Prodrugs of Thyrotropin-Releasing Hormone and Related Peptides as Central Nervous System Agents

Abstract
:1. Introduction
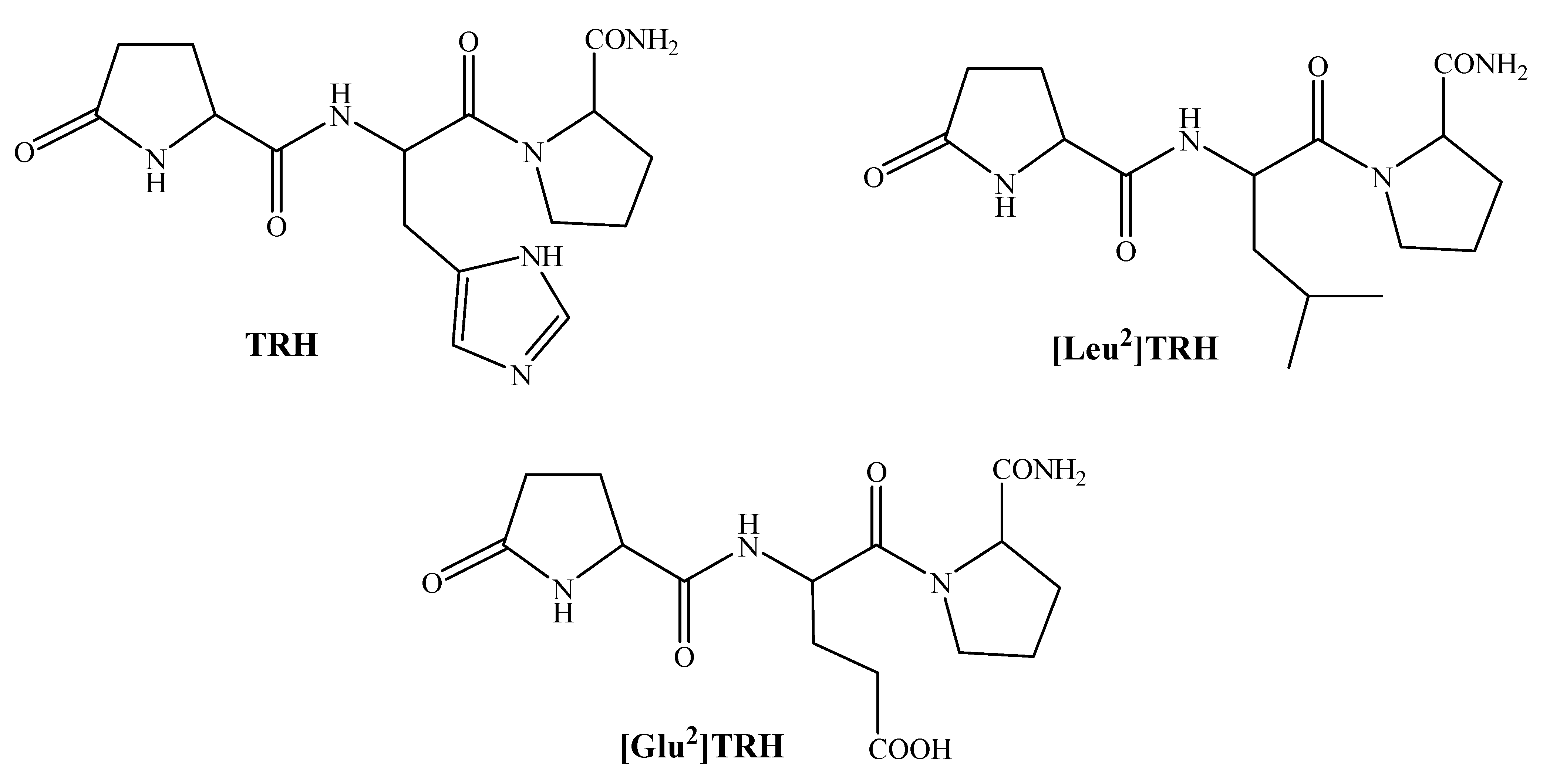
2. TRH and Related Peptides as CNS Agents
2.1. [Leu2]TRH prodrugs
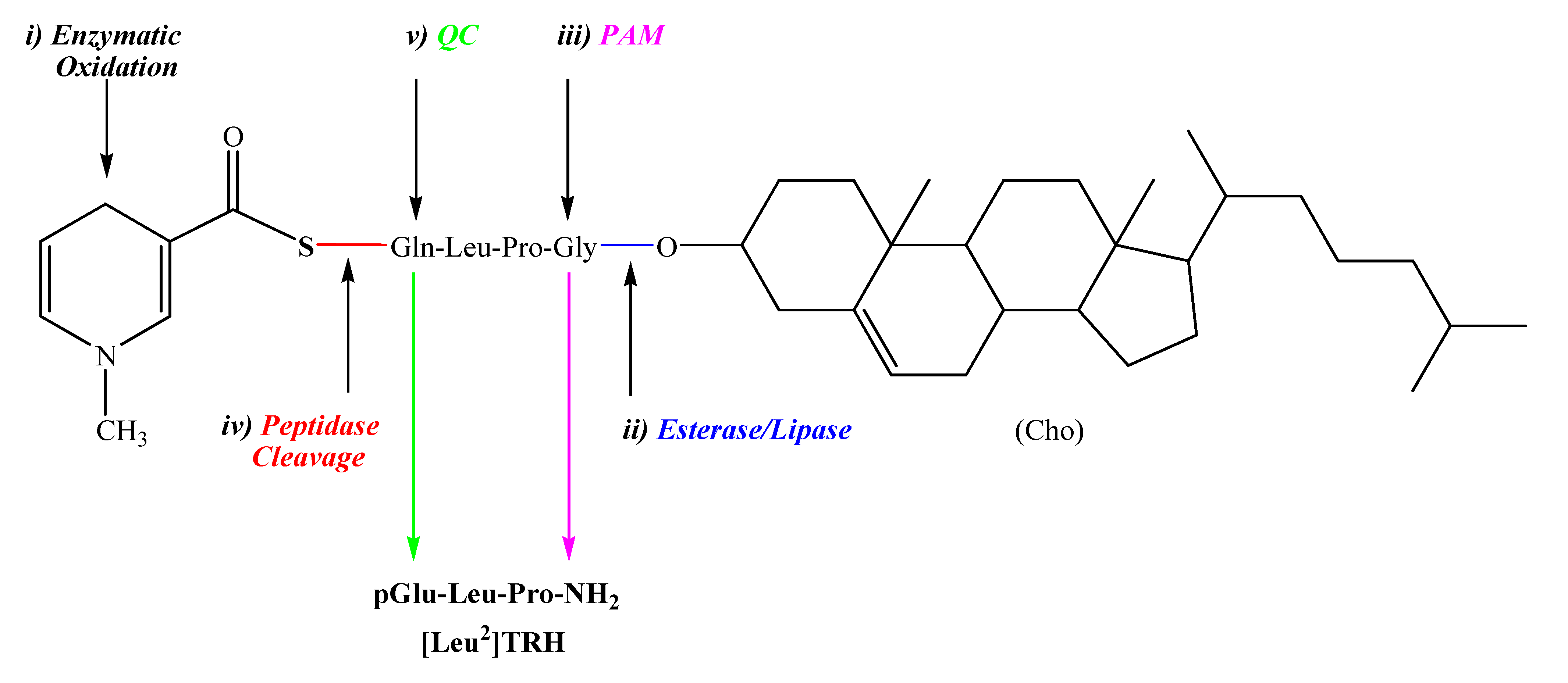

{kind=link}
{kind=link}
{kind=link}
{kind=link}
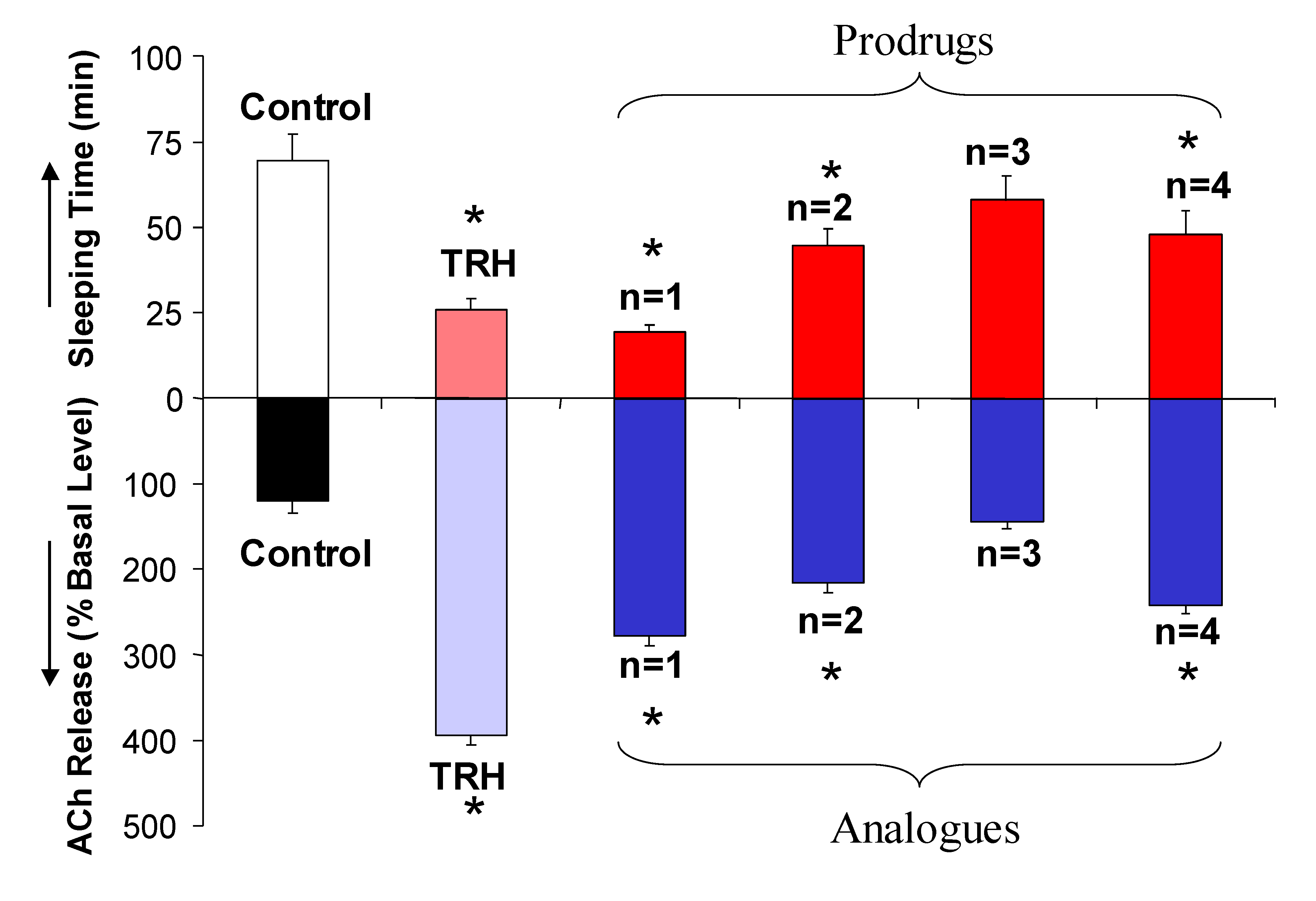{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % Decrease in Sleeping Time |
|---|---|
| [Leu2]TRH | 17 ± 7* |
| Prodrug, S= Ala | 30 ± 3* |
| Prodrug, S= Pro | 47 ± 6** |
| Prodrug, S= Ala-Ala | 32 ± 4* |
| Prodrug, S= Pro-Ala | 56 ± 4** |
| Prodrug, S= Pro-Pro | 55 ± 7** |
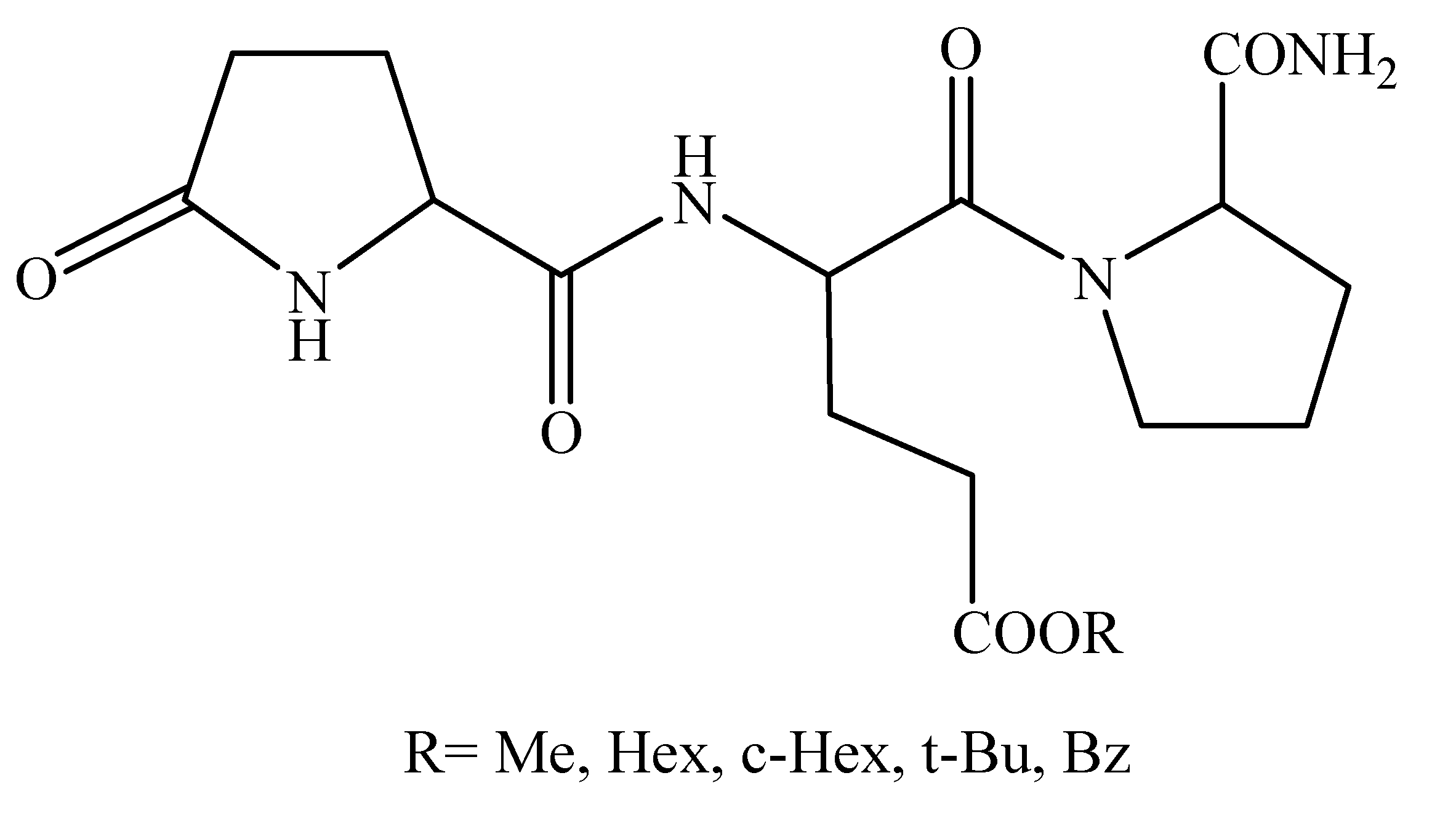
2.2. [Glu2]TRH Prodrugs
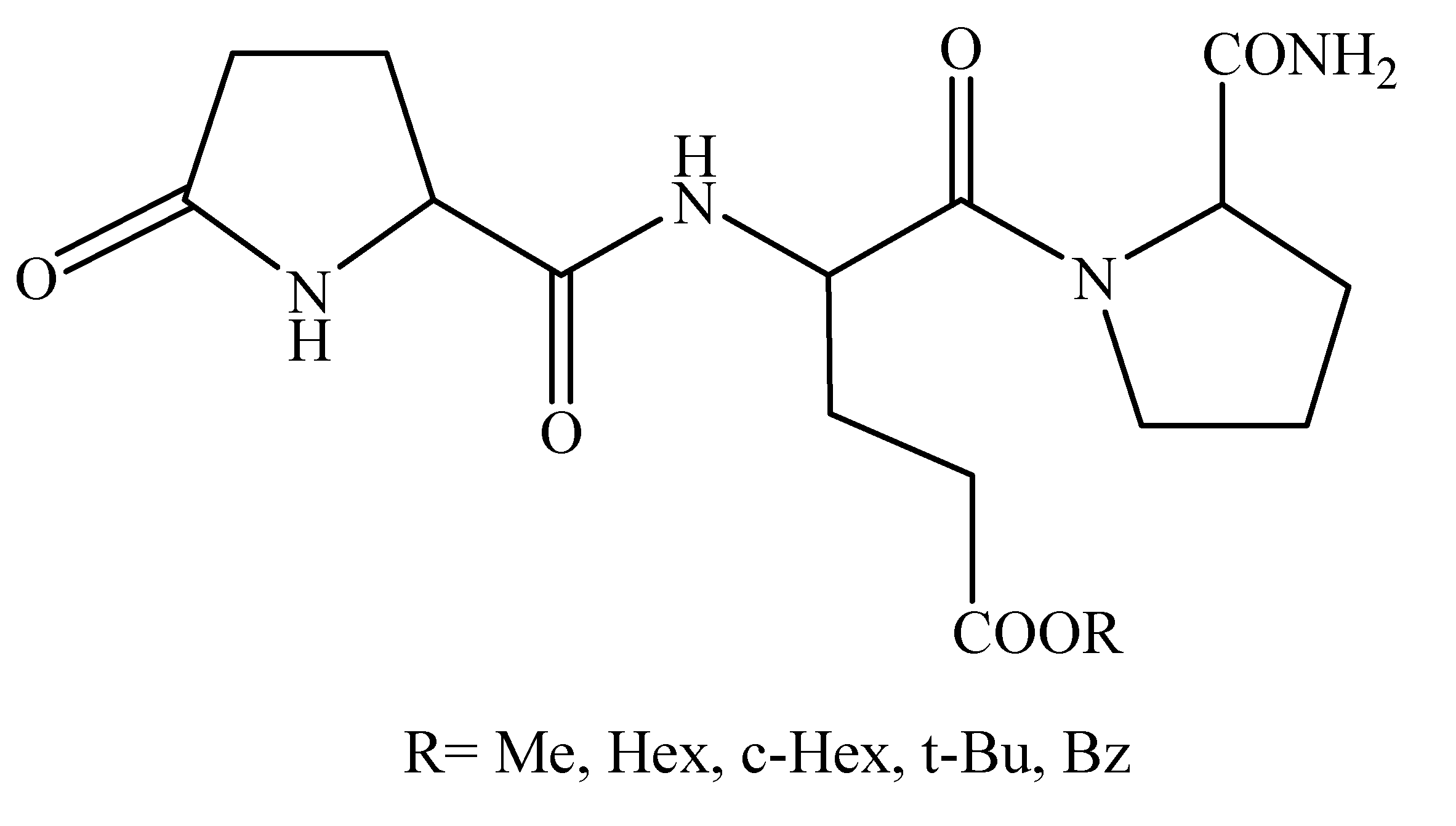
| Compound | k’IAM | t1/2 (min) | Sleeping time (min) |
|---|---|---|---|
| Vehicle | N/A | N/A | 80±2 |
| [Glu2]TRH, R=H | 0 | N/A | 65±3* |
| R=Me | 0.13 | 20 | 54±1** |
| R=Hex | 16.0 | 22 | 50±2** |
| R=cHex | 6.02 | 55 | 55±2** |
| R=tBu | 1.67 | 70 | 58±1* |
| R=Bz | 5.48 | >120 | 72±2 |
3. Prodrug-amenable analogues of TRH and related peptides as CNS Agents
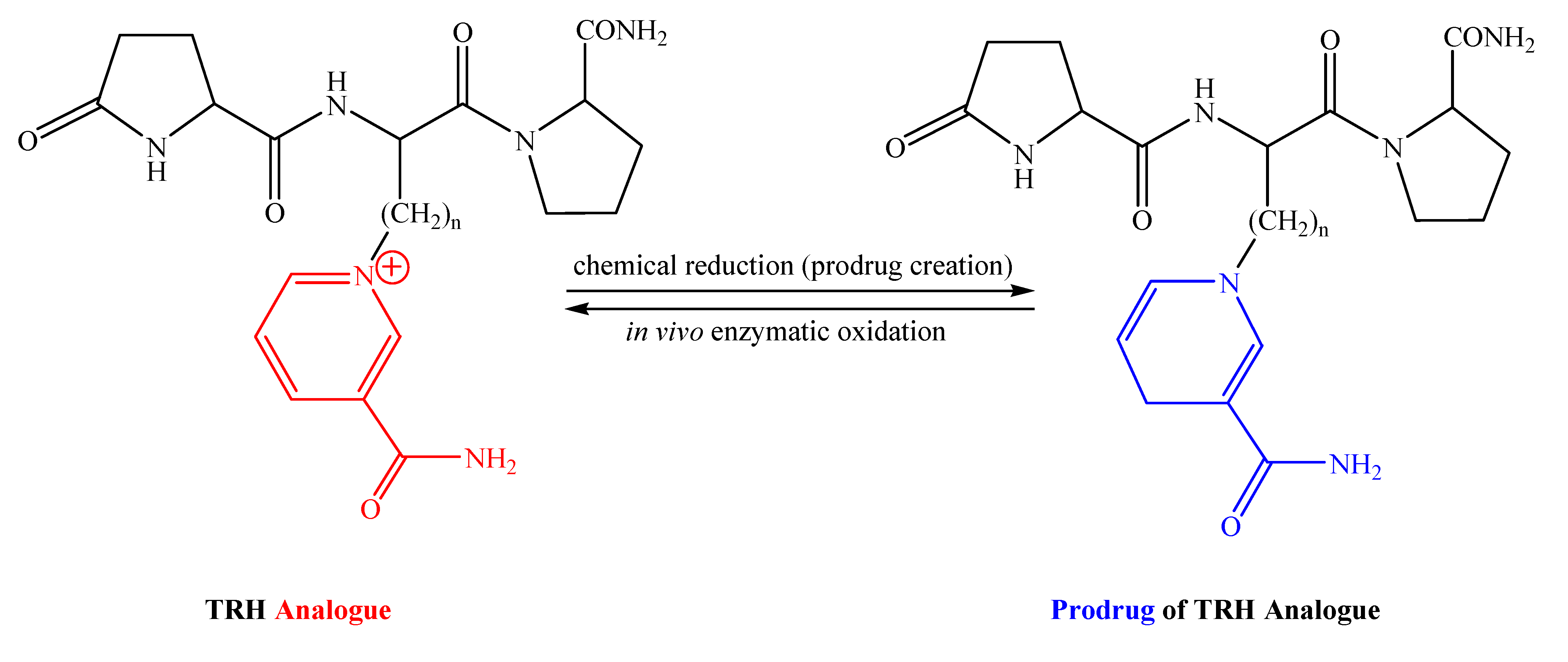
3.1. TRH analogues by replacement of the basic central residue
| Compound | k’IAM | Computed logP | ||
|---|---|---|---|---|
| Fragment-basedmethod [65] | Quantum chem. approach [66] | Volume-based method [67] | ||
| TRH | 0.51 | -4.37 | -0.27 | -1.74 |
| Pyridinium Analogue, n=1 | 0.30 | -2.67 | -2.15 | -2.64 |
| Prodrug of Analogue, n=1 | 0.55 | -4.38 | -3.14 | -3.29 |
| Pyridinium Analogue, n=2 | 0.42 | -2.61 | -1.85 | -2.18 |
| Prodrug of Analogue, n=2 | 0.80 | -4.33 | -2.92 | -2.84 |
| Pyridinium Analogue, n=3 | 0.49 | -2.16 | -2.67 | -1.74 |
| Prodrug of Analogue, n=3 | 1.47 | -3.88 | -3.30 | -2.39 |
| Pyridinium Analogue, n=4 | 0.60 | -1.77 | -2.68 | -1.29 |
| Prodrug of Analogue, n=4 | 2.30 | -3.48 | -3.55 | -1.94 |
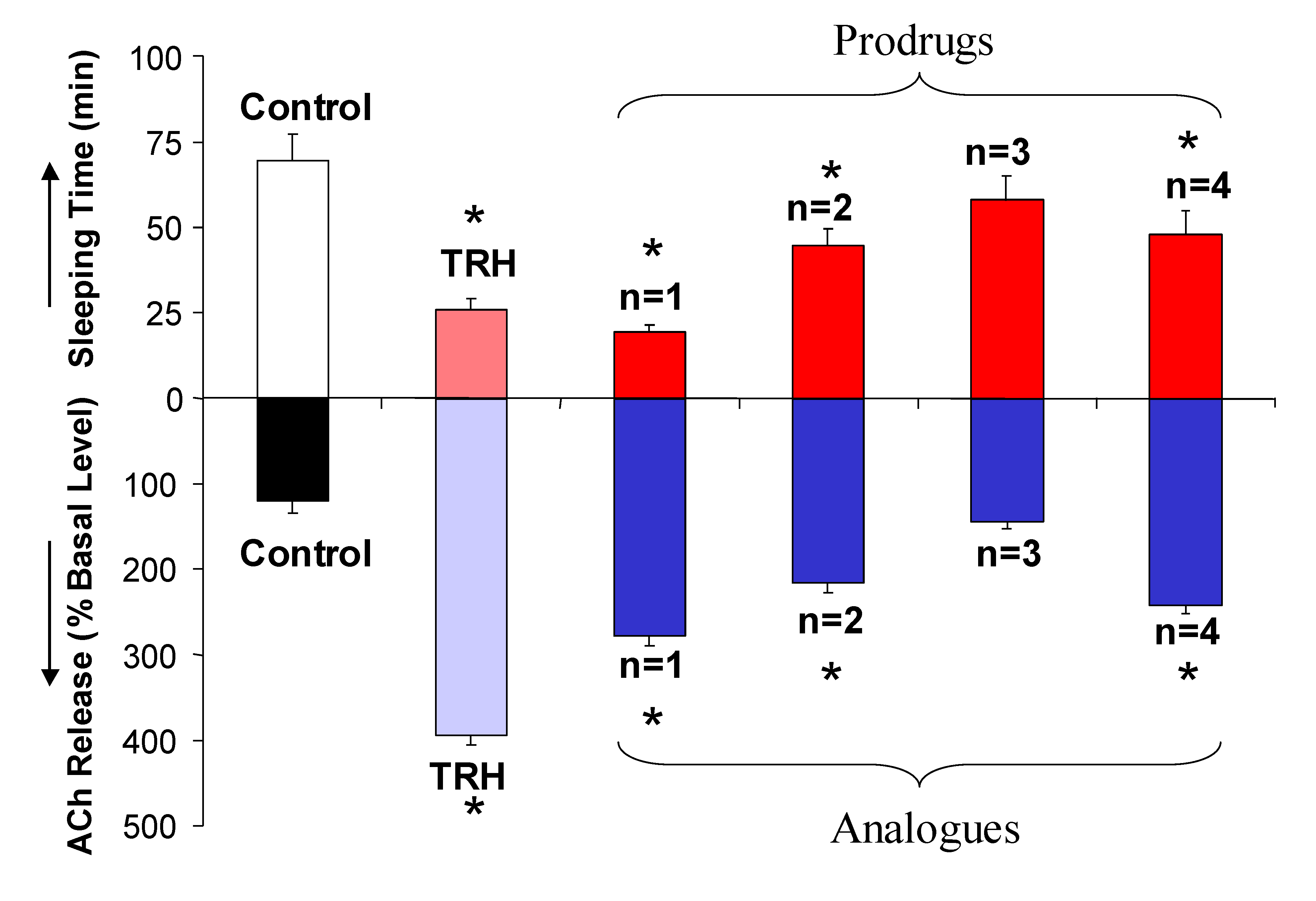
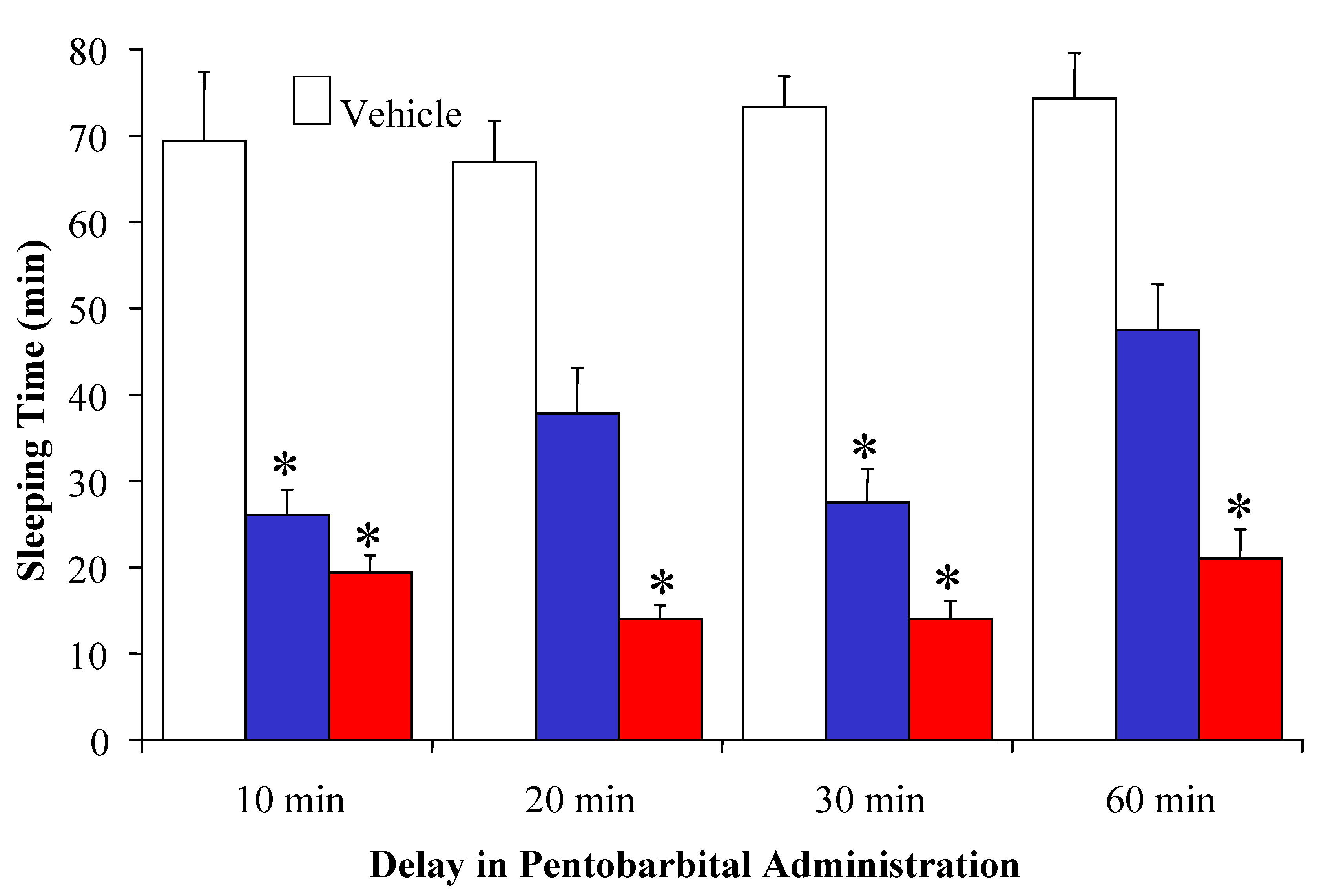
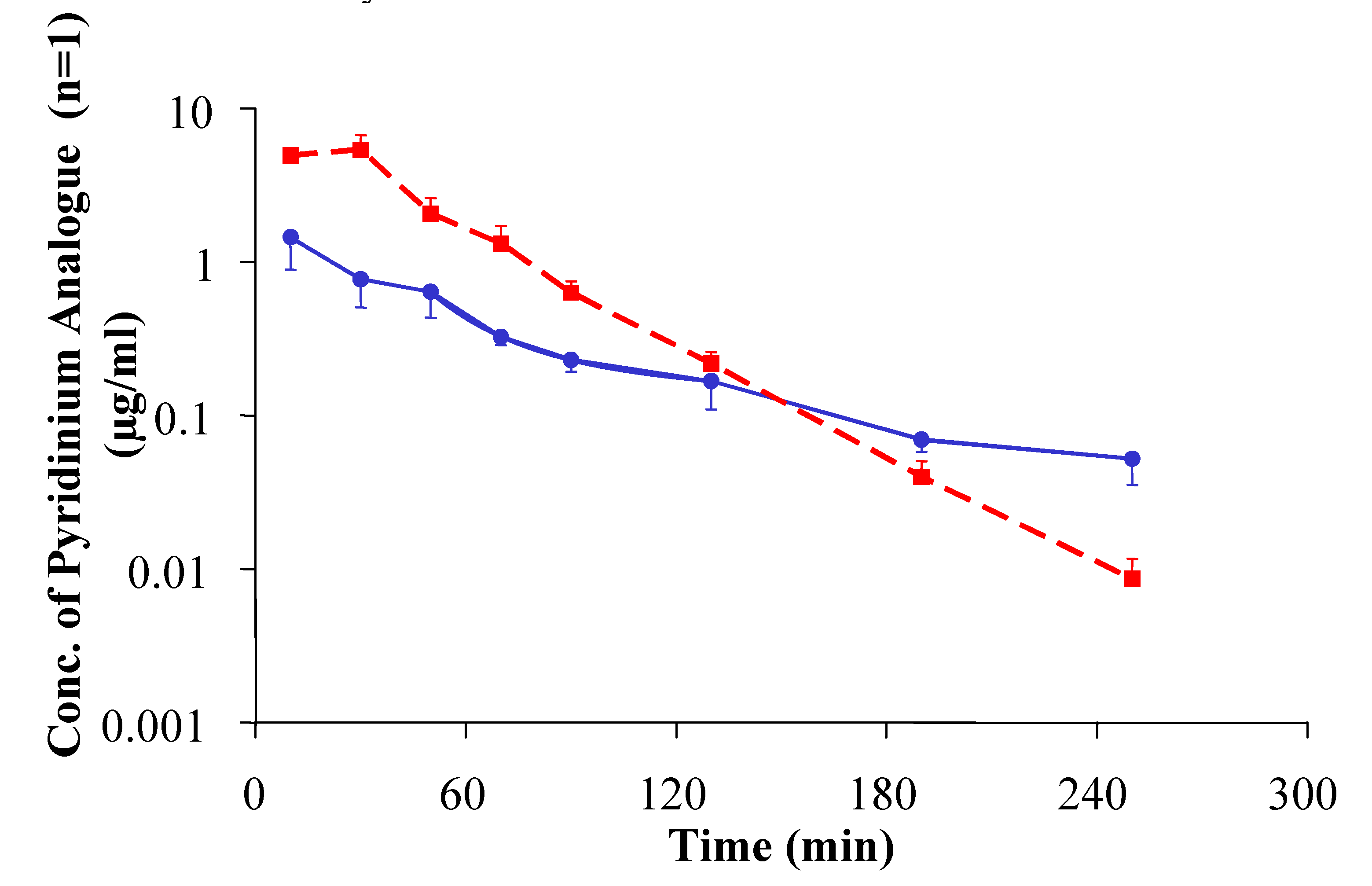
 ) and its [pyridinium2] analogue with n=1 (
) and its [pyridinium2] analogue with n=1 (  ) administered in its prodrug form upon varying the time for pentobarbital treatment (60 mg/kg body weight, i.p.) post-drug administration. * indicates statistically significant differences (ANOVA followed by Dunnett’s test, P<0.05) from vehicle control. Reprinted with permission from [63]. ©2004 American Chemical Society.
