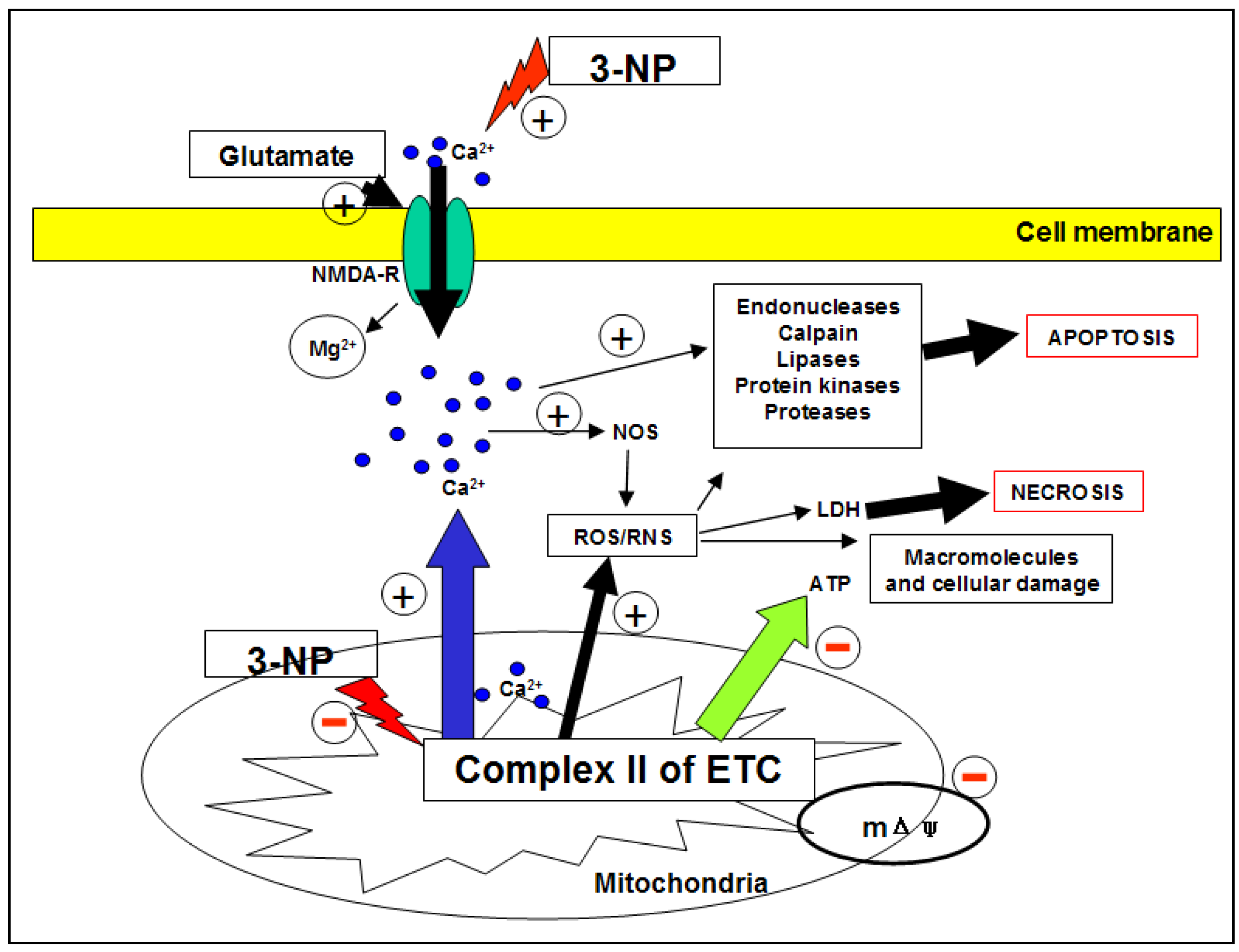
- i)
- Endothelial NOS (eNOS): also known as type III NOS. This is a calcium-dependent enzyme initially found in the endothelium.
- ii)
- Neuronal NOS (nNOS): also known as type I NOS, present in nerve tissue.
- iii)
- Inducible NOS (iNOS): also known as type II NOS. This enzyme is calcium independent, plays an important role in immune system modulation, and it is regulated by different cytokines. In addition, it produces NO in astrocytes, microglia and macrophages in response to inflammatory reactions.
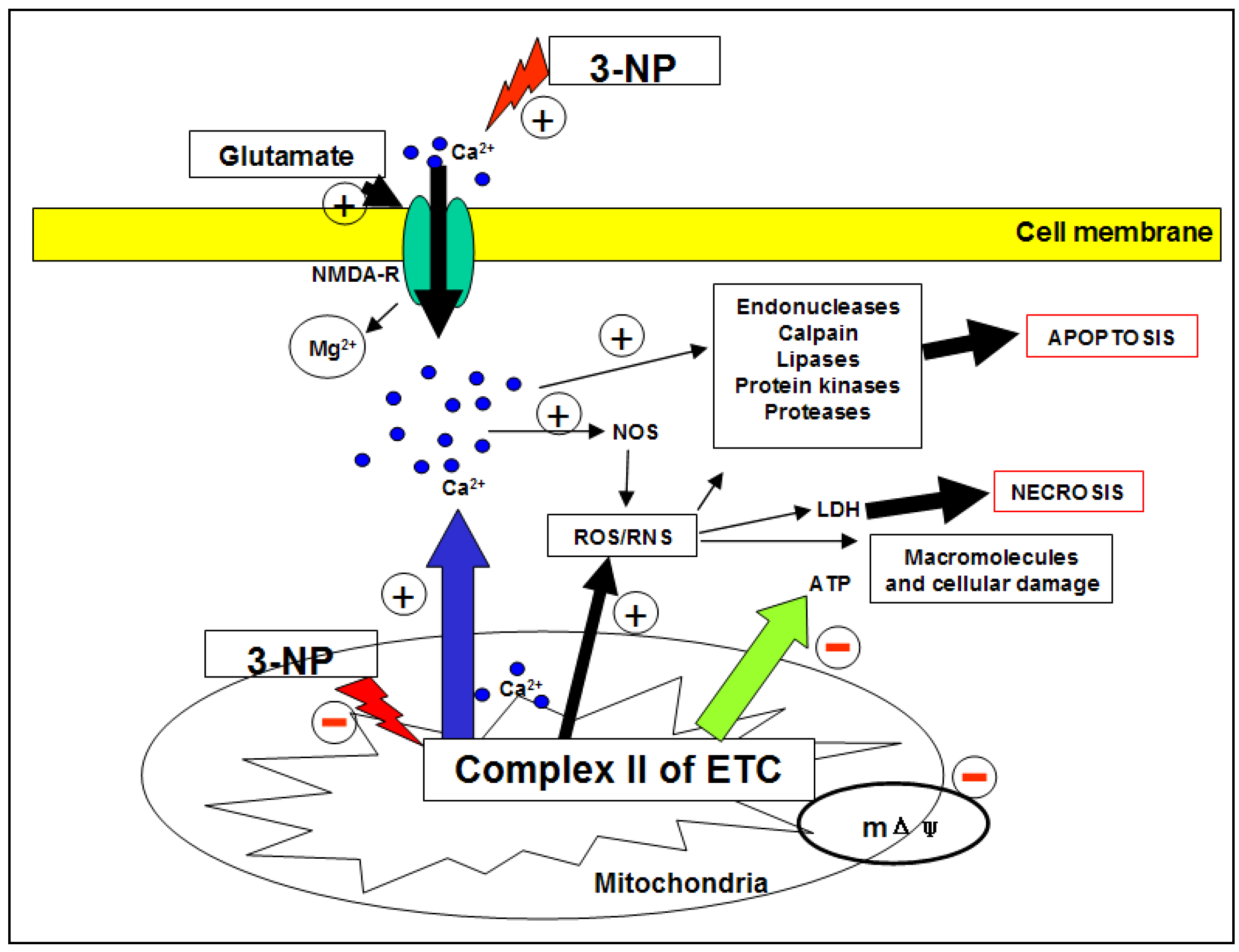
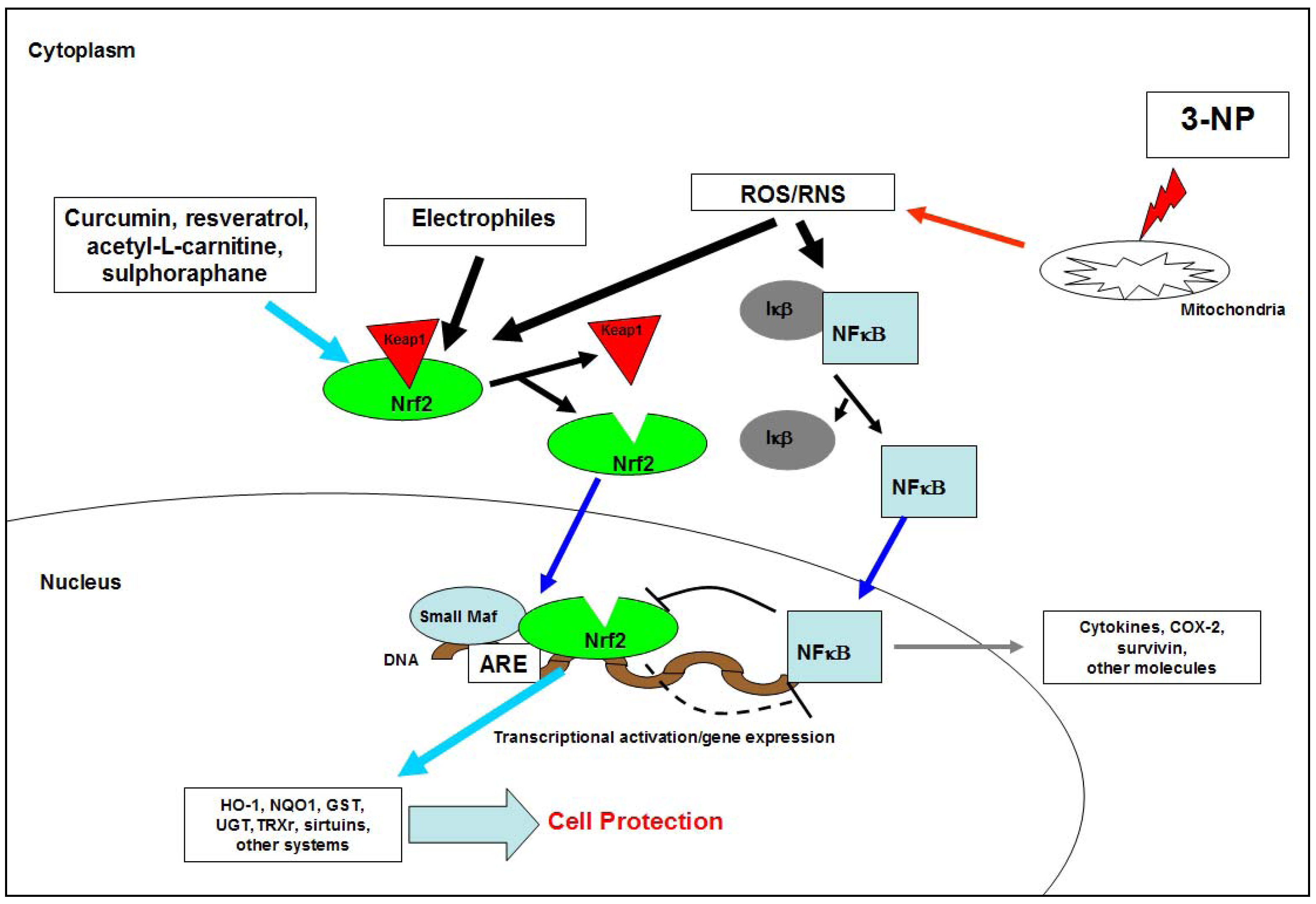
2.1.4. 3-NP and ROS/RNS
2.1.5. 3-NP, neurochemistry and neuropathology
| Adenosine release | Increased |
| ATP production | Decreased |
| Intracellular calcium levels | Increased |
| Caspase-3 activity | Increased |
| Caspase-9 activity | Increased |
| Choline acetyltransferase | Decreased |
| Citochrome c release | Increased |
| Dopamine | Increased |
| Dopamine 3,4-dihydroxyphenylacetic acid (DOPAC) | Increased |
| Endocannabinoids | Decreased |
| Enkephalin | Decreased |
| GABA | Decreased |
| Homovanillic acid (HVA) | Increased |
| LDH | Increased |
| Neuropeptide Y | Increased |
| Neurotensin | Increased |
| NMDA-R | Increased sensibility to basal levels of glutamate |
| NO | Increased |
| ROS production | Increased |
| RNS production | Increased |
| SDH activity | Decreased |
| Somatostatin | Increased |
| Substantia P | Decreased |
2.1.6. 3-NP and death cell
2.1.7. 3-NP model: advantages and disadvantages
- I)
- mHtt is not produced or folded in metabolic toxic models; cytoplasmic and neuronal inclusions are therefore not observed;
- II)
- the onset of cellular death is progressive and inversely proportional to the number of CAG triplets, a situation (especially the later) not replicated in the metabolic model since cell death is induced immediately by 3-NP through excitotoxicity and metabolic mechanisms not dependent on mHtt;
- III)
- despite the fact that the 3-NP model reproduces different cognitive and behavioral aspects of the HD phenotype, the resemblance of other behavioural aspects, such as suicidal tendencies and obsessive-compulsive conduct, has not been possible. Nonetheless, results obtained recently by our group (unpublished data) show that the administration of a daily dose of 3-NP (10 mg/kg/ip) for three days induces depression and anxiety valued by the forced swimming and open field test, respectively;
- IV)
- the model induced by 3-NP is itself limited to the bioavailability of the toxicant; once metabolized and eliminated, it ceases to have an effect, enabling tissues to respond to the insult.
