Abstract
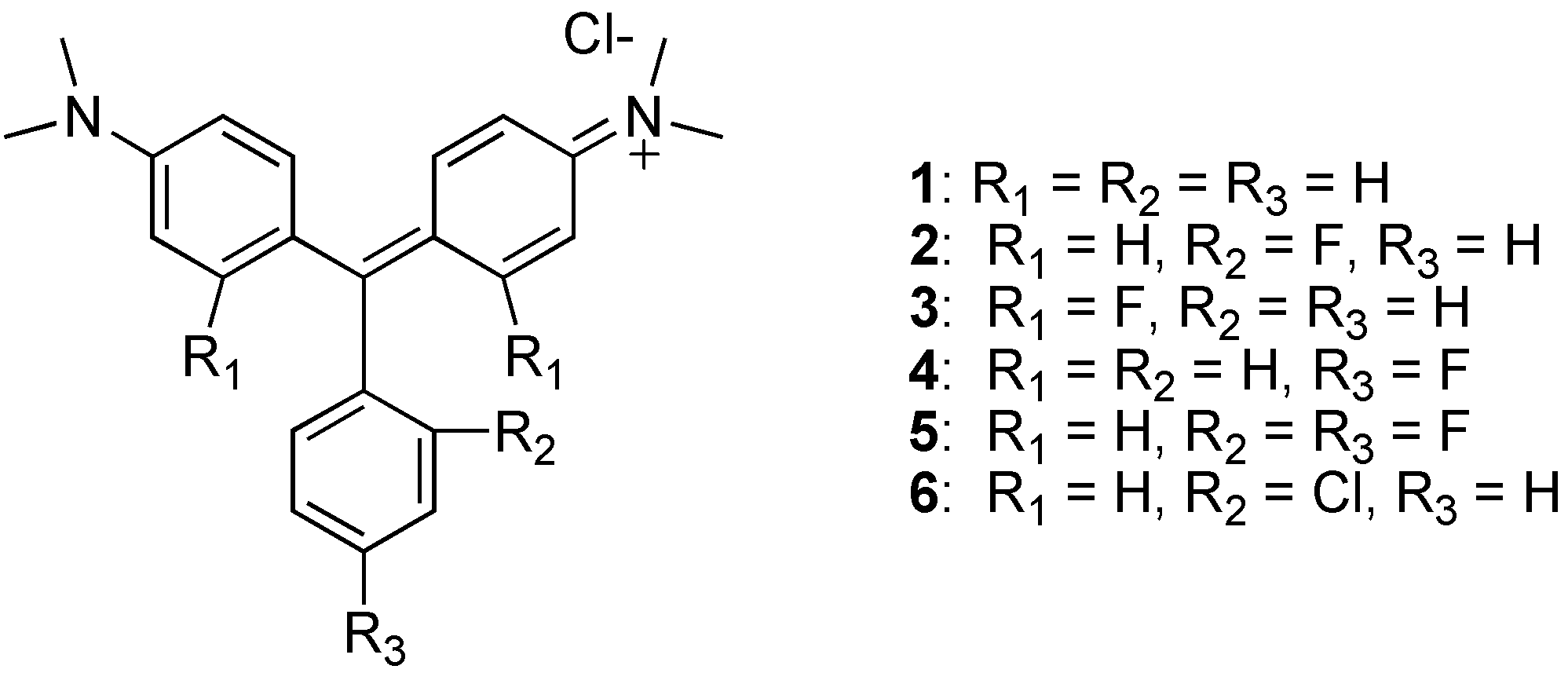
A series of fluorinated analogs of malachite green (MG) have been synthesized and their toxicity to Saccharomyces cerevisiae and a human ovarian epithelial cell line examined. The toxicity profiles were found to be different for these two species. Two analogs, one with 2,4-difluoro substitution and the other with 2-fluoro substitution seem to be the most promising analogs because they showed the lowest toxicity to the human cells.