) and its [pyridinium2] analogue with n=1 ( ) administered in its prodrug form upon varying the time for pentobarbital treatment (60 mg/kg body weight, i.p.) post-drug administration. * indicates statistically significant differences (ANOVA followed by Dunnett’s test, P<0.05) from vehicle control. Reprinted with permission from [63]. ©2004 American Chemical Society.
) administered in its prodrug form upon varying the time for pentobarbital treatment (60 mg/kg body weight, i.p.) post-drug administration. * indicates statistically significant differences (ANOVA followed by Dunnett’s test, P<0.05) from vehicle control. Reprinted with permission from [63]. ©2004 American Chemical Society.
) and its [pyridinium2] analogue with n=1 ( ) administered in its prodrug form upon varying the time for pentobarbital treatment (60 mg/kg body weight, i.p.) post-drug administration. * indicates statistically significant differences (ANOVA followed by Dunnett’s test, P<0.05) from vehicle control. Reprinted with permission from [63]. ©2004 American Chemical Society.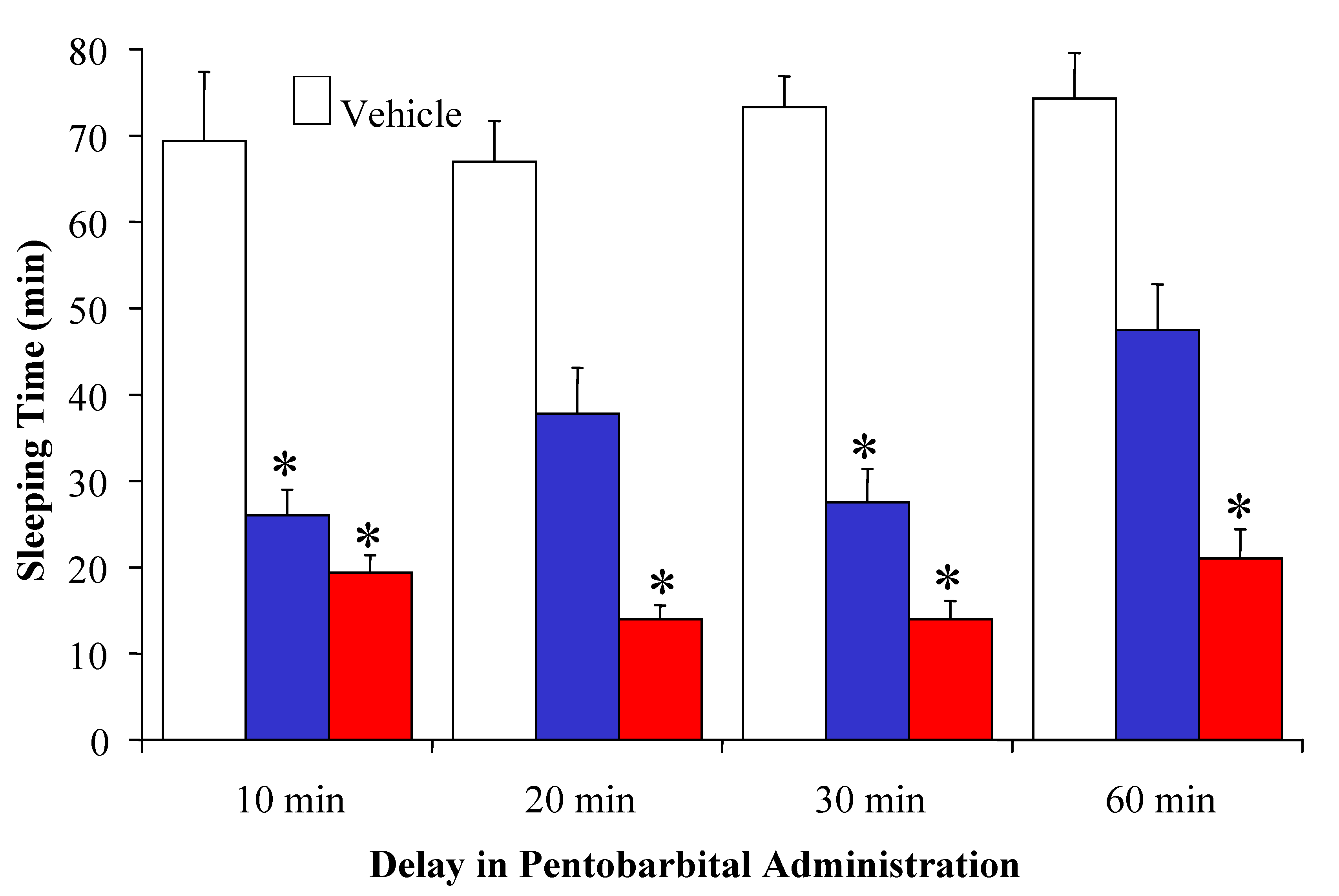
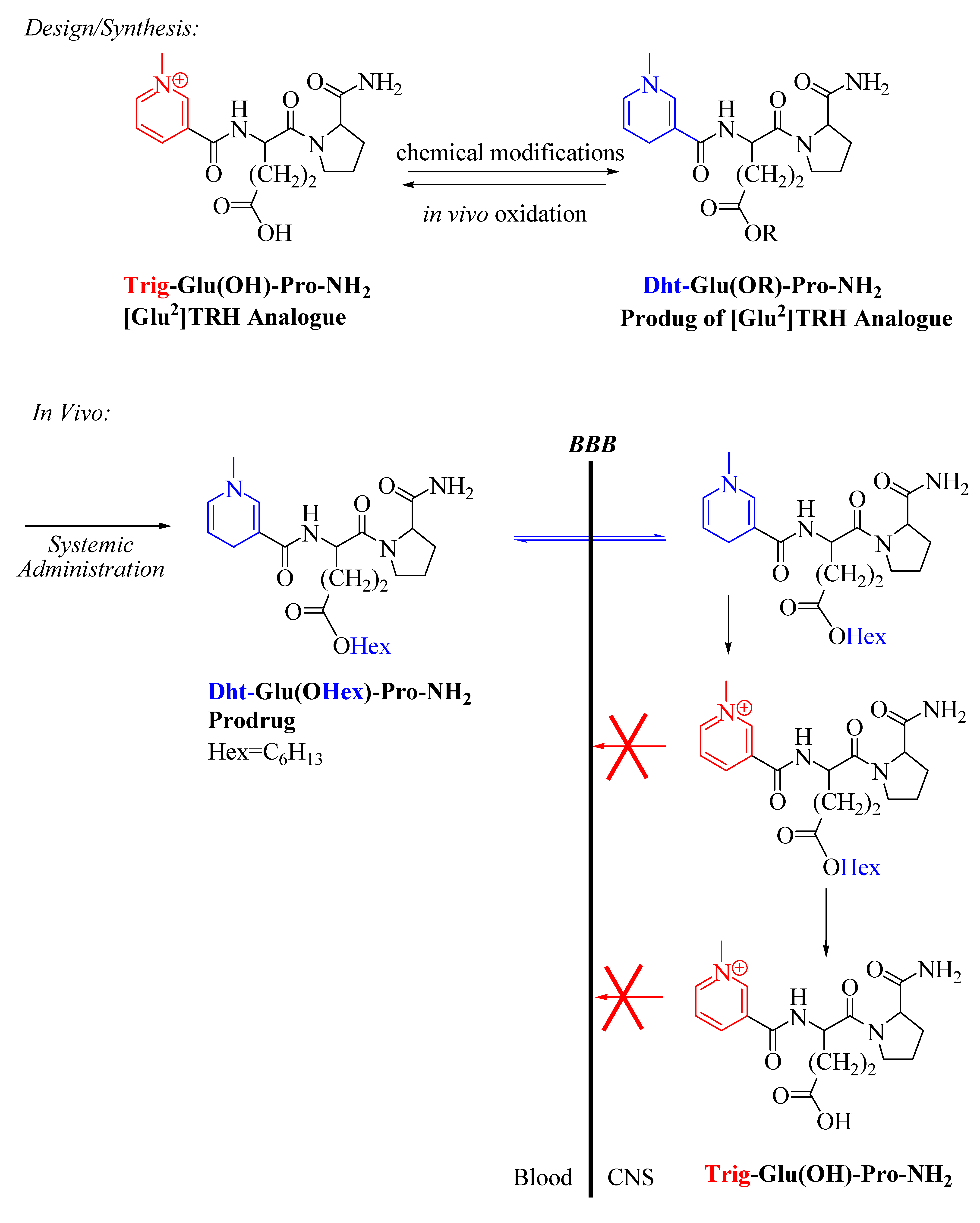
3.2. [Glu2]TRH analogues with pGlu-modification strategy
| Compound | Analeptic Effect:% Decrease in Sleeping Time | Porsolt Swim Test:% Decrease in Immobility |
|---|---|---|
| TRH: pGlu-His-Pro-NH2 | 50±2* | 36±2* |
| Glu2[TRH]: pGlu-Glu-Pro-NH2 | 17±2* | 33±4* |
| pGlu-Glu(OHex)–Pro-NH2 | 37±3* | 44±2* |
| Dht-Glu(OHex)–Pro-NH2 | 18±2* | 43±3* |
| Trig-Glu(OHex)–Pro-NH2 | 1±3 | 2±1 |
4. Conclusions
Acknowledgements
References and Notes
- Begley, D.J.; Brightman, M.W. Progress in Drug Research; Prokai, L., Prokai-Tatrai, K., Eds.; Birkhauser: Basel, Switzerland, 2003; Vol. 61, Chapter 2, pp. 39–78. [Google Scholar]
- Brownlees, J.; Williams, C.H. Peptidases, Peptides, and the Mammalian Blood-Brain Barrier. J. Neurochem. 1993, 60, 793–803. [Google Scholar]
- Tatsuta, T.; Naito, M.; Oh-Hara, T.; Sugawara, I.; Tsuruo, T. Functional Involvement of P-glycoprotein in Blood-Brain Barrier. J. Biol. Chem. 1992, 28, 20383–20391. [Google Scholar]
- Golden, P.L.; Pardridge, W.M. Brain Microvascular P-glycoprotein and a Revised Model of Multidrug Resistance in Brain. Cell Mol. Neurobiol. 2000, 20, 165–181. [Google Scholar] [CrossRef]