- I)
- massive cell death induced by the neurotoxin makes it a useful model for studying neurotoxicity phenomena;
- II)
- it is a useful model for analyzing and studying neuroprotective and neurorestoration therapies for HD patients;
- III)
- it is a useful model also for examining the synergic effect of mitochondrial alterations on Htt mutation;
- IV)
- it is,m indeed, useful for studying mechanisms involved in HD pathogenesis such as ROS formation, protease activation, astrogliosis, etc.
2.2. Other Huntington’s disease induced models: Emphasis on QA and facilitating models
3. The Present
4. The Future (Conclusion)
Acknowledgements
- Sample Availability: Not available.
References
- Gonzalez-Alegre, P.; Afifi, A.K. Clinical characteristics of childhood-onset (juvenile) Huntington disease: Report of 12 patients and review of the literature. J. Child. Neurol. 2006, 21, 223–229. [Google Scholar]
- Avila-Giron, R. Medical and social aspects of Huntington`s chorea in the state of Zulia, Venezuela. Adv. Neurol. 1973, 1, 261–266. [Google Scholar]
- González-Ferrer, S.; Pineda-Bernal, L.; Delgado-Luengo, W.; Villalobos-Cabrera, H. Medical genetics in Zulia, a State of Venezuela. Community Genet. 2004, 7, 153–156. [Google Scholar] [CrossRef]
- Okun, M.S.; Thommi, N. Americo Negrette (1924 to 2003): Diagnosing Huntington disease in Venezuela. Neurology 2004, 63, 340–343. [Google Scholar] [CrossRef]
- Paradisi, I.; Hernández, A.; Arias, S. Huntington disease mutation in Venezuela: Age of onset, haplotype analyses and geographic aggregation. J. Hum. Genet. 2008, 53, 127–135. [Google Scholar] [CrossRef]
- Pridmore, S.; Cook, A.; McCormick, G.; West, A. Trinucleotide expansion in Tasmanian HD families. Aust. N Z J. Psychiat. 1995, 29, 157. [Google Scholar]
- Pridmore, S.A. The large Huntington’s disease family of Tasmania. Med. J. Aust. 153, 593–595.
- Pridmore, S.A. The prevalence of Huntington’s disease in Tasmania. Med. J. Aust. 153, 133–134.
- Wright, H.H.; Still, C.N.; Abramson, R.K. Huntington’s disease in black kindreds in South Carolina. Arch. Neurol. 1981, 38, 412–414. [Google Scholar] [CrossRef]
- Young, A.B.; Shoulson, I.; Penney, J.B.; Starosta-Rubinstein, S.; Gomez, F.; Travers, H.; Ramos-Arroyo, M.A.; Snodgrass, S.R.; Bonilla, E.; Moreno, H.; Wexler, N.S. Huntington`s disease in Venezuela: Neurologic features and functional decline. Neurology 1986, 36, 244–249. [Google Scholar] [CrossRef]
- Aubeeluck, A.; Brewer, H. Huntington’s disease. Part 2: Treatment and management issues in juvenile HD. Br. J. Nurs. 2008, 17, 260–263. [Google Scholar]
- Biglan, K.; Shoulson, I. Juvenile-onset Huntington disease: A matter of perspective. Arch. Neurol. 2007, 64, 783–784. [Google Scholar] [CrossRef]
- Foroud, T.; Gray, J.; Ivashina, J.; Conneally, P.M. Differences in duration of Huntington’s disease based on age at onset. J. Neurol. Neurosurg. Psychiat. 1999, 66, 52–56. [Google Scholar] [CrossRef]
- Leegwater, J.; Jang-Ho, J. The paradigm of Huntington’s disease: Therapeutic opportunities in neurodegeneration. Am. Soc. Exp. Neuro.Ther. 2004, 1, 128–138. [Google Scholar]
- Nance, M.A.; Myers, R.H. Juvenile onset Huntington’s disease—clinical and research perspectives. Ment. Retard Dev. Disabil. Res. Rev. 2001, 7, 153–157. [Google Scholar] [CrossRef]
- Ribaï, P.; Nguyen, K.; Hahn-Barma, V.; Gourfinkel-An, I.; Vidailhet, M.; Legout, A.; Dodé, C.; Brice, A.; Dürr, A. Psychiatric and cognitive difficulties as indicators of juvenile huntington disease onset in 29 patients. Arch. Neurol. 2007, 64, 813–819. [Google Scholar] [CrossRef]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington`s disease chromosomes. Cell 1993, 72, 971–983. [CrossRef]
- Arnulf, I.; Nielsen, J.; Lohmann, E.; Schiefer, J.; Wild, E.; Jennum, P.; Konofal, E.; Walker, M.; Oudiette, D.; Tabrizi, S.; Durr, A. Rapid eye movement sleep disturbances in Huntington disease. Arch. Neurol. 2008, 65, 482–488. [Google Scholar] [CrossRef]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; Magnusson, A.; Woodman, B.; Landles, C.; Pouladi, M.A.; Hayden, M.R.; Khalili-Shirazi, A.; Lowdell, M.W.; Brundin, P.; Bate, G.P.; Leavitt, B.R.; Möller, T.; Tabrizi, S.J. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef]
- Eskenazi, B.R.; Wilson-Rich, N.S.; Starks, P.T. A Darwinian approach to Huntington’s disease: Subtle health benefits of a neurological disorder. Med. Hypoth. 2007, 71, 151–152. [Google Scholar]
- Gaba, A.M.; Zhang, K.; Marder, K.; Moskowitz, C.B.; Werner, P.; Boozer, C.N. Energy balance in early-stage Huntington disease. Am. J. Clin. Nutr. 2005, 81, 1335–1341. [Google Scholar]
- Folstein, S.E.; Leigh, R.J.; Parhad, I.M.; Folstein, M.F. The diagnosis of Huntington’s disease. Neurology 1986, 36, 1279–1283. [Google Scholar] [CrossRef]
- Johnson, S.A.; Stout, J.C.; Solomon, A.C.; Langbehn, D.R.; Aylward, E.H.; Cruce, C.B.; Ross, C.A.; Nance, M.; Kayson, E.; Julian-Baros, E.; Hayden, M.R.; Kieburtz, K.; Guttman, M.; Oakes, D.; Shoulson, I.; Beglinger, L.; Duff, K.; Penziner, E.; Paulsen, J.S. Predict-HD Investigators of the Huntington Study Group. Beyond disgust: Impaired recognition of negative emotions prior to diagnosis in Huntington’s disease. Brain 2007, 130, 1732–1744. [Google Scholar] [CrossRef]
- Martin, J.B.; Gusella, J.F. Huntington’s disease. Pathogenesis and management. N. Engl. J. Med. 1986, 315, 1267–1276. [Google Scholar] [CrossRef]
- Morton, A.J.; Wood, N.I.; Hastings, M.H.; Hurelbrink, C.; Barker, R.A.; Maywood, E.S. Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J. Neurosci. 2005, 25, 3994. [Google Scholar]
- Phillips, W.; Shannon, K.M.; Barker, R.A. The current clinical management of Huntington’s disease. Mov. Disord. 2008, 23, 1491–1504. [Google Scholar] [CrossRef]
- Purdon, S.E.; Mohr, E.; Ilivitsky, V.; Jones, B.D. Huntington’s disease: Pathogenesis, diagnosis and treatment. J. Psychiatry Neurosci. 1994, 19, 359–367. [Google Scholar]
- Roze, E.; Saudou, F.; Caboche, J. Pathophysiology of Huntington’s disease: From huntingtin functions to potential treatments. Curr. Opin. Neurol. 2008, 21, 497–503. [Google Scholar]
- Sandyk, R. Pineal and habenula calcification in schizophrenia. Int. J. Neurosci. 1992, 67, 19–30. [Google Scholar] [CrossRef]
- Shiwach, R.S.; Norbury, C.G. A controlled psychiatric study of individuals at risk for Huntington’s disease. Br. J. Psychiatry 1994, 165, 500–505. [Google Scholar] [CrossRef]
- Van Duijn, E.; Kingma, E.M.; van der Mast, R.C. Psychopathology in verified Huntington’s disease gene carriers. J. Neuropsychiatry. Clin. Neurosci. 2007, 19, 441–448. [Google Scholar] [CrossRef]
- Walker, F. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Cha, J.H. Transcriptional dysregulation in Huntington’s disease. Trends Neurosci. 2000, 23, 387–392. [Google Scholar] [CrossRef]
- La Spada, A.R.; Roling, D.B.; Harding, A.E.; Warner, C.L.; Spiegel, R.; Hausmanowa- Petrusewicz, I.; Yee, W.C.; Fischbeck, K.H. Meiotic stability and genotype-phenotype correlation of the trinucleotide repeat in X-linked spinal and bulbar muscular atrophy. Nat. Genet. 1992, 2, 301–304. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Rangone, H.; Humbert, S.; Saudou, F. Huntington’s disease: How does huntingtin, an anti-apoptotic protein, become toxic? Pathol. Biol. (Paris) 2004, 52, 338–342. [Google Scholar] [CrossRef]
- Brouillet, E.; Condé, F.; Beal, M.F.; Hantraye, P. Replicating Huntington’s disease phenotype in experimental animals. Prog. Neurobiol. 1999, 59, 427–468. [Google Scholar] [CrossRef]
- Chiarugi, A.; Calvani, M.; Meli, E.; Traggiai, E.; Moroni, F. Synthesis and release of neurotoxic kynurenine metabolites by human monocyte-derived macrophages. J. Neuroimmunol. 120, 190–198.