Introduction
We are developing a new procedure for real-time imaging of gene expression. It involves a nucleic acid probe that can be used in conjunction with a 18F-labeled target molecule to image gene expression in vivo [1]. The probe has the basic feature that its terminal segments hybridize to form a stem in the absence of the target mRNA. One of the terminal segments is an aptamer and its activity is inhibited by hybridization with the other terminal segment (the attenuator). The presence of the target mRNA will result in hybridization of the probe via the intervening sequence (located between the aptamer and attenuator) with the target. Hybridization will release of the constraint on the aptamer with the result that the aptamer will bind its target and localize the 18F-labeled malachite green (MG) to the tissue site of gene expression.
In considering the aptamer to use for developing this imaging approach, we chose the malachite green one for our initial studies. This aptamer has a high affinity for its ligand and has been shown to function inside yeast cells. The use of MG in an imaging context would also constitute an entirely new application for this compound, which could easily be linked to 18F for imaging purposes. In addition, the fluorescent output of MG is increased over 2000 fold when it binds to the aptamer, which would allow imaging in cell culture without the use of a radiolabeled MG [2].
Fluorinated derivatives of MG have the potential for being labeled with 18F for in vivo tracking and are likely to substitute for MG in binding the MG aptamer. However, a critical question is whether these MG derivatives are toxic to human cells. In this study we have tested this question and compared the toxicity levels for human cells with those for yeast cells in which the MG aptamer has been previously shown to function [3].
The general usefulness of this study is: 1) the intrinsic value of the defined toxicity range of MG and some new derivatives for cells from humans and yeast, 2) specific information for groups working with the MG aptamer to provide information regarding potential intracellular ligands for probe development, and 3) the report of new chemical syntheses and the properties of new MG analogs.
Only two isolated reports of malachite green analogs bearing fluorines directly attached to the aromatic rings have been published [4,5]. Compared with MG (1), substitution of a fluorinated moiety reduced electron-donating capability resulting in a hypsochromic shift [6]. Here we present the chemical syntheses of a series of analogs with fluorine substituents located at various positions on the aromatic rings of MG. Their spectral properties and toxicities in yeast and humans are described.