- Taylor, E.M. The Impact of Efflux Transporters in the Brain on the Development of Drugs for CNS Disorders. Clin. Pharmacokinet. 2002, 41, 81–92. [Google Scholar] [CrossRef]
- Temsamani, J.; Scherrmann, J.M.; Reesa, A. R.; Kaczore, M. Brain Drug Delivery Technologies: Novel Approaches for Transporting Therapeutics. Pharm. Sci. Technolo. Today 2000, 3, 155–162. [Google Scholar] [CrossRef]
- Fortin, D. Progress in Drug Research; Prokai, L., Prokai-Tatrai, K., Eds.; Birkhauser: Basel, Switzerland, 2003; Vol. 61, Chapter 4; pp. 125–154. [Google Scholar]
- Pardridge, W.M.; Eisenberg, J.; Yamada, T. Human Blood-Brain Barrier Insulin Receptor. J. Neurochem. 1985, 44, 1171–1178. [Google Scholar]
- Tsuji, A. Small Molecular Drug Transfer Across the Blood–Brain Barrier via Carrier-Mediated Transport Systems. NeuroRx 2005, 1, 54–62. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood–Brain Barrier Delivery of Protein and Non-Viral Gene Therapeutics with Molecular Trojan Horses. J. Controll. Release 2007, 122, 345–348. [Google Scholar] [CrossRef]
- Kreuter, J. Application of Nanoparticles for the Delivery of Drugs to the Brain. Int. Congress Series 2005, 1277, 85–94. [Google Scholar] [CrossRef]
- Huynh, G.H.; Deen, D. F.; Szoka, F.C. Barriers to Carrier Mediated Drug and Gene Delivery to Brain. J. Controll. Release 2006, 110, 236–259. [Google Scholar] [CrossRef]
- Albert, A. Chemical Aspects of Selective Toxicity. Nature 1958, 182, 421–423. [Google Scholar] [CrossRef]
- Anand, B.S.; Dey, S.; Mitra, A.K. Current Prodrug Strategies via Membrane Transporters/Receptors. Exp. Opin. Ther. Biol. 2002, 2, 607–620. [Google Scholar] [CrossRef]
- Levin, V.A. Relationship of Octanol/Water Partition Coefficient, and Molecular Weight to Rat Brain Capillary Permeability. J. Med. Chem. 1980, 23, 682–684. [Google Scholar] [CrossRef]
- Partridge, W.M. Blood-Brain Barrier Biology, and Methodology. J. Neurovirol. 2000, 5, 556–569. [Google Scholar] [CrossRef]
- Abraham, M.H.; Chadha, H.S.; Mitchell, R.C. Hydrogen-Bonding. Part 36. Determination of Blood Brain Distribution Using Octanol-Water Partition Coefficients. Drug Dis. Discov. 1995, 13, 123–131. [Google Scholar]
- Habgood, M.D.; Begley, D.J.; Abbott, N.J. Determination of Passive Drug Entry into the Central Nervous System. Cell Mol. Neurobiol. 2000, 20, 231–253. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J. Peptides and the Blood-Brain Barrier: Lipophilicity as a Predictor of Permeability. Brain Res. Bull. 1985, 15, 287–292. [Google Scholar] [CrossRef]