- Chiarugi, A.; Meli, E.; Moroni, F. Similarities and differences in the neural death processes activated by 3OH-Kynurenine and quinolinic acid. J. Neurochem. 77, 2001b, 1310–1318. [Google Scholar]
- Estrada-Sánchez, A.M.; Mejía-Toiber, J.; Massieu, L. Excitotoxic neuronal death and the pathogenesis of Huntington’s disease. Arch. Med. Res. 2008, 39, 265–276. [Google Scholar] [CrossRef]
- Dawson, R.; Beal, M.F.; Bondy, S.C.; Di Monte, D.A.; Isom, G.E. Excitotoxins, aging, and environmental neurotoxins: Implications for understanding human neurodegenerative diseases. Toxicol. Appl. Pharmacol. 1995, 134, 1–17. [Google Scholar] [CrossRef]
- Guidetti, P.; Bates, G.P.; Graham, R.K.; Hayden, M.R.; Leavitt, B.R.; MacDonald, M.E.; Slow, E.J.; Wheeler, V.C.; Woodman, B.; Schwarcz, R. Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease mice. Neurobiol. Dis. 2006, 23, 190–197. [Google Scholar] [CrossRef]
- Pérez-de la Cruz, V.; Santamaría, A. Integrative hypothesis for Huntington’s disease: A brief review of experimental evidence. Physiol. Res. 56, 513–526.
- Brouillet, E.; Jacquard, C.; Bizat, N.; Blum, D. 3-Nitropropionic acid: A mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington's disease. J. Neurochem. 2005, 95, 1521–1540. [Google Scholar] [CrossRef]
- Brennan, W.A., Jr.; Bird, E.D.; Aprille, J.R. Regional mitochondiral respieratoy activity in Huntington’s disease brain. J. Neurochem. 1985, 44, 1948–1950. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Baoyson, S.J.; Luder, A.S.; Parks, J.K. Evidence for a defect in NADH: Ubiquinone oxidoreductase (complex I) in Huntington’s disease. Neurology 1990, 40, 1231–1234. [Google Scholar] [CrossRef]
- Arenas, J.; Campos, Y.; Ribacoba, R.; Martín, M.A.; Rubio, J.C.; Ablanedo, P.; Cabello, A. Complex I defect in muscle from patients with Huntington’s disease. Ann. Neurol. 1998, 43, 397–400. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A. Protective effect of rivastigmine against 3-nitropropionic acid-induced Huntington’s disease like symptoms: Possible behavioural, biochemical and cellular alterations. Eur. J. Pharmacol. 2009, 615, 91–101. [Google Scholar] [CrossRef]
- Túnez, I.; Santamaría, A. Model of Huntington’s disease induced with 3-nitropropionic acid. Rev. Neurol. 2009, 48, 430–434. [Google Scholar]
- Tasset, I.; Sánchez, F.; Túnez, I. The molecular bases of Huntington’s disease: The role played by oxidative stress. Rev. Neurol. 2009, 49, 424–429. [Google Scholar]
- Gil, J.M.; Rego, A.C. Mechanisms of neurodegeneration in Huntington’s disease. Eur. J. Neurosci. 2008, 27, 2803–2820. [Google Scholar] [CrossRef]
- Beal, M.F. Aging, energy, and oxidative stress in neurodegenerative disease. Ann. Neurol. 1995, 38, 357–366. [Google Scholar] [CrossRef]
- Banoei, M.M.; Houshmand, M.; Panahi, M.S.; Shariati, P.; Rostami, M.; Manshadi, M.D.; Majidizadeh, T. Huntington’s disease and mitochondrial DNA deletions: Event or regular mechanism for mutant huntingtin protein and CAG repeats expansion?! Cell. Mol. Neurobiol. 2007, 27, 867–875. [Google Scholar] [CrossRef]
- Lim, D.; Fedrizzi, L.; Tartari, M.; Zuccato, C.; Cattaneo, E.; Brini, M.; Carafoli, E. Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J. Biol. Chem. 2008, 283, 5780–5789. [Google Scholar]
- Kalonia, H.; Kumar, P.; Kumar, A.; Nehru, B. Effects of caffeic acid, rofecoxib, and their combination against quinolinic acid-induced behavioral alterations and disruption in glutathione redox status. Neurosci. Bull. 2009, 25, 343–352. [Google Scholar] [CrossRef]
- Kumar, P.; Kalonia, H.; Kumar, A. Lycopene modulates nitric oxide pathways against 3-nitropropioni acid-induced neurotoxicity. Life Sci. 2009, 85, 711–718. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A. Protective effect of hesperidin and naringin against 3-nitropropionic acid induced Huntington’s like symptoms in rats: Possible role of nitric oxide. Behav. Brain Res. 2010, 38–46. [Google Scholar] [CrossRef]
- Browne, S.E. Mitochondrial and Huntingon’s disease pathogenesis: Insight from genetic and chemical models. Ann. N.Y. Acad. 2008, 1147, 358–382. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef]
- Finkbeiner, S.; Mitra, S. The ubiquitin-proteasome pathway in Huntington’s disease. Sci.Word J. 2008, 8, 421–433. [Google Scholar]
- Gafni, J.; Hermel, E.; Young, J.E.; Wellington, C.L.; Hayden, M.R.; Ellerby, L.M. Inhibition of calpain cleavage of huntingtin reduces toxicity: Accumulation of calpain/caspase fragments in the nucleus. J. Biol. Chem. 2004, 279, 20211–20220. [Google Scholar]
- Lunkes, A.; Lindenberg, K.S.; Ben-Haïem, L.; Weber, C.; Devys, D.; Landwehrmeyer, G.B.; Mandel, J.L.; Trottier, Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell. 2002, 10, 259–269. [Google Scholar] [CrossRef]
- Sánchez, I.; Mahlke, C.; Yuan, J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 2003, 421, 373–379. [Google Scholar] [CrossRef]
- Poirier, M.A.; Li, H.; Macosko, J.; Cai, S.; Amzel, M.; Ross, C.A. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 2002, 277, 41032–41037. [Google Scholar]
- Poirier, M.A.; Jiang, H.; Ross, C.A. A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: Evidence for a compact beta-sheet structure. Hum. Mol. Genet. 2005, 14, 765–774. [Google Scholar] [CrossRef]
- Ross, C.A.; Pickart, C.M. The ubiquitin-proteasome pathway in Parkinson’s disease and other neurodegenerative diseases. Trends Cell Biol. 2004, 14, 703–711. [Google Scholar] [CrossRef]
- Venkatraman, P.; Wetzel, R.; Tanaka, M.; Nukina, N.; Goldberg, A.L. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell. 2004, 14, 95–104. [Google Scholar] [CrossRef]
- Canals, J.M.; Pineda, J.R.; Torres-Peraza, J.F.; Bosch, M.; Martín-Ibáñez, R.; Muñoz, M.T.; Mengod, G.; Ernfors, P.; Alberch, J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington's disease. J. Neurosci. 2004, 24, 7727–7739. [Google Scholar]
- Cattaneo, E. Dysfunction of wild-type huntingtin in Huntington disease. News Physiol. Sci. 2003, 18, 34–37. [Google Scholar]
- Jiang, H.; Poirier, M.A.; Liang, Y.; Pei, Z.; Weiskittel, C.E.; Smith, W.W.; DeFranco, D.B.; Ross, C.A. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis. 2006, 23, 543–551. [Google Scholar] [CrossRef]
- Kegel, K.B.; Meloni, A.R; Yi, Y.; Kim, Y.J.; Doyle, E.; Cuiffo, B.G.; Sapp, E.; Wang, Y.; Qin, Z.H.; Chen, J.D.; Nevins, J.R.; Aronin, N.; DiFiglia, M. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J. Biol. Chem. 2002, 277, 7466–7476. [Google Scholar]
- Lee, S.T.; Kim, M. Aging and neurodegeneration. Molecular mechanisms of neuronal loss in Huntington’s disease. Mech Ageing Dev. 2006, 127, 432–435. [Google Scholar] [CrossRef]
- Liu, Y.F.; Deth, R.C.; Devys, D. SH3 domain-dependent association of huntingtin with epidermal growth factor receptor signalling complexes. J. Biol. Chem. 1997, 272, 8121–8124. [Google Scholar]
- Martindale, D.; Hackam, A.; Wieczorek, A.; Ellerby, L.; Wellington, C.; McCutcheon, K.; Singaraja, R.; Kazemi-Esfarjani, P.; Devon, R.; Kim, S.U.; Bredesen, D.E.; Tufaro, F.M ; Hayden, M.R. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genet. 1998, 18, 150–154. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Scappini, E.; Koh, T.W.; Martin, N.P.; O’Bryan, J.P. Intersectin enhances huntingtin aggregation and neurodegeneration through activation of c-Jun-NH2-terminal kinase. Hum. Mol. Genet. 2007, 16, 1862–1871. [Google Scholar] [CrossRef]
- Steffan, J.S.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.C.; Slepko, N.; Illes, K; Lukacsovich, T.; Zhu, Y.Z.; Cattaneo, E.; Pandolfi, P.P.; Thompson, L.M.; Marsh, J.L. SUMO modification of Huntingtin and Huntington’s disease pathology. Science 2004, 304, 100–104. [Google Scholar]
- Sugars, K.L.; Rubinsztein, D.C. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003, 19, 233–238. [Google Scholar] [CrossRef]
- Zuccato, C.; Liber, D.; Ramos, C.; Tarditi, A.; Rigamonti, D.; Tartari, M.; Valenza, M.; Cattaneo, E. Progressive loss of BDNF in a mouse model of Huntington’s disease and rescue by BDNF delivery. Pharmacol. Res. 2003, 52, 133–139. [Google Scholar]
- Coyle, J.T.; Schwarcz, R. Lesion of striatal neurones with kainic acid provides a model for Huntington’s chorea. Nature 1976, 263, 244–246. [Google Scholar] [CrossRef]
- Rothman, S.M.; Olney, J.W. Excitotoxicity and the NMDA receptor-still lethal after eight years. Trends Neurosci. 1995, 18, 57–58. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; Bates, G.P. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Parker, J.A.; Connolly, J.B.; Wellington, C.; Hayden, M.; Dausset, J.; Neri, C. Expanded poluglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc. Natl. Acad. Sci. USA 2001, 98, 13318–13323. [Google Scholar]
- Marsh, J.L.; Pallos, J.; Thompson, L.M. Fly models of Huntington’s disease. Hum. Mol. Genet. 2003, 2, R187–R193. [Google Scholar]
- Powers, W.J.; Videen, T.O.; Markham, J.; McGee-Minnich, L.; Antenor-Dorsey, J.V.; Hershey, T.; Perlmutter, J.S. Selective defect of in vivo glycolysis in early Huntington’s disease striatum. Proc. Natl. Acad. Sci. USA 2007, 104, 2945–2949. [Google Scholar]
- Beal, M.F. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann. Neurol. 2007, 31, 119–130. [Google Scholar] [CrossRef]
- Hamilton, B.F.; Gould, D.H. Nature and distribution of brain lesions in rats intoxicated with 3-nitropropionic acid: A type of hypoxic (energy deficient) brain damage. Acta Neuropathol. 1987, 72, 286–297. [Google Scholar] [CrossRef]
- Liu, X.; Luo, X.; Hu, W. Studies on the epidemiology and etiology of moldy sugarcane poisoning in China. Biomed. Environ. Sci. 1992, 5, 161–177. [Google Scholar]
- Ramaswamy, S.; McBrid, J.L.; Kordower, J.H. Animal models of Huntington’s disease. ILAR. J. 2007, 48, 356–373. [Google Scholar]
- Borlongan, C.V.; Koutouzis, T.K.; Freeman, T.B.; Hauser, R.A.; Cahill, D.W.; Sanberg, R. Hyperactivity and hypoactivity in a rat model of Huntington’s disease: The systemic 3-nitropropionic acid model. Brain Res. Brain Res. Protoc. 1, 253–257.