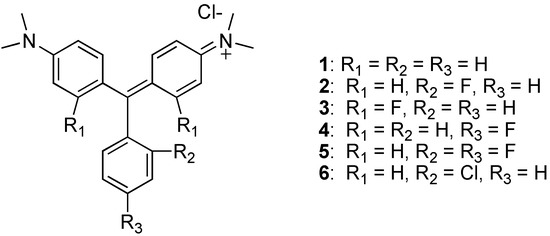
Figure 1.
Figure 1.
Results and Discussion
Preparation and Characteristics of MG analogs
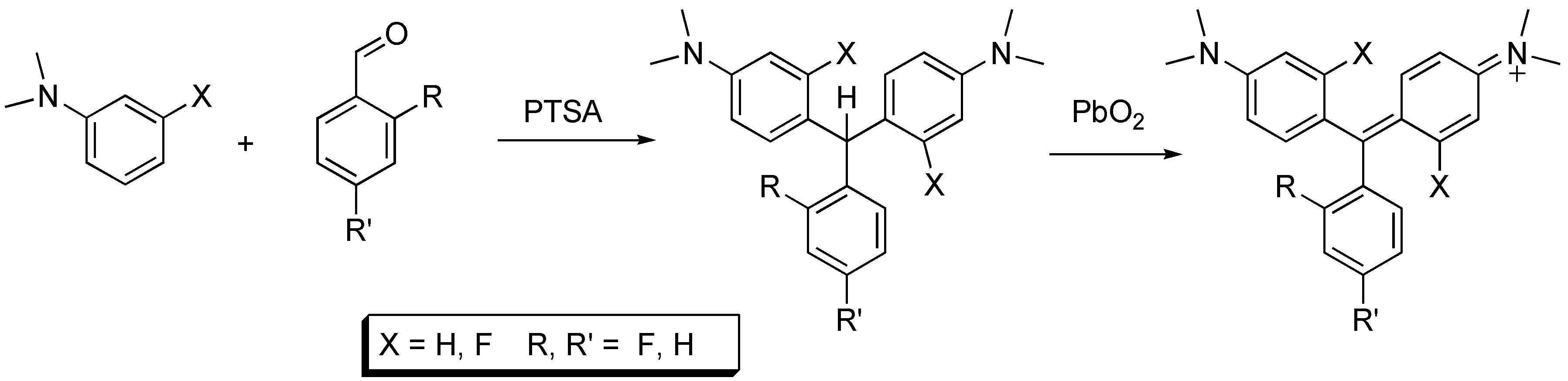
Analogs 2, 4, and 5 were synthesized from the reaction of dimethyl aniline and the requisite benzaldehyde in the presence of para-toluenesulfonic acid, as shown in Scheme 1. Oxidation to the MG analog was achieved using lead dioxide. This oxidant provided the MG analogs in almost quantitative yields. The oxidation of leucomalachite green using manganese dioxide, potassium permanganate and potassium persulfate has been reported [7]. Our oxidation procedure is rapid and convenient. Analog 3 was prepared from 3-fluoro dimethylaniline and benzaldehyde.
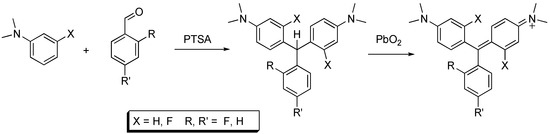
Scheme 1.
Scheme 1.
The absorption spectra in the visible range of each analog dissolved in water were determined in the range of 300 to 700 nm. (Figure 2). All analogs were measured at 20 μM except analog 3, which was measured at 500 μM. The figure inset shows the wavelengths of maximal absorption (λmax) in nm and the molar extinction coefficient in units of M-1cm-1. All analogs showed a strong hypochromatic shift compared with MG as demonstrated by the extinction coefficients at the wavelength of maximum absorbance.
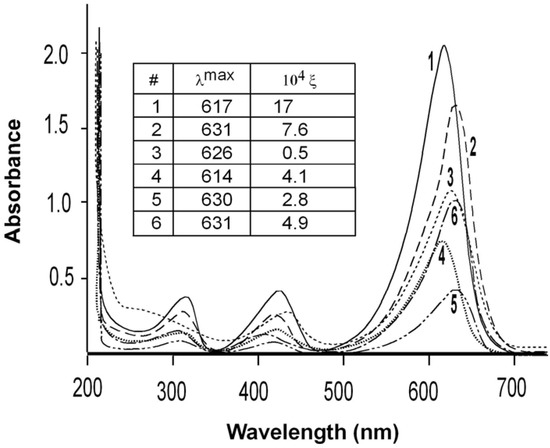
Figure 2.
Visible absorption spectra.
Figure 2.
Visible absorption spectra.
The extinction coefficients for MG (1) are comparable to two literature values of 186,100 M-1 cm-1 determined in 20 mM sodium acetate buffer pH 4.0 [8], and 148,900 M-1 cm-1 determined in water [9]. The λmax of 617 nm for MG is the same as has been previously published [8,9]. The three ortho-substituted rings as well as the 3′,3″ analog showed a red-shift in λmax of over 10 nm (9 nm for the 3′,3″ analog). It has been reported recently that MG also showed a red-shift in the maximum absorption frequency by 14 nm after binding to the RNA MG aptamer [10]. In association with the aptamer, the MG rings adopt a more coplanar relationship, compared with their orientation in untethered MG. The more planar conformation leads to a more extended π-system in the aptamer-bound MG and a red-shift in the MG λmax. A substitution in the 2-position of the 4′,4″-di-(N-methyl-N-2,2,2-trifluoroethyl-amino)-triphenylmethyl cation was also reported to result in bathochromic shifts of λmax [6]. Charge delocalization by the fluorine substituents would result in rotation of the phenyl rings to produce a more planar overall conformation of the analogs compared with MG. The observed red-shift in λmax of analogs 2, 3, and 5, can be similarly explained by charge delocalization that require the rings to adopt a coplanar orientation.