- Hansch, C.; Björkroth, J.P.; Leo, A. Hydrophobicity and Central Nervous System Agents: On the Principle of Minimal Hydrophobicity in Drug Design. J. Pharm. Sci. 1987, 76, 663–687. [Google Scholar] [CrossRef]
- Tatsuta, T.; Naito, M.; Oh-Hara, T.; Sugawara, I.; Tsuruo, T. Functional Involvement of P-glycoprotein in Blood-Brain Barrier. J. Biol. Chem. 1992, 28, 20383–20391. [Google Scholar]
- Bak, A.; Gudmundsson, O.S.; Friis, G.J.; Shiahaan, T.J.; Borchardt, R.T. Acyloxy-Alkoxy-Based Cyclic Prodrugs of Opioid Peptides: Evaluation of the Chemical and Enzymatic Stability as well as Their Transport Properties Across Caco-2 Cell Monolayers. Pharm. Res. 1999, 16, 24–29. [Google Scholar] [CrossRef]
- Begley, D. J. The Blood-Brain Barrier: Principles for Targeting Peptides and Drugs to the Central Nervous System. J. Pharm. Pharmacol. 1996, 48, 136–146. [Google Scholar] [CrossRef]
- Prokai, L.; Prokai-Tatrai, K.; Ouyang, X.; Kim, H.S.; Wu, W.M.; Zharikova, A.D.; Bodor, N. Metabolism-Based Brain-Targeting System for a Thyrotropin-Releasing Hormone Analogue. J. Med. Chem. 1999, 42, 4563–4571. [Google Scholar] [CrossRef]
- Prokai, L.; Prokai-Tatrai, K.; Bodor, N. Targeting Drugs to the Bain by Redox Chemical Delivery Systems. Med. Res. Rev. 2000, 20, 367–416. [Google Scholar]
- Bodor, N.; Farag, H.H.; Brewster, M. E. Site-Specific, Sustained Release of Drugs to the Brain. Science 1981, 214, 1370–1372. [Google Scholar]
- Zlokovic, B.V.; Segal, M.B.; Begley, D.J.; Davson, H.; Rakic, L. Permeability of the Blood-Cerebrospinal Fluid and the Blood-Brain Barriers to Thyrotropin-Releasing Hormone. Brain Res. 1985, 358, 191–199. [Google Scholar] [CrossRef]
- Prokai, L. Progress in Drug Research; Jucker, E., Ed.; Birkhauser: Basel, Switzerland, 2002; Vol. 59, Chapter 5, pp. 133–170. [Google Scholar]
- Schally, A.V.; Bowers, C.Y.; Redding, T.W.; Barrett, J.F. Isolation of Thyrotropin Releasing Factor (TRF) from Porcine Hypothalamus. Biochem. Biophys. Res. Commun. 1966, 25, 165–169. [Google Scholar] [CrossRef]
- Böler, J.; Enzmann, F.; Folkers, K.; Bowes, C.Y.; Schally, A.V. The Identity of Chemical and Hormonal Properties of the Thyrotropin Releasing Hormone and Pyroglutamyl-Histidyl-Proline Amide. Biochem. Biophys. Res. Commun. 1969, 37, 705–710. [Google Scholar] [CrossRef]