- Ouary, S.; Bizat, N.; Altairac, S.; Ménétrat, H.; Mittoux, V.; Condé, F.; Hantraye, P.; Brouillet, E. Major strain differences in response to chronic systemic administration of the mitochondrial toxin 3-nitropropionic acid in rats: Implications for neuroprotection studies. Neuroscience 2000, 97, 521–530. [Google Scholar] [CrossRef]
- Ahuja, M.; Bishnoi, M.; Chopra, K. Protective effect of minocycline, a semi-synthetic second-generation tetracycline against 3-nitropropionic acid (3-NP)-induced neurotoxicity. Toxicology 2008, 244, 111–122. [Google Scholar] [CrossRef]
- Deshpande, S.B.; Hida, H.; Takei-Io, N.; Masuda, T.; Baba, H.; Nishino, H. Involvement of nitric oxide in 3-nitropropionic acid-induced striatal toxicity in rats. Brain Res. 2006, 1108, 205–215. [Google Scholar] [CrossRef]
- Dhir, A.; Akula, KK.; Kulkarni, S.K.; Tiagabine, A. GABA uptake inhibitor, attenuates 3-nitropropionic acid-induced alterations in various behavioral and biochemical parameters in rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 835–843. [Google Scholar] [CrossRef]
- Lukács, A.; Szabó, A.; Vezér, T.; Papp, A. The acute effects of 3-nitropropionic acid on the behavior and spontaneous cortical electrical activity of rats. Acta Neurobiol. Exp. (Wars). 2006, 66, 227–233. [Google Scholar]
- McBride, J.L.; During, M.J.; Wuu, J.; Chen, E.Y.; Leurgans, S.E.; Kordower, J.H. Structural and functional neuroprotection in a rat model of Huntington`s disease by viral gene transfer of GDNF. Exp. Neurol. 2003, 181, 213–223. [Google Scholar] [CrossRef]
- Mogami, M.; Hayashi, Y.; Masuda, T.; Kohri, K.; Nishino, H.; Hida, H. Altered striatal vulnerability to 3-nitropropionic acid in rats due to sex hormone levels during late phase of brain development. Neurosci. Lett. 2008, 436, 321–325. [Google Scholar] [CrossRef]
- Przybyla-Zawislak, B.D.; Thorn, B.T.; Ali, S.F.; Dennis, R.A.; Amato, A.; Virmani, A.; Binienda, Z.K. Identification of rat hippocampal mRNAs altered by the mitochondrial toxicant, 3-NPA. Ann. N. Y. Acad. Sci. 2006, 1053, 162–173. [Google Scholar]
- Túnez, I.; Collado, J.A.; Medina, F.J.; Peña, J.; Muñoz, M.C.; Jimena, I.; Franco, F.; Rueda, I.; Feijóo, M.; Muntané, J.; Montilla, P. 17 Beta-estradiol may affect vulnerability of striatum in a 3-nitropropionic acid-induced experimental model of Huntington's disease in ovariectomized rats. Neurochem. Int. 48, 367–373.
- Túnez, I.; Drucker-Colín, R.; Jimena, I.; Medina, F.J.; Muñoz, M.C.; Peña, J.; Montilla, P. Transcranial magnetic stimulation attenuates cell loss and oxidative damage in the striatum induced in the 3-nitropropionic model of Huntington's disease. J. Neurochem. 97, 619–630.
- Túnez, I.; Feijóo, M.; Collado, J.A.; Medina, F.J.; Peña, J.; Muñoz, M.C.; Jimena, I.; Franco, F.; Rueda, I.; Muntané, J.; Montilla, P. Effect of testosterone on oxidative stress and cell damage induced by 3-nitropropionic acid in striatum of ovariectomized rats. Life Sci. 2007, 80, 1221–1227. [Google Scholar] [CrossRef]
- Túnez, I.; Montilla, P.; Muñoz, M.C.; Feijóo, M.; Salcedo, M. Protective effect of melatonin on 3-nitropropionic acid-induced oxidative stress in synaptosomes in an animal model of Huntington’s disease. J. Pineal Res. 37, 252–256.
- Túnez, I.; Montilla, P.; Muñoz, M.C.; Drucker-Colín, R. Effect of nicotine on 3-nitropropionic acid-induced oxidative stress in synaptosomes. Eur. J. Pharmacol. 504, 169–175.
- Túnez, I.; Muñoz, M.C.; Montilla, P. Treatment with dehydroepiandrosterone prevents oxidative stress induced by 3-nitropropionic acid in synaptosomes. Pharmacology 2005, 74, 113–118. [Google Scholar]
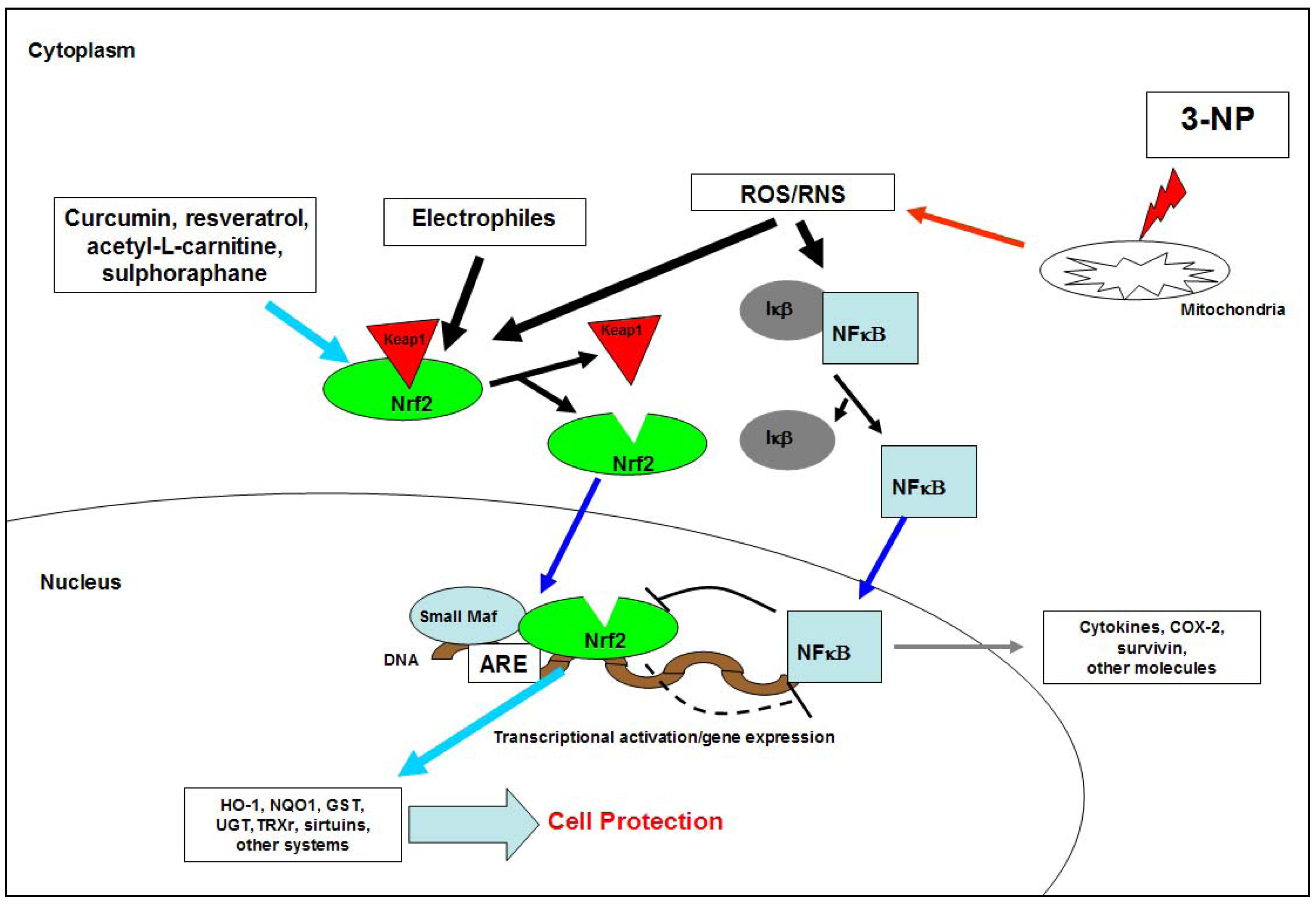
- Shih, A.Y.; Imbeault, S.; Barakauskas, V.; Erb, H.; Jiang, L.; Li, P.; Murphy, T.H. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J. Biol. Chem. 2005, 280, 22925–22936. [Google Scholar]
- Borlongan, C.V.; Koutouzis, T.K.; Sanberg, P.R. 3-Nitropropionic acid animal model and Huntington’s disease. Neurosci. Biobehav. Rev. 1997, 21, 289–293. [Google Scholar] [CrossRef]
- Beal, M.F.; Brouillet, E.; Jenkins, B.G.; Ferrante, R.J.; Kowall, N.W.; Miller, J.M.; Storey, E.; Srivasta, R.; Rosen, B.R.; Hyman, B.T. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-ntropropionic acid. J. Neurosci. 1993, 13, 4181–4192. [Google Scholar]
- Guyot, M.C.; Hantraye, P.; Dolan, R.; Palfi, S.; Maziére, M.; Brouillet, E. Quantifiable bradykinesia, gait abnormalities and Huntington's disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience 1997, 79, 45–56. [Google Scholar] [CrossRef]
- Hamilton, B.F.; Gould, D.H. Nature and distribution of brain lesions in rats intoxicated with 3-nitropropionic acid: A type of hypoxic (energy deficient) brain damage. Acta Neuropathol. 1987, 72, 286–297. [Google Scholar] [CrossRef]
- Beal, M.F. Neurochemistry and toxin models in Huntington’s disease. Curr. Opin. Neurol. 1994, 7, 542–547. [Google Scholar] [CrossRef]
- Brouillet, E.; Guyot, M.C.; Mittoux, V.; Altairac, S.; Conde, F.; Palfi, S.; Hantraye, P. Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J. Neurochem. 1998, 70, 794–805. [Google Scholar]
- Borlongan, C.V.; Koutouzis, T.K.; Freeman, T.B.; Cahill, D.W.; Sanberg, P.R. Behavioral pathology induced by repeated systemic injections of 3-nitropropionic acid mimics the motoric symptoms of Huntington’s disease. Brain Res. 697, 254–257.