By contrast with the other analogs, the para-substituted ring in the analog 4 shows a small hypsochromic shift (from 617 to 614 nm). This small shift can be attributed to reduction of the negative charge on the phenyl ring produced by introduction of the electron withdrawing fluorine atom and resulting in less intense π→π* transitions.
Toxicity of MG analogs
The synthesized analogs were tested for their toxicity to yeast as a representative fungus and to cultured A2780 human ovarian epithelial cells as representative mammalian cells (Figure 3).
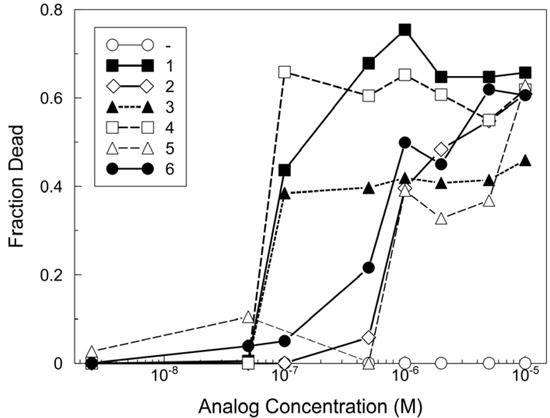
Figure 3.
Comparison of MG analog toxicities for mammalian cells.
Figure 3.
Comparison of MG analog toxicities for mammalian cells.
A2780 human ovarian epithelial cells were treated with the indicated analogs at concentrations from 6.3 x 10‑12 M to 1 x 10-4 M. The results represent the average of results from nine independently performed experiments, which provided compiled data such that the effect of each analog was measured in triplicate in each of two or three independent experiments. Shown are the data from 5 x 10-8 M to 1 x 10-5 M. Within each experiment, the fraction of dead cells from the DMSO vehicle controls were subtracted from each analog treatment. On average over all experiments, the fraction of dead cells (with the vehicle addition alone) was 0.12 ± 0.04 and the average coefficient of variation for all values was 33%. The limiting toxic concentrations determined from these experiments are shown in Table 1. The toxicity of each of the analogs to A2780 cells was determined as described in Experimental Procedures. Thirteen concentrations of each analog were tested including the highest (10-4 M) and lowest (6.25 x 10-12 M). The limiting toxic concentration is the lowest concentration at which toxicity of 10% or more above the control (vehicle) was detected. The differences of 5-10-fold in limiting toxic concentration between analogs are well outside the estimated average range of error (33%) of these estimates.

Table 1.
Limiting toxic concentration for malachite green analogs.
| Analog | Limiting toxic concentration |
|---|---|
| 1 | 1 x 10-7 |
| 2 | 1 x 10-6 |
| 3 | 1 x 10-7 |
| 4 | 1 x 10-7 |
| 5 | 1 x 10-6 |
| 6 | 5 x 10-7 |
First, the limiting toxic concentration of the analogs for A2780 cells was determined using a YO-PRO®-1-based assay with automated image analysis. The limiting toxic concentration was defined as the lowest concentration at which the percentage of dead cells was increased above the basal level. The basal level was established by a control series to which the vehicle, DMSO, alone was added at the same concentrations as included with the analogs. The limiting toxic concentrations determined from these studies showed that 1 and analogs 3 and 4 were equally toxic as MG for human cells, whereas analogs 2, 5 and 6 were less toxic (Table 1).
The toxicity profile for these analogs was different for yeast and humans (Figure 4). Saccharomyces cerevisiae, strain Y2158 and A2780 human ovarian epithelial cells were treated with the indicated analogs at concentrations of 0, 0.1, 0.5, 1 and 5 μM. In particular, analog 3, one of the most toxic for animal cells, was not toxic to yeast in the concentration range tested. On the other hand, human cells were less sensitive to analog 5 than yeast cells. These studies have revealed one analog of malachite green that can be used in yeast up to at least 5 μM and two analogs that can be used with mammalian cells at concentrations below 1 μM without toxicity.