- Griffiths, E.C. Thyrotrophin Releasing Hormone: Endocrine and Central Effects. Psychoneuroendocrinology 1985, 10, 225–235. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Matsushita, H.; Kawano, H.; Sakai, H.; Yoshimoto, K.; Sawada, T. TRH Increases Cerebrospinal Fluid Concentration of Kynurenine. Neuroreport 1999, 10, 3601–3603. [Google Scholar] [CrossRef]
- Breese, G.; Cott, J.; Cooper, B.; Prange, A.; Lippton, M.; Plotnikoff, N. Effects of Thyrotropin-Releasing Hormone (TRH) on Actions of Actions of Pentobarbital and Other Centrally Active Drugs. J. Pharmacol. Exp. Ther. 1975, 193, 11–22. [Google Scholar]
- Hinkle, P.M.; Pekary, A.E.; Senanayaki, S.; Sattin, A. Role of TRH Receptors as Possible Mediators of Analeptic Actions of TRH-Like Peptides. Brain Res. 2002, 935, 59–64. [Google Scholar] [CrossRef]
- Schmidt, D. Effects of Thyrotropin Releasing Hormone (TRH) on Pentobarbital-Induced Decrease in Cholinergic Neuronal Activity. Commun. Psychopharm. 1977, 1, 469–473. [Google Scholar]
- Miyamoto, M.; Nagai, Y.; Narumi, S.; Saji, Y.; Nagawa, Y. TRH and its Novel Analog (DN-1417): Antipentobarbital Action and Involvement of Cholinergic Mechanisms. Pharmacol. Biochem. Behav. 1982, 17, 797–806. [Google Scholar] [CrossRef]
- Bassiri, R. M.; Utiger, R.D. Metabolism and Excretion of Exogenous Thyrotropin-Releasing Hormone in Humans. J. Clin. Invest. 1981, 52, 1616–1619. [Google Scholar] [CrossRef]
- Redding, T.W.; Schally, A.V. On the Half Life of Thyrotropin-Releasing Hormone in Rats. Neuroendocrinology 1972, 9, 250–256. [Google Scholar] [CrossRef]
- Kelly, J.A.; Slator, G.R.; Tipton, K.F.; Williams, C.H.; Karl, Bauer. Kinetic Investigation of the Specificity of Porcine Brain Thyrotropin-Releasing Hormone-Like Peptides. J. Biol. Chem. 2000, 275, 16746–16751. [Google Scholar]
- Zlokovic, B.V.; Segal, M.B.; Begley, D.J.; Davson, H.; Rakic, L. Permeability of the Blood-Cerebrospinal Fluid and the Blood-Brain Barriers to Thyrotropin-Releasing Hormone. Brain Res. 1985, 358, 191–199. [Google Scholar] [CrossRef]
- Pekary, A.E.; Sattina, A.; Meyerhoff, J.L.; Chilingar, M. Valproate Modulates TRH Receptor, TRH and TRH-like Peptide Levels in Rat Brain. Peptides 2004, 25, 647–658. [Google Scholar]