- Borlongan, C.V.; Koutouzis, T.K.; Randall, T.S.; Freeman, T.B.; Cahill, D.W.; Sanberg, P.R. Systemic 3-nitropropionic acid: Behavioral deficits and striatal damage in adult rats. Brain Res. Bull. 1995, 6, 549–556. [Google Scholar]
- Brouillet, E.; Hantraye, P.; Ferrante, R.J.; Dolan, R.; Leroy-Willig, A.; Kowall, N.W.; Beal, M.F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl. Acad. Sci. USA 1995, 92, 7105–7109. [Google Scholar]
- Mettler, F.A. Choreoathetosis and striopallidonigral necrosis due to sodium azide. Exp. Neurol. 1972, 34, 291–308. [Google Scholar] [CrossRef]
- Palfi, S.; Ferrante, R.J.; Brouillet, E.; Beal, M.F.; Dolan, R.; Guyot, M.C.; Peschanski, M.; Hantraye, P. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of Huntington’s disease. J. Neurosci. 1996, 16, 3019–3025. [Google Scholar]
- Stober, T.; Wussow, W.; Schimrigk, K. Bicaudate diameter the most specific and simple CT parameter in the diagnosis of Huntington’s disease. Neuroradiology 1984, 26, 25–28. [Google Scholar] [CrossRef]
- Ming, L. Moldy sugarcane poisoning-a case report with a brief review. J. Toxicol. Clin. Toxicol. 1995, 33, 363–367. [Google Scholar] [CrossRef]
- Ludolph, A.C.; He, F.; Spencer, P.S.; Hammerstad, J.; Sabri, M. 3-Nitropropinic acid-exogenous animal neurotoxin and possible human striatal toxin. Can. J. Neurol. Sci. 1991, 18, 492–498. [Google Scholar]
- Alston, T.A.; Mela, L.; Bright, H.J. Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc. Natl. Acad. Sci. USA 1977, 74, 3767–3771. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.; Bird, E.D.; Beal, M. Oxidative damage and metabolic dysfunction in Huntington's disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 199, 39, 385–389. [Google Scholar]
- Tabrizi, S.J.; Cleeter, M.W.; Xuereb, J.; Taanman, J.W.; Cooper, J.M.; Schapira, A.H. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann. Neurol. 1999, 45, 25–32. [Google Scholar] [CrossRef]
- Lee, J.M.; Shih, A.Y.; Murphy, T.H.; Johnson, J.A. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J. Biol. Chem. 2003, 278, 37948–37956. [Google Scholar]
- Nasr, P.; Gursahani, H.I.; Pang, Z.; Bondada, V.; Lee, J.; Hadley, R.W.; Geddes, J.W. Influence of cytosolic and mitochondrial Ca2+, ATP, mitochondrial membrane potential, and calpain activity on the mechanism on neuron death induced by 3-nitropropionic acid. Neurochem. Int. 2003, 43, 89–99. [Google Scholar] [CrossRef]
- Maciel, E.N.; Kowaltowski, A.J.; Schwalm, F.D.; Rodrigues, J.M.; Souza, D.O.; Vercesi, A.E.; Wajner, M.; Castilho, R.F. Mitochondrial permeability transition in neuronal damage promoted by Ca2+ and respiratory chain complex II inhibition. J. Neurochem. 2004, 90, 1025–1035. [Google Scholar] [CrossRef]
- Galas, M.C.; Bizat, N.; Cuvelier, L.; Bantubungi, K.; Brouillet, E.; Schiffmann, S.N.; Blum, D. Death of cortical and striatal neurons induced by mitochondrial defect involves differential molecular mechanisms. Neurol. Dis. 2004, 15, 152–159. [Google Scholar] [CrossRef]
- Alexi, T.; Hughes, P.E.; Faull, R.L.; Williams, C.E. 3-Nitropropionic acid’s lethal triplet: Cooperative pathways of neurodegeneration. NeuroReport 1998, 9, R57–R64. [Google Scholar] [CrossRef]
- Deshpande, S.B.; Fukuda, A.; Nishino, H. 3-Nitropropionic acid increases the intracellular Ca2+ in cultured astrocytes by reverse operation of the Na+-Ca2+ exchanger. Exp. Neurol. 1997, 145, 38–45. [Google Scholar] [CrossRef]
- Fukuda, A.; Deshpande, S.B.; Shimano, Y.; Nishino, H. Astrocytes are more vulnerable than neurons to cellular Ca2+ overload induced by a mitochondrial toxin, 3-nitropropionic acid. Neuroscience 1998, 87, 497–507. [Google Scholar] [CrossRef]
- Montilla, P.; Túnez, I.; Muñoz, M.C.; Salcedo, M.; Feijóo, M.; Muñoz-Castañeda, J.R.; Bujalance, I. Effect of glucocorticoids on 3-nitropropionic acid-induced oxidative stress in synaptosomes. Eur. J. Pharmacol. 2004, 488, 19–25. [Google Scholar] [CrossRef]
- Lafon-Cazal, M.; Pietri, S.; Culcasi, M.; Bockaert, J. NMDA-dependent superoxide production and neurotoxicity. Nature 1993, 364, 535–537. [Google Scholar]
- La Fontaine, M.A.; Geddes, J.W.; Banks, A.; Butterfield, D.A. 3-Nitropropionic acid induced in vivo protein oxidation in striatal and cortical synaptosomes: Insights into Huntington`s disease. Brain Res. 2000, 858, 356–362. [Google Scholar] [CrossRef]
- Marti, M.; Mela, F.; Ulazzi, L.; Hanau, S.; Stocchi, S.; Paganini, F.; Beani, L.; Bianchi, C.; Morari, M. Differential responsiveness of rat striatal nerve endings to the mitochondrial toxin 3-nitropropionic acid: Implications for Huntington’s disease. Eur. J. Neurosci. 2003, 18, 759–767. [Google Scholar]
- Storgaard, J.; Kornblit, B.T.; Zimmer, J.; Gramsbergen, J.B. 3-Nitropropionic acid neurotoxicity in organotypic striatal and corticostriatal slice cultures is dependent on glucose and glutamate. Exp. Neurol. 2000, 164, 227–235. [Google Scholar] [CrossRef]
- Novelli, A.; Reilly, A.; Lysk, P.G.; Henneberry, R.C. Glutamate becomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988, 451, 205–212. [Google Scholar] [CrossRef]
- Beal, M.F.; Kowall, N.W.; Ellison, D.W.; Mazurek, M.F.; Swartz, K.J.; Martin, J.B. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature 1986, 321, 168–171. [Google Scholar]
- Nasr, P.; Carbery, T.; Geddes, J.W. N-methyl-D-aspartate receptor antagonists have variable affect in 3-nitropropionic acid toxicity. Neurochem. Res. 2009, 34, 490–498. [Google Scholar] [CrossRef]
- Moro, M.A.; De Alba, J.; Cárdenas, A.; De Cristóbal, J.; Leza, J.C.; Lizasoain, I.M.; Díaz-Guerra, M.J.; Boscá, L.; Lorenzo, P. Mechanisms of the neuroprotective effect of aspirin after oxygen and glucose deprivation in rat forebrain slices. Neuropharmacology 2000, 39, 1309–1318. [Google Scholar] [CrossRef]
- Ryu, J.K.; Nagai, A.; Kim, J.; Lee, M.C.; McLarnon, J.G.; Kim, S.U. Microglial activation and cell death induced by the mitochondial toxin 3-nitropropionic acid: In vitro and in vivo studies. Neurobiol. Dis. 2003, 12, 121–132. [Google Scholar] [CrossRef]
- Ohgoh, M.; Shimizu, H.; Ogura, H.; Nishizawa, Y. Astroglial trophic support and neuronal cell death: influence of cellular energy level on type of cell death induced by mitochondrial toxin in cultured rat cortical neurons. J. Neurochem. 2000, 75, 925–933. [Google Scholar]
- Mittoux, V.; Ouary, S.; Monville, C.; Lisovoski, F.; Poyot, T.; Conde, F.; Escartin, C.; Robichon, R.; Brouillet, E.; Peschanski, M.; Hantraye, P. Corticostriatopallidal neuroprotection by adenovirus-mediated ciliary neurotrophic factor gene transfer in a rat model of progressive striatal degeneration. J. Neurosci. 2002, 22, 4478–4486. [Google Scholar]
- Villarán, R.F.; Tomás-Camardiel, M.; de Pablos, R.M.; Santiago, M.; Herrera, A.J.; Navarro, A.; Machado, A.; Cano, J. Endogenous dopamine enhances the neurotoxicity of 3-nitropropionic acid in the striatum through the increase of mitochondrial respiratory inhibition and free radicals production. Neurotoxicology 2008, 29, 244–258. [Google Scholar]
- Nishino, H.; Kumazaki, M.; Fukuda, A.; Fujimoto, I.; Shimano, Y.; Hida, H.; Sakurai, T.; Deshpande, S.B.; Shimizu, H.; Morikawa, S.; Inubushi, T. Acute 3-nitropropionic acid intoxication induces striatal astrocytic cell death and dysfunction of the blood-brain barrier: Involvement of dopamine toxicity. Neurosci. Res. 1997, 27, 343–355. [Google Scholar] [CrossRef]
- McCracken, E.; Dewar, D.; Hunter, A.J. White matter damage following systemic injection of the mitochondrial inhibitor 3-nitropropionic acid in rat. Brain Res. 2001, 892, 329–335. [Google Scholar] [CrossRef]
- Sato, S.; Gobbel, G.T.; Honkaniemi, J.; Li, Y.; Kondo, T.; Murakami, K.; Sato, M.; Copin, J.C.; Chan, P.H. Apoptosis in the striatum of rats following intraperitoneal injection of 3-nitropropionic acid. Brain Res. 1997, 745, 343–347. [Google Scholar] [CrossRef]
- Sato, S.; Gobbel, G.T. Blood-brain barrier disruption, HSP70 expression and apoptosis due to 3-nitropropionic acid, a mitochondrial toxin. Acta Neurochir. Suppl. 1997, 70, 237–239. [Google Scholar]
- Bantubungi, K.; Jacquard, C.; Greco, A.; Pintor, A.; Chtarto, A.; Tai, K.; Galas, M.C.; Tenenbaum, L.; Déglon, N.; Popoli, P.; Minghetti, L.; Brouillet, E.; Brtchi, J.; Levivier, M.; Schiffmann, S.N.; Blum, D. Minocycline in phenotypic models of Huntington’s disease. Neurobiol. Dis. 2005, 18, 206–217. [Google Scholar] [CrossRef]
- Nishino, H.; Hida, H.; Kumazaki, M.; Shimano, Y.; Nakajima, K.; Shimizu, H.; Ooiwa, T.; Baba, H. The striatum is the most vulnerable region in the brain to mitochondrial energy compromise: A hypothesis to explain its specific vulnerability. J. Neurotrauma 2000, 17, 251–260. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef]
- Moncada, S.; Bolaños, J.P. Nitric oxide, cell bioenergetics and neurodegeneration. J. Neurochem. 2006, 97, 1676–1689. [Google Scholar] [CrossRef]
- Coles, C.J.; Edmondson, D.E.; Singer, T.P. Inactivation of succinate dehydrogenase by 3-nitropropionate. J. Biol. Chem. 1979, 254, 5161–5167. [Google Scholar]
- Beal, M.F.; Hyman, B.T.; Koroshetz, W. Do defects in mitochondrial energy metabolism underlie the pathology of neurodegenerative diseases? Trends Neurosci. 1993, 16, 125–131. [Google Scholar] [CrossRef]