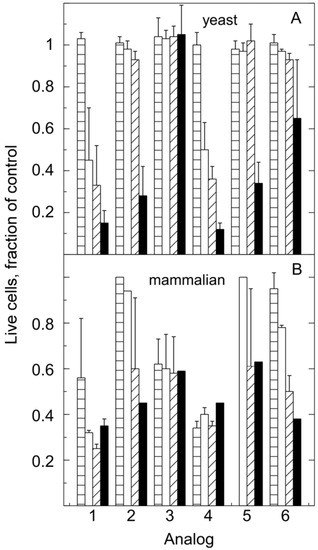
Figure 4.
Comparison of toxicity of MG analogs for yeast and human cells. All cell numbers are expressed as fractions of the DMSO control. Bars: 0.1 μM (parallel slash), 0.5 μM (white), 1 μM (45° slash), 5 μM (black). The error bars show standard deviations. Each yeast point is the average of 3 independent sets of experiments. The mammalian data is the average of two to three experiments with triplicate independent estimates in each experiment. Error bars are only shown where the values for more than one experiment have been averaged.
Figure 4.
Comparison of toxicity of MG analogs for yeast and human cells. All cell numbers are expressed as fractions of the DMSO control. Bars: 0.1 μM (parallel slash), 0.5 μM (white), 1 μM (45° slash), 5 μM (black). The error bars show standard deviations. Each yeast point is the average of 3 independent sets of experiments. The mammalian data is the average of two to three experiments with triplicate independent estimates in each experiment. Error bars are only shown where the values for more than one experiment have been averaged.
Experimental
General
YO-PRO®-1 iodide was from Invitrogen and DMSO was from Fisher. Hoechst 33342, YPD broth and malachite green oxalate were from Sigma. 2Cl-MG was purchased from TCI. Spectra were measured using a UV/VIS-S2100 Diode Array Spectrophotometer (Biochrom Ltd). Toxicity for yeast was measured by growing the yeast in a New Brunswick Scientific C24 shaker incubator set for 220 rpm. Toxicity for human cells was measured using a Cellomics Arrayscan II HCS system (Zeiss). 1H- and 13C-NMR experiments were performed with a Varian 300 MHz instrument operating at 300 and 75 MHz, respectively. All chemical shifts are reported relative to the solvent (CDCl3) peak (7.27 ppm for 1H and 77.23 ppm for 13C). Coupling constants (J) are reported in Hz with peak multiplicities indicated by the following abbreviaions: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. High resolution mass spectra were recorded on a Kratos model MS-50 spectrometer. Standard grade silica gel (60 A, 32-63 μm) was used for flash column chromatography.
General procedure for the synthesis of fluorinated malachite green analogs
Aldehyde (10 mmol), N,N-dimethylaniline (40 mmol), p-toluenesulfonic acid (10 mmol), and benzene (20 mL) were placed in a flask fitted with a Dean-Stark trap and boiled for 9 h. The reaction mixture was diluted with benzene (40 mL) and washed twice with 10% sodium bicarbonate solution and brine. After solvent evaporation, the residue was purified by column chromatography on silica gel (10:1 hexane-ethyl acetate) to afford 95 – 100 % yield of the fluorinated leucomalachite green analog. This compound (0.5 mmol) in H2O (3 mL) was then treated with PbO2 (0.65 mmol) and 15 drops of concentrated HCl. The reaction was stirred at room temperature overnight. The mixture was poured into a separatory funnel containing methanol (30 mL) and HCl and methylene chloride. After extraction, drying and concentration in vacuo, the residue was purified by chromatography on silica gel (CH2Cl2 -methanol = 9:1) to afford pure MG analogs (100% yield).