- Szirtes, T.L.; Kisfaludi, L.; Palosi, E.; Szporny, L. Synthesis of Thyrotropin-Releasing Hormone Analogues. 1. Complete Dissociation of Central Nervous System Effect from Thyrotropin-Releasing Activity. J. Med. Chem. 1984, 27, 741–745. [Google Scholar] [CrossRef]
- Nillni, E.A.; Sevarino, K.A. The Biology of Pro-Thyrotropin-Releasing Hormone-Derived Peptides. Endocr. Rev. 1999, 20, 599–648. [Google Scholar]
- Stenly Jr., S.; Prokai-Tatrai, K.; Prokai, L. Rapid Screening of Combinatorial Libraries by Mass Spectrometry: A Novel Approach for Monitoring Substrate Specificity of Enzymes. Anal. Chem. 2005, 7, 698–701. [Google Scholar]
- Fischer, W.; Spiess, J. Identification of a Mammalian Glutaminyl Cyclase Converting Glutaminyl into Pyroglutaminyl Peptides. Proc. Natl. Acad. Sci. USA 1987, 84, 3628–3632. [Google Scholar] [CrossRef]
- Lambeir, A.M.; Durinx, C.; Scharpé, S.; De Meester, I. Dipeptidyl-Peptidase IV from Bench to Bedside: An Update on Structural Properties, Functions, and Clinical Aspects of the Enzyme DPP IV. Crit. Rev. Clin. Lab. Sci. 2003, 40, 209–294. [Google Scholar] [CrossRef]
- Polgar, L. Structure-Function of Prolyl Oligopeptidase and its Role in Neurological Disorders. Curr. Med. Chem. CNS Agents 2002, 2, 251–257. [Google Scholar]
- Kenny, A. J.; Booth, A. G.; George, S. G.; Ingram, J.; Kershaw, D.; Wood, E. J.; Young, A. R. Dipeptidyl Peptidase IV, a Kidney Brush-Border Serine Peptidase. Biochem. J. 1965, 157, 169–182. [Google Scholar]
- Prokai-Tatrai, K.; Prokai, L.; Bodor, N. Brain-Targeted Delivery of a Leucine-Enkephalin Analogue by Retrometabolic Design. J. Med. Chem. 1996, 39, 4775–4782. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Kim, H.S.; Prokai., L. The Utility of Oligopeptidase in Brain-Targeting Delivery of an Enkephalin Analogue by Prodrug Design. Open Med. Chem. J. 2008, 2, 97–100. [Google Scholar] [CrossRef]
- Cocle, S. M.; Aitken, A.; Beg, F.; Smyth, D.G. A Novel Peptide, Pyroglutamylglutamylproline, in the Rabbit Prostate Complex Structurally Related to Thyrotropin-Releasing Hormone. J. Biol. Chem. 1989, 264, 7788–7791. [Google Scholar]
- Rondeel, J.M.M.; Klootwijk, W.; Linkels, E.; van de Greef, W.J.; Visser, T. Neural Differentiation of the Human Neuroblastoma Cell-Line IMR32 Induces Production of a Thyrotropin-Releasing Hormone-Like Peptide. Brain Res. 1994, 665, 262–268. [Google Scholar] [CrossRef]