- Palfi, S.; Leventhal, L.; Goetz, C.G.; Hantraye, T.; Roitberg, B.Z.; Sramek, J.; Emborg, M.; Kordower, J.H. Delayed onset of progressive dystonia following subacute 3-nitropropionic acid treatment in Cebus apella monkeys. Mov. Disord. 2000, 15, 524–530. [Google Scholar] [CrossRef]
- Yang, L.; Calingasan, N.Y.; Chen, J.; Ley, J.J.; Becker, D.A.; Beal, M.F. A novel azulenyl nitrone antioxidant protects against MPTP and 3-nitropropionic acid neurotoxicities. Exp. Neurol. 2005, 191, 86–93. [Google Scholar] [CrossRef]
- Binienda, Z.; Simmons, C.; Hussain, S.; Slikker, W., Jr.; Ali, S.F. Effect of acute exposure to 3-nitropropionic acid on activities of endogenous antioxidants in the rat brain. Neurosci. Lett. 1998, 251, 173–176. [Google Scholar] [CrossRef]
- Herrera-Mundo, M.N.; Silva-Adaya, D.; Maldonado, P.D.; Galván-Arzate, S.; Andrés-Martínez, L.; Pérez-de la Cruz, V.; Pedraza-Chaverrí, J.; Santamaría, A. S-Allyslcysteine prevents the rat from 3-nitropropionic acid-induced hypeactivity, early markers of oxidative stress and mitochondrial dysfunction. Neurosci. Res. 2006, 56, 39–44. [Google Scholar] [CrossRef]
- Nam, E.; Lee, S.M.; Koh, S.E.; Joo, W.S.; Maeng, S.; Im, H.I.; Kim, Y.S. Melatonin protects against neuronal damage induced by 3-nitropropionic acid in rat striatum. Brain Res. 2005, 1046, 90–96. [Google Scholar] [CrossRef]
- Pérez-de la Cruz, V.; González-Cortés, C.; Pedraza-Chaverrí, J.; Maldonado, P.D.; Andrés-Martínez, L.; Santamaría, A. Protective effect of S-allylcysteine on 3-nitropropionic acid-induced lipid peroxidation and mitochondrial dysfunction in rat brain synaptosomes. Brain Res. Bull. 2006, 68, 379–383. [Google Scholar] [CrossRef]
- Schulz, J.B.; Henshaw, D.R.; MacGarvey, U.; Beal, M.F. Involvement of oxidative stress in 3-nitropropionic acid neurotoxicity. Neurochem. Int. 1996, 29, 167–171. [Google Scholar] [CrossRef]
- Pérez-de la Cruz, V.; Konigsberg, M.; Pedraza-Chaverri, J.; Herrera-Mundo, N.; Díaz-Muñoz, M.; Morán, J.; Fortoul-van der Goes, T.; Rondán-Zárate, A.; Maldonado, P.D.; Ali, S.F.; Santamaría, A. Cytoplasmic calcium mediates oxidative damage in an excitotoxic/energetic deficit synergic model in rats. Eur. J. Neurosci. 2008, 27, 1075–1085. [Google Scholar]
- Ferdinandy, P.; Schulz, R. Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br. J. Pharmacol. 2003, 138, 532–543. [Google Scholar] [CrossRef]
- Blum, D.; Gall, D.; Cuvelier, L.; Schiffmann, S.N. Topological analysis of striatal lesions induced by 3-nitropropionic acid in the Lewis rat. Neuroreport 2001, 12, 1769–1772. [Google Scholar] [CrossRef]
- Pang, Z.; Geddes, J.W. Mechanisms of cell death induced by the mitochondrial toxin 3-nitropropionic acid: Acute excitotoxic necrosis and delayed apoptosis. J. Neurosci. 1997, 17, 3064–3073. [Google Scholar]
- Bossi, S.R.; Simpson, J.R.; Isacson, O. Age dependence of striatal neuronal death caused by mitochondrial dysfunction. Neuroreport 1993, 4, 73–76. [Google Scholar] [CrossRef]
- Koutouzis, T.K.; Borlongan, C.V.; Scorcia, T.; Creese, I.; Cahill, D.W.; Freeman, T.B.; Sanberg, P.R. Systemic 3-nitropropionic acid: Long-term effects on locomotor behavior. Brain Res. 1994, 646, 242–246. [Google Scholar] [CrossRef]
- Filloux, F.; Townsend, J.J. Pre- and postsynaptic neurotoxic effects of dopamine demostrated by intrastriatal injection. Exp. Neuro. 1993, 119, 79–88. [Google Scholar] [CrossRef]
- Reynolds, D.S.; Carter, R.J.; Morton, A.J. Dopamine modulates the susceptibility of striatal neurons to 3-nitropropionic acid in the rat model of Huntington`s disease. J. Neurosci. 1998, 18, 10116–10127. [Google Scholar]
- Maragos, W.F.; Young, K.L.; Altman, C.S.; Pocernich, C.B.; Drake, J.; Butterfield, D.A.; Seif, I.; Holschneider, D.P.; Chen, K.; Shih, J.C. Striatal damage and oxidative stress induced by the mitochondrial toxin malonate are reduced in clorgyline-treated rats and MAO-A deficient mice. Neurochem. Res. 2004, 29, 741–746. [Google Scholar]
- Blum, D.; Hourez, R.; Galas, M.C.; Popoli, P.; Schiffmann, S.N. Adenosine receptors and Huntington’s disease: Implications for pathogenesis and therapeutics. Lancet Neurol. 2003, 2, 366–374. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Fezza, F.; Cebeira, M.; Bisogno, T.; Ramos, J.A.; Milone, A.; Fernández-Ruiz, J.; Di Marzo, V. Changes in endocannabinoid transmission in the basal ganglia in a rat model of Huntington’s disease. Neuroreport 2001, 12, 2125–2129. [Google Scholar]
- Lastres-Becker, I.; Gómez, M.; De Miguel, R.; Ramos, J.A.; Fernández-Ruiz, J. Loss of Cannabinoid CB(1) receptors in the basal ganglia in the late akinetic phase of rats with experimental Huntington’s disease. Neurotox Res. 2002, 4, 601–608. [Google Scholar] [CrossRef]
- Canals, J.M.; Checa, N.; Marco, S.; Akerud, P.; Michels, A.; Pérez-Navarro, E.; Tolosa, E.; Arenas, E.; Alberch, J. Expression of brain-derived neurotrophic factor in cortical neurons is regulated by striatal target area. J. Neurosci. 2001, 21, 117–124. [Google Scholar]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbertm, S.; Saudou, F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E; Friedlander, R.M.; Silani, V.; Hayden, M.R.; Timmusk, T.Ç.; Sipione, S.; Cattaneo, E. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar]
- Zuccato, C.; Tartari, M.; Crotti, A.; Goffredo, D.; Valenza, M.; Conti, L.; Cataudella, T.; Leavitt, B.R.; Hayden, M.R.; Timmusk, T.; Rigamonti, D.; Cattaneo, E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003, 35, 76–83. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington's disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef]
- Altar, C.A.; Cai, N.; Bliven, T.; Juhasz, M.; Conner, J.M.; Acheson, A.L.; Lindsay, R.M.; Wiegand, S.J. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 1997, 389, 856–860. [Google Scholar] [CrossRef]
- Kovalchuk, Y.; Hanse, E.; Kafitz, KW.; Konnerth, A. Postsynaptic induction of BDNF-mediated long-term potentiation. Science 2002, 295, 1729–1734. [Google Scholar] [CrossRef]
- Ferrer, I.; Goutan, E.; Marín, C.; Rey, M.J.; Ribalta, T. Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000, 866, 257–261. [Google Scholar] [CrossRef]
- Ernfors, P.; Bramham, C.R. The coupling of a trkB tyrosine residue to LTP. Trends Neurosci. 2003, 26, 171–173. [Google Scholar] [CrossRef]
- Almli, C.R.; Levy, T.J.; Han, B.H.; Shah, A.R.; Gidday, J.M.; Holtzman, D.M. BDNF protects against spatial memory deficits following neonatal hypoxia-ischemia. Exp. Neurol. 2000, 166, 99–114. [Google Scholar] [CrossRef]
- Hock, C.; Heese, K.; Hulette, C.; Rosenberg, C.; Otten, U. Region-specific neurotrophin imbalances in Alzheimer disease: Decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch. Neurol. 2000, 57, 846–851. [Google Scholar] [CrossRef]
- Hellweg, R.; von Arnim, C.A.; Büchner, M.; Huber, R.; Riepe, M.W. Neuroprotection and neuronal dysfunction upon repetitive inhibition of oxidative phosphorylation. Exp. Neurol. 2003, 183, 346–354. [Google Scholar] [CrossRef]
- Cho, H.Y.; Jedlicka, A.E.; Reddy, S.P.; Kensler, T.W.; Yamamoto, M.; Zhang, L.Y.; Kleeberger, S.R. Role of NRF2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2002, 26, 175–182. [Google Scholar]
- Gao, X.; Talalay, P. Induction of phase 2 genes by sulforaphane protects retinal pigment epithelial cells against photooxidative damage. Proc. Natl. Acad. Sci. USA 2004, 101, 10446–10451. [Google Scholar]
- Calkins, M.J.; Jakel, R.J.; Johnson, D.A.; Chan, K.; Kan, Y.W.; Johnson, J.A. Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc. Natl. Acad. Sci. USA 2005, 102, 244–249. [Google Scholar]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar]
- Kotlo, K.U.; Yehiely, F.; Harasty, H.; Hesabi, B.; Shchors, K.; Einat, P.; Rozen, A.; Berent, E.; Deiss, L.P. Nrf2 is an inhibitor of the Fas pathway as identified by Achilles`Hell Method, a new function-based approach to gene identification in human cells. Oncogene 2003, 22, 797–806. [Google Scholar] [CrossRef]
- Almeida, S.; Brett, A.C.; Góis, I.N.; Oliveira, C.R.; Rego, A.C. Caspase-dependent and -independent cell death induced by 3-nitropropionic acid in rat cortical neurons. J. Cell Biochem. 2006, 98, 93–101. [Google Scholar] [CrossRef]
- Almeida, S.; Domingues, A.; Rodrigues, L.; Oliveira, C.R.; Rego, A.C. FK506 prevents mitochondrial-dependent apoptotic cell death induced by 3-nitropropionic acid in rat primary cortical cultures. Neurobiol. Dis. 2004, 17, 435–444. [Google Scholar] [CrossRef]
- Enoksson, M.; Robertson, J.D.; Gogvadze, V.; Bu, P.; Kropotov, A.; Zhivotovsky, B.; Orrenius, S. Caspase-2 permeabilizes the outer mitochondrial membrane and disrupts the binding of cytochrome c to anionic phospholipids. J. Biol. Chem. 2004, 279, 49575–49578. [Google Scholar]
- Garcia, M.; Vanhoutte, P.; Pages, C.; Besson, M.J.; Brouillet, E.; Caboche, J. The mitochondrial toxin 3-nitropropionic acid induces striatal neurodegeneration via a c-jun N-terminal kinase/c-Jun module. J. Neurosci. 2002, 22, 2174–2184. [Google Scholar]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar]
- Minn, Y.; Cho, K.J.; Kim, H.W.; Kim, H.J.; Suk, S.H.; Lee, B.I.; Kim, G.W. Induction of apoptosis signal-regulating kinase 1 and oxidative stress mediate age dependent vulnerability to 3-nitropropionic acid in the mouse striatum. Neurosci. Lett. 2008, 430, 142–146. [Google Scholar]