(2) 2-F-MG: 1H-NMR: δ 7.65-7.58 (m, 1H), 7.36 (d, J = 9.0 Hz, 4H), 7.32-7.13 (m, 3H), 6.95 (d, J = 9.3 Hz, 4H), 3.37 (s, 12H); 13C-NMR: δ 169.02, 162.92, 159.54, 157.14, 140.13, 135.11, 134.59, 134.47, 127.41, 127.39, 127.12, 126.96, 124.65, 124.60, 116.72, 116.43, 114.34, 41.43; EI-HRMS calcd for C23H24FN2+ m/z [M+] 347.19235, found 347.19278.
(3) 3′, 3″ DiF-MG: 1H-NMR: δ 7.69 (t, J = 7.5 Hz, 1H), 7.52 (t, J = 7.8 Hz, 2H), 7.31 (d, J = 7.2 Hz, 2H), 7.02 (t, J = 9.0 Hz, 2H), 6.77 (dd, J = 9.3, 2.4 Hz, 2H), 6.54 (dd, J = 14.7, 2.4 Hz, 2H), 3.40 (s, 12H); 13C-NMR: δ 169.01, 167.64, 167.62, 164.13, 164.10, 159.31, 159.13, 140.29, 140.27, 140.24, 139.97, 134.54, 133.98, 129.01, 118.55, 118.37, 110.83, 100.17, 99.80, 41.74; EI-HRMS calcd for C23H23F2N2+ m/z [M+] 365.18293, found 365.18340.
(4) 4F-MG: 1H-NMR: δ 7.36-7.20 (m, 8H), 6.97 (d, J = 9.3 Hz, 4H), 3.38 (s, 12H); 13C-NMR: δ 175.86, 167.72, 164.31, 157.14, 140.88, 137.15, 137.03, 135.70, 135.66, 127.31, 116.42, 116.13, 114.13, 41.38; EI-HRMS calcd for C23H24FN2+ m/z [M+] 347.19235, found 347.19278.
(5) 2,4-DiF-MG: 1H-NMR: δ 7.40 (d, J = 9.3 Hz, 4H), 7.24-7.19 (m, 1H), 7.12-7.01 (m, 6H), 3.42 (s, 12H); 13C-NMR: δ 167.45, 163.96, 163.80, 163.46, 163.29, 160.10, 160.04, 159.88, 159.29, 157.03, 139.86, 136.57, 136.53, 136.44, 136.40, 127.10, 123.28, 123.24, 123.12, 123.07, 114.29, 112.50, 112.46, 112.22, 112.17, 105.38, 105.04, 104.70, 41.27; EI-HRMS calcd for C23H23F2N2+ m/z [M+] 365.18293, found 365.18340.
Procedure for Measuring Toxicity of MG Analogs to Yeast
Saccharomyces cerevisiae, strain Y2158 were grown at 30°C in YPD culture medium (Sigma-Aldrich, St. Louis MO) for 18 h and then diluted to OD600=0.4 into YPD medium with 0.032% DMSO and the indicated concentration of the MG analog. The cells were incubated with shaking (18 mL/flask) at 30°C for 24h with samples being removed at 4, 6, 8, 10, and 24 h. The samples were analyzed for OD600 and growth curves plotted. For most cases, all cell cultures had reached a growth plateau by 24 h. The 10 h time points were chosen for comparison of toxicity because the cultures were still in the log phase of growth at that time.