- Pekary, A.E.; Meyerhoff, J. L.; Sattin, A. Electroconvulsive Seizures Modulate Levels of Thyrotopin Releasing Hormone and Related Peptides in Rat Hypothalamus, Cingulate and Lateral Cerebellum. Brain Res. 2000, 884, 174–183. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Nguyen, V.; Zharikova, A.D.; Braddy, A.C.; Stevens, S.M., Jr.; Prokai, L. Prodrugs to Enhance Central Nervous System Effects of the TRH-like Peptide pGlu-Glu-Pro-NH2 Bioorg. Med. Chem. Lett. 2003, 13, 1011–1014. [Google Scholar] [CrossRef]
- Ong, S.W.; Liu, H.L.; Pidgeon, C. Immobilized-Artificial-Membrane Chromatography: Measurements of Membrane Partition Coefficient and Predicting Drug Membrane Permeability. J. Chromatogr. A 1996, 728, 113–128. [Google Scholar] [CrossRef]
- Braddy, A.C.; Janáky, T.; Prokai, L. Immobilized Artificial Membrane Chromatography Coupled with Atmospheric Pressure Ionization Mass Spectrometry. J. Chromatogr. A 2002, 966, 81–87. [Google Scholar] [CrossRef]
- Prokai, L.; Zharikova, A.D.; Janáky, T.; Li, X.; Braddy, A.C.; Perjési, P.; Matveeva, L.; Powell, D.H.; Prokai-Tatrai, K. Integration of Mass Spectrometry into Early-Phase Discovery and Development of Central Nervous System Agents. J. Mass. Spectrom. 2001, 36, 1211–1219. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Setting. Adv. Drug Deliv. Rev. 1997, 23, 3–26. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Liu, Q; Nakagome, I.; Matsushita, Y. Simple Method of Calculating Octanol/Water Partition Coefficients. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef]
- Rivier, J.; Burgus, R.; Vale, W.; Ling, N.; Monahan, M. Synthetic Thyrotropin-Releasing Factor Analogs. 3. Effect of Replacement or Modification of Histidine Residue on Biological-Activity. J. Med. Chem. 1972, 15, 479–482. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Perjesi, P.; Zharikova, A.D.; Li, X.; Prokai, L. Design, Synthesis and Biological Evaluation of Novel, Centrally-Acting Thyrotropin-Rleasin Hormone Analogues. Bioorg. Med. Chem. Lett. 2002, 12, 2171–2174. [Google Scholar] [CrossRef]
- Prokai, L.; Prokai-Tatrai, K.; Zharikova, A.D.; Nguyen, V.; Stevens, S.M., Jr. Centrally-Acting and Metabolically Stable Thyrotropin-Releasing Hormone Analogues by Replacement of Histidine with Substituted Pyridinium. J. Med. Chem. 2004, 47, 6025–6033. [Google Scholar] [CrossRef]
- Eda, M. E.; Kurth, M. J. Polymer Site-Site Interactions: Mechanistic Implication in the Solid-Phase Zincke Reaction. Chem. Commun. 2001, 723–724. [Google Scholar]