- Pelegrí, C.; Duran-Vilaregut, J.; del Valle, J.; Crespo-Biel, N.; Ferrer, I.; Pallàs, M.; Camins, A.; Vilaplana, J. Cell cycle activation in striatal neurons from Huntington’s disease patients and rats treated with 3-nitropropionic acid. Int. J. Dev. Neurosci. 2008, 26, 665–671. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Breunig, J.J.; Depboylu, C.; Rouaux, C.; Michel, P.P.; Alvarez-Fischer, D.; Boutillier, A.L.; Degregori, J.; Oertel, W.H.; Rakic, P.; Hirsch, E.C.; Hunot, S. The pRb/E2F cell-cycle pathway mediates cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA. 2007, 104, 3585–3590. [Google Scholar]
- Crespo-Biel, N.; Camins, A.; Pelegrí, C.; Vilaplana, J.; Pallàs, M.; Canudas, A.M. 3-Nitropropionic acid activates calpain/cdk5 pathway in rat striatum. Neurosci. Lett. 2007, 421, 77–81. [Google Scholar]
- Park, D.S.; Obeidat, A.; Giovanni, A.; Greene, L.A. Cell cycle regulators in neuronal death evoked by excitotoxic stress: Implications for neurodegeneration and its treatment. Neurobiol. Aging 2000, 21, 771–781. [Google Scholar] [CrossRef]
- Seidel, B.; Jiang, L.; Wolf, G. Differentially displayed genes in neuroblastoma cells treated with a mitochondrial toxin: Evidence for possible involvement of ICAM-1 in 3-nitropropionic acid-mediated neurodegeneration. Toxicol. Lett. 2000, 115, 213–222. [Google Scholar] [CrossRef]
- Napolitano, M.; Zei, D.; Centonze, D.; Palermo, R.; Bernardi, G.; Vacca, A.; Calabresi, P.; Gulino, A. NF-kB/NOS cross-talk induced by mitochondrial complex II inhibition: Implications for Huntington`s disease. Neurosci. Lett. 2008, 434, 241–246. [Google Scholar]
- García-Ramos, R.; del Val-Fernández, J.; Catalán-Alonso, M.J.; Barcia-Albacar, J.A.; Matías-Guiu, J. Experimental models of Huntington`s disease. Rev. Neurol. 2007, 45, 437–441. [Google Scholar]
- Qin, Z.H.; Wang, J.; Gu, Z.L. Development of novel therapies for Huntington’s disease: Hope and challenge. Acta Pharmacol. Sin. 2005, 26, 129–142. [Google Scholar] [CrossRef]
- Wang, L.H.; Qin, Z.H. Animal models of Huntington`s disease: Implications in uncovering pathogenic mechanisms and developing therapies. Acta Pharmacol. Sin. 2006, 27, 1287–1302. [Google Scholar] [CrossRef]
- Leavitt, B.R.; van Raamsdonk, J.M.; Shehadeh, J.; Fernandes, H.; Murphy, Z.; Graham, R.K.; Wellington, C.L.; Raymond, L.A.; Hayden, M.R. Wild-type huntingtin protects neurons from excitotoxicity. J. Neurochem. 2006, 96, 1121–1129. [Google Scholar] [CrossRef]
- Sanberg, P.R.; Coyle, J.T. Scientific approaches to Huntington’s disease. CRC Crit. Rev. Clin. Neurobiol. 1984, 1, 1–44. [Google Scholar]
- Santamaría, A.; Jiménez, M.E. Oxidative/nitrative stress, a common factor in different neurotoxic paradigms: An overview. Curr. Topics Neurochem. 2006, 1–20. [Google Scholar]
- Schwarcz, R.; Foster, A.C.; French, E.D.; Whetsell, W.O., Jr.; Köhler, C. Excitotoxic models for neurodegenerative disorders. Life Sci. 1984, 35, 19–32. [Google Scholar] [CrossRef]
- Isacson, O.; Brundin, P.; Gage, F.H.; Björklund, A. Neural grafting in a rat model of Huntington’s disease: Progressive neurochemical changes after neostriatal ibotenate lesions and striatal tissue grafting. Neuroscience 1985, 16, 799–817. [Google Scholar] [CrossRef]
- Hantraye, P.; Riche, D.; Maziere, M.; Isacson, O. A primate model of Huntington’s disease: Behavioral and anatomical studies of unilateral excitotoxic lesions of the caudate-putamen in the baboon. Exp. Neurol. 1990, 108, 91–104. [Google Scholar] [CrossRef]
- Pérez-de la Cruz, V.; Königsberg, M.; Santamaría, A. Kynurenine pathway and disease: An overview. CNS Neurol. Disord. Drug Targets 6, 398–410.
- Dang, Y.; Dale, W.E.; Brown, O.R. Comparative effects of oxygen on indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase of the kynurenine pathway. Free Radic. Biol. Med. 2000, 15, 615–624. [Google Scholar]
- Guidetti, P.; Luthi-Carter, R.E.; Augood, S.J.; Schwarcz, R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease. Neurobiol. Dis. 2004, 17, 455–461. [Google Scholar] [CrossRef]
- González-Cortés, C.; Santamaría, A. New Perspectives on Brain Cell Damage, Neurodegeneration and Neuroprotective Strategies. Research Signpost, 2007. [Google Scholar]
- Santamaría, A.; Jiménez-Capdeville, M.E.; Camacho, A.; Rodríguez-Martínez, E.; Flores, A.; Galván-Arzate, S. In vivo hydroxyl radical formation after quinolinic acid infusion into rat corpus striatum. Neuroreport 2001, 12, 2693–2696. [Google Scholar] [CrossRef]
- Stone, T.W. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 1993, 45, 309–379. [Google Scholar]
- Heyes, M.P.; Lackner, A. Increased cerebrospinal fluid quinolinic acid, kynurenic acid, and L-kynurenine in acute septicemia. J. Neurochem. 1990, 55, 338–341. [Google Scholar]
- Heyes, M.P.; Mefford, I.N.; Quearry, B.J.; Dedhia, M.; Lackner, A. Increased ratio of quinolinic acid to kynurenic acid in cerebrospinal fluid of D retrovirus-infected rhesus macaques: Relationship to clinical and viral status. Ann. Neurol. 1990, 27, 666–675. [Google Scholar] [CrossRef]
- Heyes, MP.; Saito, K.; Crowley, JS.; Davis, LE.; Demitrack, M.A.; Der, M.; Dilling, L.A.; Elia, J.; Kruesi, M.J.; Lackner, A.; Larsen, SA.; Lee, K.; Leonard, HL.; Markey, SP.; Martin, A.; Milstein, S.; Mouradian, MM.; Pranzatelli, MR.; Quearry, BJ.; Salazar, A.; Smith, M.; Strauss, SE.; Sunderland, T.; Swedo, SW.; Tourtellotte, WW. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain 1992, 115, 1249–1273. [Google Scholar]
- Moroni, F.; Lombardi, G.; Carlà, V.; Pellegrini, D.; Carassale, G.L.; Cortesini, C. Content of quinolinic acid and of other tryptophan metabolites increases in brain regions of rats used as experimental models of hepatic encephalopathy. J. Neurochem. 1986, 46, 869–874. [Google Scholar] [CrossRef]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology 1992, 42, 1702–1706. [Google Scholar] [CrossRef]
- Schwarcz, R.; Okuno, E.; White, R.J.; Bird, E.D.; Whetsell, W.O., Jr. 3-Hydroxyanthranilate oxygenase activity is increased in the brains of Huntington disease victims. Proc. Natl. Acad. Sci. USA 1988, 85, 4079–4081. [Google Scholar] [CrossRef]
- Stone, T.W. Endogenous neurotoxins from tryptophan. Toxicology 2001, 39, 61–73. [Google Scholar]
- Stone, T.W.; Connick, J.H. Quinolinic acid and other kynurenines in the central nervous system. Neuroscience 1985, 15, 597–617. [Google Scholar] [CrossRef]
- During, M.J.; Heyes, M.P.; Freese, A.; Markey, S.P.; Martin, J.B.; Roth, R.H. Quinolinic acid concentrations in striatal extracellular fluid reach potentially neurotoxic levels following systemic L-tryptophan loading. Brain Res. 1989, 476, 384–387. [Google Scholar] [CrossRef]
- Foster, A.C.; Collins, J.F.; Schwarcz, R. On the excitotoxic properties of quinolinic acid, 2,3-piperidine dicarboxylic acids and structurally related compounds. Neuropharmacology 1983, 22, 1331–1342. [Google Scholar] [CrossRef]
- Santamaría, A.; Ríos, C. MK-801, an N-methyl-D-aspartate receptor antagonist, blocks quinolinic acid-induced lipid peroxidation in rat corpus striatum. Neurosci. Lett. 1993, 159, 51–54. [Google Scholar] [CrossRef]
- Shear, D.A.; Dong, J.; Gundy, C.D.; Haik-Creguer, K.L.; Dunbar, G.L. Comparison of intrastriatal injections of quinolinic acid and 3-nitropropionic acid for use in animal models of Huntington's disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 1998, 22, 1217–1240. [Google Scholar] [CrossRef]
- Behan, W.M.; McDonald, M.; Darlington, L.G.; Stone, T.W. Oxidative stress as a mechanism for quinolinic acid-induced hippocampal damage: Protection by melatonin and deprenyl. Br. J. Pharmacol. 1999, 128, 1754–1760. [Google Scholar]
- Pérez-De La Cruz, V.; González-Cortés, C.; Galván-Arzate, S.; Medina-Campos, O.N.; Pérez-Severiano, F.; Ali, S.F.; Pedraza-Chaverrí, J.; Santamaría, A. Excitotoxic brain damage involves early peroxynitrite formation in a model of Huntington's disease in rats: Protective role of iron porphyrinate 5,10,15,20-tetrakis (4-sulfonatophenyl)porphyrinate iron (III). Neuroscience 2005, 135, 463–474. [Google Scholar] [CrossRef]
- Rodríguez-Martínez, E.; Camacho, A.; Maldonado, P.D.; Pedraza-Chaverrí, J.; Santamaría, D.; Galván-Arzate, S.; Santamaría, A. Effect of quinolinic acid on endogenous antioxidants in rat corpus striatum. Brain Res. 2000, 858, 436–439. [Google Scholar] [CrossRef]
- Stone, T.W.; Behan, W.M.; MacDonald, M.; Darlington, L.G. Possible mediation of quinolinic acid-induced hippocampal damage by reactive oxygen species. Amino Acids. 2000, 19, 275–281. [Google Scholar] [CrossRef]
- del Río, P.; Montiel, T.; Chagoya, V.; Massieu, L. Exacerbation of excitotoxic neuronal death induced during mitochondrial inhibition in vivo: Relation to energy imbalance or ATP depletion? Neuroscience 2007, 146, 1561–1570. [Google Scholar] [CrossRef]
- del Río, P.; Massieu, L. Mild mitochondrial inhibition in vivo enhances glutamate-induced neuronal damage through calpain but not caspase activation: Role of ionotropic glutamate receptors. Exp. Neurol. 2008, 212, 179–188. [Google Scholar] [CrossRef]
- Henneberry, R.C.; Novelli, A.; Cox, J.A.; Lysko, P.G. Neurotoxicity at the N-methyl -D- aspartate receptor in energy-compromised neurons. An hypothesis for cell death in aging and disease. Ann. N.Y. Acad. Sci. 1989, 568, 225–233. [Google Scholar] [CrossRef]
- Henneberry, R.C.; Novelli, A.; Vigano, M.A.; Reilly, J.A.; Cox, J.A.; Lysko, P.G. Energy-related neurotoxicity at the NMDA receptor: A possible role in Alzheimer's disease and related disorders. Prog. Clin. Biol. Res. 317, 143–156.