Procedure for Measuring Toxicity of MG Analogs to Human Cells
Human A2780 ovarian epithelial cancer cells were plated in RPMI (GIBCO) containing 10% fetal bovine serum at 2,000 cells per well in a 96-well plate (100 μL/well) and grown for 24 h at 37°C in a 5% CO2 atmosphere. The MG analogs were added at the indicated concentrations and the cells incubated for another 48 h. The medium was removed and fresh medium containing 1 μM YO-PRO®-1 iodide (Invitrogen) and 10 μg/mL Hoechst 33342 (Sigma) stain was added to the cells. The cells were incubated for 30 min at 37°C in a 5% CO2 atmosphere in the dark before counting the YO-PRO®-1 and Hoechst stained nuclei using the optimal wavelengths YO-PRO®-1of λem=485 nm and λex=530 nm. Ten fields were counted in each well at a magnification of 10X which resulted in 2,000-6,000 nuclei being counted in control wells. The number of live cells per well were determined by subtracting the number of nuclei labeled with YO-PRO®-1 from the number of nuclei labeled with Hoechst in each well. Treatments were done in triplicate in each experiment. Cells were considered to be dead if they stained with YO-PRO®-1 or were not attached to the substratum. Because higher concentrations of drugs resulted in loss of significant numbers of cells from the substratum, the averages of triplicates (Hoechst minus YO-PRO®-1) for each treatment were normalized to the total number of cells in the control wells (no addition) to determine the fraction of live cells. These values were subtracted from 1.0 to determine the fraction of dead cells. Before averaging the results from different experiments, the average % dead cells in the vehicle control was subtracted from each value.
Acknowledgements
The authors extend their thanks to Dr. Nicolas Wang for running Cellomic assays for the mammalian cell toxicity tests. This work is supported by the Director, Office of Energy Science Research, Office Medical Sciences, Life Sciences of the U. S. Department of Energy under Contracts W-7405-Eng-82 with the Ames National Laboratories and DE-AC03-76SF00098 with the University of California. This is publication number LBNL-59259.
References
- Cong, X.; Nilsen-Hamilton, M. Allosteric aptamers: targeted reversibly attenuated probes. Biochemistry 2005, 44, 7945–7954. [Google Scholar]
- Nguyen, D. H.; DeFina, S. C.; Fink, W. H.; Dieckmann, T. Binding to an RNA aptamer changes the charge distribution and conformation of malachite green. J. Am. Chem. Soc. 2002, 124, 15081–15084. [Google Scholar]
- Grate, D.; Wilson, C. Inducible regulation of the S. cerevisiae cell cycle mediated by an RNA aptamer-ligand complex. Bioorg. Med. Chem. 2001, 9, 2565–2570. [Google Scholar]
- Inukai, K.; Maki, Y.; Ueda, T. Fluoro- and trifluoromethyl-substituted malachite green. Nippon Kagaku Ryoho Gakkai Zasshi 1956, 59, 515–517. [Google Scholar]
- Inukai, K.; Maki, Y. Difluoro-substituted malachite green. Nippon Kagaku Ryoho Gakkai Zasshi 1956, 59, 1160–1163. [Google Scholar]
- Armstrong, L.; Jones, A. M. Triphenylmethane dyes containing the N-methyl-N-2,2,2-trifluoroethyl group. Dyes Pigments 1999, 42, 65–70. [Google Scholar]
- Agunwa, U.; Okonkwo, E. Production of malachite green by oxidation of its leuco base using potassium persulphate, potassium permanganate and manganese dioxide. J. Pure Appl. Sci. 2004, 10, 143–146. [Google Scholar]
- Shin, K.; Oh, I.; Kim, C. Production and Purification of Remazol Brilliant Blue R Decolorizing Peroxidase from the Culture Filtrate of Pleurotus ostreatus. Appl. Environ. Microbiol. 1997, 63, 1744–1748. [Google Scholar]
- Green, F. The Sigma-Aldrich Handbook of Stains, Dyes and Indicators; Aldrich Chemical Company: Milwaukee, WI, USA, 1990. [Google Scholar]
- Nguyen, D.; Dieckmann, T.; Colvin, M.; Fink, W. Dynamics Studies of a Malachite Green-RNA Complex Revealing the Origin of the Red-Shift and Energetic Contributions of Stacking Interactions. J. Phys. Chem. B 2004, 108, 1279–1286. [Google Scholar]
- Sample availabilty: Contact the authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.