- Ghose, A. K.; Pritchett, A.; Crippen, G. M. Atomic Physicochemical Parameters for 3-Dimensional Structure Directed Quantitative Structure-Activity-Relationships. 3. Modeling Hydrophobic Interactions. J. Comput. Chem. 1988, 9, 80–90. [Google Scholar] [CrossRef]
- Bodor, N.; Gabanyi, Z.; Wong, C. K. A New Method for the Estimation of Partition-Coefficient. J. Am. Chem. Soc. 1989, 111, 3783–3786.67. [Google Scholar] [CrossRef]
- Buchwald, P.; Bodor, N. Octanol-Water Partition of Nonzwitterionic Peptides: Predictive Power of a Molecular Size-Based Model. Proteins 1998, 30, 86–99. [Google Scholar] [CrossRef]
- Prokai, L.; Zharikova, A.D. Neuropharmacodynamic Evaluation of Centrally Active Thyrotropin Releasing Hormone Analogue [Leu2]TRH and its Chemical Brain Targeting System. Brain Res. 2002, 952, 268–274. [Google Scholar] [CrossRef]
- Brown, W. M. Taltirelin (Tanabe Seiyaku). IDrugs 2001, 4, 1389–1400. [Google Scholar]
- Prokai-Tatrai, K.; Teixido, M.; Nguyen, V.; Zharikova, A.D.; Prokai, L. A Pyridinium-Substituted Analogue of the TRH-Like Tripeptide pGlu-Glu-Pro-NH2 and its Prodrugs as Central Nervous System Agents. Med. Chem. 2005, 1, 141–152. [Google Scholar] [CrossRef]
- Porsolt, R.D.; Le Pichon, M.; Jalfre, M. Depression: A New Animal Model Sensitive to Antidepressant Treatment. Nature 1977, 266, 730–732. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds presented in this review are available from the authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Prokai-Tatrai, K.; Prokai, L. Prodrugs of Thyrotropin-Releasing Hormone and Related Peptides as Central Nervous System Agents. Molecules 2009, 14, 633-654. https://doi.org/10.3390/molecules14020633
Prokai-Tatrai K, Prokai L. Prodrugs of Thyrotropin-Releasing Hormone and Related Peptides as Central Nervous System Agents. Molecules. 2009; 14(2):633-654. https://doi.org/10.3390/molecules14020633
Chicago/Turabian StyleProkai-Tatrai, Katalin, and Laszlo Prokai. 2009. "Prodrugs of Thyrotropin-Releasing Hormone and Related Peptides as Central Nervous System Agents" Molecules 14, no. 2: 633-654. https://doi.org/10.3390/molecules14020633
APA StyleProkai-Tatrai, K., & Prokai, L. (2009). Prodrugs of Thyrotropin-Releasing Hormone and Related Peptides as Central Nervous System Agents. Molecules, 14(2), 633-654. https://doi.org/10.3390/molecules14020633