- Greene, J.G.; Greenamyre, J.T. Manipulation of membrane potential modulates malonate-induced striatal excitotoxicity in vivo. J. Neurochem. 1996, 66, 637–643. [Google Scholar]
- Ikonomidou, C.; Turski, L. Neurodegenerative disorders: Clues from glutamate and energy metabolism. Crit. Rev. Neurobiol. 1996, 10, 239–263. [Google Scholar] [CrossRef]
- Massieu, L.; García, O. The role of excitotoxicity and metabolic failure in the pathogenesis of neurological disorders. Neurobiology (Bp). 1998, 6, 99–108. [Google Scholar]
- Jacquard, C.; Trioulier, Y.; Cosker, F.; Escartin, C.; Bizat, N.; Hantraye, P.; Cancela, J.M.; Bonvento, G.; Brouillet, E. Brain mitochondrial defects amplify intracellular [Ca2+] rise and neurodegeneration but not Ca2+ entry during NMDA receptor activation. FASEB J. 2006, 20, 1021–1023. [Google Scholar]
- Bazzett, T.J.; Falik, R.C.; Becker, J.B.; Albin, R.L. Synergistic effects of chronic exposure to subthreshold concentrations of quinolinic acid and malonate in the rat striatum. Brain Res. 1996, 718, 228–232. [Google Scholar] [CrossRef]
- Ryu, J.K.; Kim, S.U.; McLarnon, J.G. Blockade of quinolinic acid-induced neurotoxicity by pyruvate is associated with inhibition of glial activation in a model of Huntington's disease. Exp. Neurol. 2004, 187, 150–159. [Google Scholar] [CrossRef]
- Guidetti, P.; Schwarcz, R. 3-Hydroxykynurenine potentiates quinolinate but not NMDA toxicity in the rat striatum. Eur. J. Neurosci. 1999, 11, 3857–3863. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Stone, T.W.; Smith, R.A. Prolonged exposures of cerebellar granule neurons to S-nitroso-N-acetylpenicillamine (SNAP) induce neuronal damage independently of peroxynitrite. Brain Res. 2008, 1230, 265–272. [Google Scholar] [CrossRef]
- Márquez-Valadez, B.; Lugo-Huitrón, R.; Valdivia-Cerda, V.; Miranda-Ramírez, L.R.; Pérez-De La Cruz, V.; González-Cuahutencos, O.; Rivero-Cruz, I.; Mata, R.; Santamaría, A.; Pedraza-Chaverrí, J. The natural xanthone alpha-mangostin reduces oxidative damage in rat brain tissue. Nutr. Neurosci. 2009, 12, 35–42. [Google Scholar] [CrossRef]
- Sudati, J.H.; Fachinetto, R.; Pereira, R.P.; Boligon, A.A.; Athayde, M.L.; Soares, F.A.; de Vargas-Barbosa, N.B.; Rocha, J.B. In vitro antioxidant activity of Valeriana officinalis against different neurotoxic agents. Neurochem. Res. 2009, 34, 1372–1379. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A. Neuroprotective effect of cyclosporine and FK506 against 3-nitropropionic acid induced cognitive dysfunction and glutathione redox in rat: Possible role of nitric oxide. Neurosci. Res. 2009, 63, 302–314. [Google Scholar] [CrossRef]
- Ubhi, K.; Lee, P.H.; Adame, A.; Inglis, C.; Mante, M.; Rockeinstein, E.; Stefanova, N.; Wenning, G.K.; Masliah, E. Mitochondrial inhibitor 3-nitropropionic acid enhances oxidative modifications of alpha-synuclein in a transgenic mouse model of multiple system atrophy. J. Neurosci. Res. 2009, 87, 2728–2739. [Google Scholar] [CrossRef]
- Pandey, M.; Borah, A.; Varghese, M.; Barman, P.K.; Mohanakumar, K.P.; Usha, R. Strital dopamine level contributes vto hydroxyl radical generation and subsequent neurodegeneration in the striatum in 3-nitropropionic acid-induced Huntington’s disease in rats. Neurochem. Int. 2009, 55, 431–437. [Google Scholar] [CrossRef]
- Medina-Navarro, R.; Guerrero-Linares, I. Whole body hyperthermia reduced oxidative stress in the striatum of rats in an animal model of mitochondrial toxicity with 3-nitropropionic acid. Int. J. Hyperthermia 2009, 25, 280–288. [Google Scholar] [CrossRef]
- Yang, L.; Calingasan, N.Y.; Wille, E.J.; Cormier, K.; Smith, K.; Ferrante, R.J.; Beal, M.F. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J. Neurochem. 2009, 109, 1427–1439. [Google Scholar] [CrossRef]
- Kumar, P.; Kalonia, H.; Kumar, A. Sesamol attenuate 3-nitropropionic acid-induced Huntington-like behavioral, biochemical, and cellular atlerations in rats. J. Asian Nat. Prod. Res. 2009, 11, 439–450. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A. Effect of lycopene and epigallocatechin-3-gallate against 3-nitropropionic acid induced cognitive dysfunction and glutathione depletion in rat: A novel nitric oxide mechanism. Food Chem. Toxicol. 2009, 47, 2522–2530. [Google Scholar]
- Kumar, P.; Kumar, A. Possible neuroprotective effect of Withania somnifera root extract against 3-nitropropionic acid-induced behavioral, biochemical, and mitochondrial dysfunction in an animal model of Huntington’s disease. J. Med. Food 2009, 12, 591–600. [Google Scholar] [CrossRef]
- Lagoa, R.; López-Sánchez, C.; Samhan-Arias, A.K.; Gañan, C.M.; García-Martínez, V.; Gutíerrez-Merino, C. Kaempferol protects against rat striatal degeneration induced by 3-nitropropionic acid. J. Neurochem. 2009, 111, 473–487. [Google Scholar]
- Al Mutairy, A.; Al Kadasah, S.; Elfaki, I.; Arshaduddin, M.; Malik, D.; Al Moutaery, K.; Tariq, M. Trolox ameliorates 3-nitropropionic acid -induced neurotoxicity in rats. Neurotoxicol. Teratol. 2009. In press. [Google Scholar]
- Tasset, I.; Pérez-De La Cruz, V.; Elinos-Calderón, D.; Carrillo-Mora, P.; González-Herrera, I.G.; Luna-López, A.; Königsberg, M.; Pedraza-Chaverrí, J.; Maldonado, P.D.; Ali, S.F.; Túnez, I.; Santamaría, A. Protective effect of tert-butylhydroquinone on the quinolinic acid-induced toxicity in rat striatal slices: Role of the Nrf2-antioxidant response element pathway. Neurosignals 2010, 18, 24–31. [Google Scholar] [CrossRef]
- Elinos-Calderón, D.; Robledo-Arratia, Y.; Pérez-De La Cruz, V.; Padraza-Chaverrí, J.; Ali, S.F.; Santamaría, A. Early nerve ending rescue from oxidative damage and energy failure by L-carnitine as post-treatment in two neurotoxic models in rat: Recovery of antioxidant and reductive capacities. Exp. Brain Res. 2009, 197, 287–296. [Google Scholar] [CrossRef]
- Kada, S.; Nakagawa, T.; Ito, J. A mouse model for degeneration of the spiral ligament. J. Assoc. Res. Otolaryngol. 2009, 10, 161–172. [Google Scholar] [CrossRef]
- Uo, T.; Veenstra, T.D.; Morrison, R.S. Histone deacetylase inhibitors prevent p53-dependent and p53-independent Bax-mediated neuronal apoptosis through two disctinct mechanisms. J. Neurosci. 2009, 29, 2824–2832. [Google Scholar]
- Liot, G.; Bossy, B.; Lubitz, S.; Kushnareva, Y.; Sejbuk, N.; Bossy-Wetzel, E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ. 2009, 16, 899–909. [Google Scholar] [CrossRef]
- Zhang, X.D.; Wang, Y.; Wang, Y.; Zhang, X.; Han, R.; Wu, J.C.; Liang, Z.Q.; Gu, Z.L.; Han, F.; Fukunaga, K.; Qin, Z.H. p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy 2009, in press. [Google Scholar]
- Singh, S.; Misiak, M.; Beyer, C.; Arnold, S. Cytochrome c oxidase isoform IV-2 is involved in 3-nitropropionic acid-induced toxicity in striatal astrocytes. Glia 2009, 57, 1480–1491. [Google Scholar] [CrossRef]
- Kraft, J.C.; Osterhaus, G.L.; Ortiz, A.N.; Garriz, P.A.; Johnson, M.A. In vivo dopamine release and uptake impairments in rats treated with 3-nitropropionic acid. Neuroscience 2009, 161, 940–949. [Google Scholar]
- Diwakarla, S.; Mercer, L.D.; Kardashsyan, L.; Chu, P.W.; Shin, Y.S.; Lau, C.L.; Hughes, M.L.; Nagley, P.; Beart, P.M. GABAergic striatal neurons exhibit caspase-independent, mitochondrially mediated programmed cell death. J. Neurochem. 2009, 109, 198–206. [Google Scholar] [CrossRef]
- Wu, C.L.; Hwang, C.S.; Yang, D.I. Protective effects of brain-derived neurotrophic factor against neurotoxicity of 3-nitropropionic acid in rat cortical neurons. Neurotoxicology 2009, 30, 718–726. [Google Scholar]
- Wu, C.L.; Chen, S.D.; Hwang, C.S.; Yang, D.I. Sonic hedgehog mediates BDNF-induced neuroprotection against mitochondrial inhibitor 3-nitropropionic acid. Biochim. Biophys. Res. Commun. 2009, 385, 112–117. [Google Scholar] [CrossRef]
- Almeida, S.; Laço, M.; Cunha-Oliveira, T.; Oliveira, C.R.; Rego, A.C. BDNF regulates BIM expression levels in 3-nitropropionic acid-treated cortical neurons. Nuerobiol. Dis. 2009, 35, 448–456. [Google Scholar]
- Wei, Z.; Chigurupati, S.; Bagsiyao, P.; Henriquez, A.; Chan, S.L. The brain uncoupling protein UCP4 attenuates mitochondrial toxin-induced cell death: Role of extracellular signal-regulated kinases in bioenergetics adaptation and cell survival. Neurotox. Res. 2009, 16, 14–29. [Google Scholar] [CrossRef]
© 2010 by the authors;
Share and Cite
Túnez, I.; Tasset, I.; Pérez-De La Cruz, V.; Santamaría, A. 3-Nitropropionic Acid as a Tool to Study the Mechanisms Involved in Huntington’s Disease: Past, Present and Future. Molecules 2010, 15, 878-916. https://doi.org/10.3390/molecules15020878
Túnez I, Tasset I, Pérez-De La Cruz V, Santamaría A. 3-Nitropropionic Acid as a Tool to Study the Mechanisms Involved in Huntington’s Disease: Past, Present and Future. Molecules. 2010; 15(2):878-916. https://doi.org/10.3390/molecules15020878
Chicago/Turabian StyleTúnez, Isaac, Inmaculada Tasset, Verónica Pérez-De La Cruz, and Abel Santamaría. 2010. "3-Nitropropionic Acid as a Tool to Study the Mechanisms Involved in Huntington’s Disease: Past, Present and Future" Molecules 15, no. 2: 878-916. https://doi.org/10.3390/molecules15020878
APA StyleTúnez, I., Tasset, I., Pérez-De La Cruz, V., & Santamaría, A. (2010). 3-Nitropropionic Acid as a Tool to Study the Mechanisms Involved in Huntington’s Disease: Past, Present and Future. Molecules, 15(2), 878-916. https://doi.org/10.3390/molecules15020878