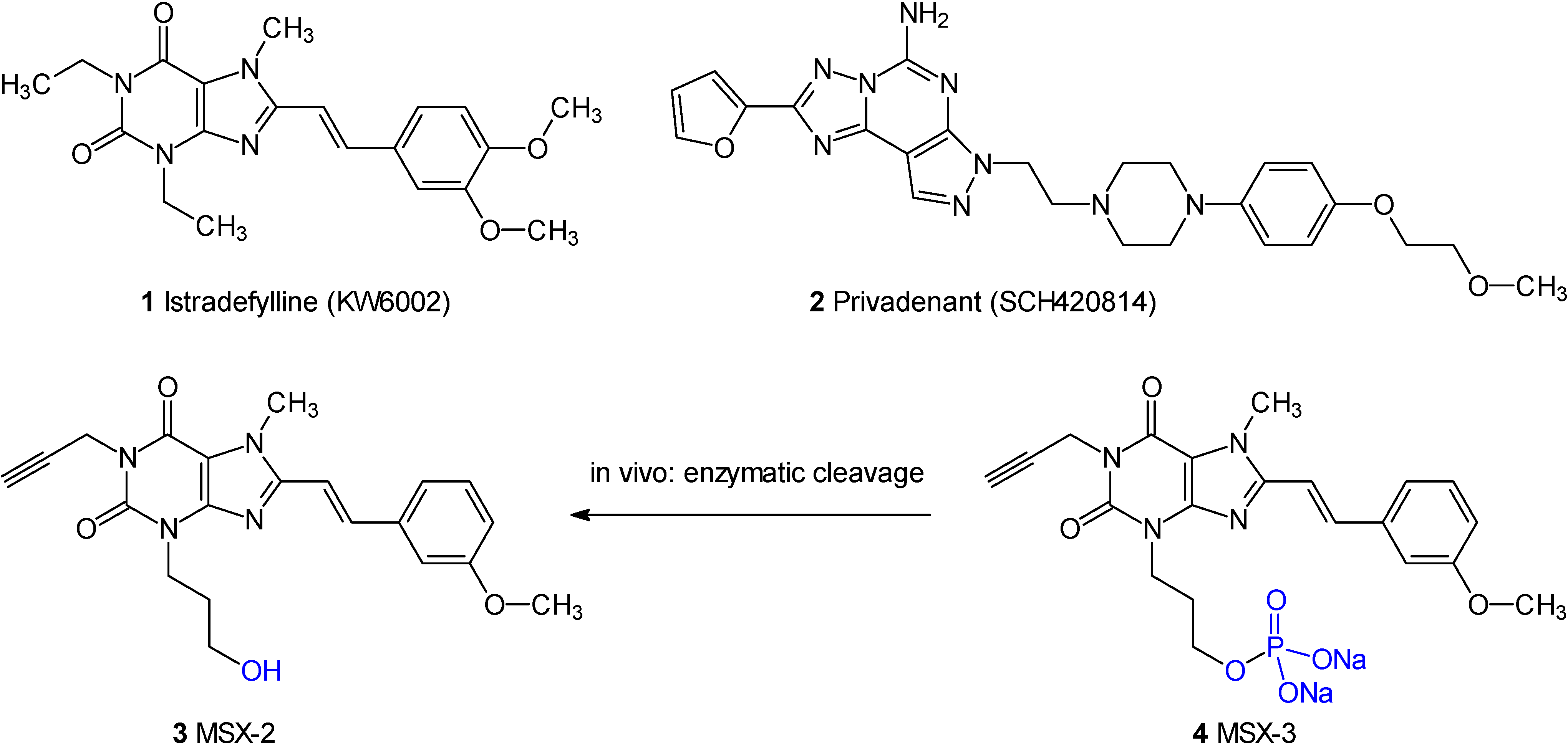
Introduction
Adenosine A
2A receptor antagonists, such as istradefylline (
1) and privadenant (
2) are currently in clinical trials as novel therapeutics for the treatment of Parkinson’s disease [
1]. Such compounds have also been proposed to possess activity as cognition enhancers, neuroprotectives, antidepressant agents, and against neuropathic pain [
1,
2,
3]. Different chemical classes of A
2A antagonists have been developed, including xanthine derivatives derived from the alkaloid caffeine, and adenine derivatives and analogs [
1,
5,
6]. MSX-2 (
3, 3-(3-hydroxypropyl)-8-(
m-methoxystyryl)-7-methyl-1-propargyl- xanthine (
3) is one of the most potent and selective adenosine A
2A receptor antagonists [
7,
8]. It is used in tritiated form ([N7-methyl-
3H]MSX-2) as a radioligand for the labelling of A
2A receptors [
9]. However, a major problem of the high-affinity A
2A antagonist is its low water-solubility, which limits its usefulness in
in vivo studies and for therapeutic application.
Figure 1.
Structures of selected adenosine A2A receptor antagonists.
Figure 1.
Structures of selected adenosine A2A receptor antagonists.
An approach to increase water-solubility is the preparation of prodrugs, in which a polar moiety is attached to the drug, but can be cleaved off
in vivo by an enzymatic reaction to release the active drug [
10,
11]. Thus, we had developed a phosphate prodrug of MSX-2, named MSX-3 (
4) [
7,
8]. It was found that MSX-3 is stable in aqueous solution at pH 7, but readily cleaved by phosphatases present in biological membranes and fluids to liberate the drug MSX-2 [
7]. The phosphate prodrug MSX-3 has proven to be a very useful pharmacological tool allowing parenteral application in aqueous solutions due to its high water-solubility [e.g.
12,
13,
14,
15]. However, peroral application of phosphate prodrugs is less favourable because it is highly unlikely that the intact – highly polar – prodrug can be absorbed. Phosphoric acid esters usually undergo enzymatic hydrolysis prior to absorption and therefore phosphate prodrugs are typically used for intravenous administration. There is a recent example, however, showing that phosphate prodrugs can also increase the peroral bioavailability of slightly soluble drugs: fosamprenavir, the phosphate prodrug of the HIV protease inhibitor amprenavir, is readily dissolved in the intestine due to its high water-solubility, in contrast to its hardly soluble parent drug, and the phosphate is cleaved by phosphatases in the intestinal cell membranes allowing the drug to be absorbed [
16]. Another approach to increase water-solubility would be to introduce polar moieties into the molecule, however this has been shown to cause a decrease in potency as adenosine receptor antagonists and/or central nervous system (CNS) bioavailability of styrylxanthine derivatives [
17].
As an alternative to the phosphate prodrug approach, we considered introducing a different prodrug moiety into MSX-2 (
3) which would increase water-solubility, enhance peroral absorption, and release the drug not in the intestine already, but preferably after absorption. An amino acid ester prodrug approach was selected. Amino acid ester prodrugs of drugs which bear an alcoholic function have been reported to show enhanced bioavailability (i) because of increased water-solubility, and (ii) due to absorption by active amino acid carrier mechanisms [
18,
19,
20]. Valaciclovir is an example of such an amino acid ester prodrug with improved bioavailability. It has been reported that the
L-valine ester of acyclovir provided the best bioavailability among a series of several different amino acid esters [
21]. Since acyclovir and MSX-2 are structurally related molecules, both being N-substituted purine derivatives, we synthesized the
L-valine ester of MSX‑2,
L-valine-3-{8-[(
E)-2-[3-methoxyphenyl)-ethenyl]-7-methyl-1-propargylxanthine-3-yl}propyl ester hydrochloride (MSX-4,
9) and investigated its properties.
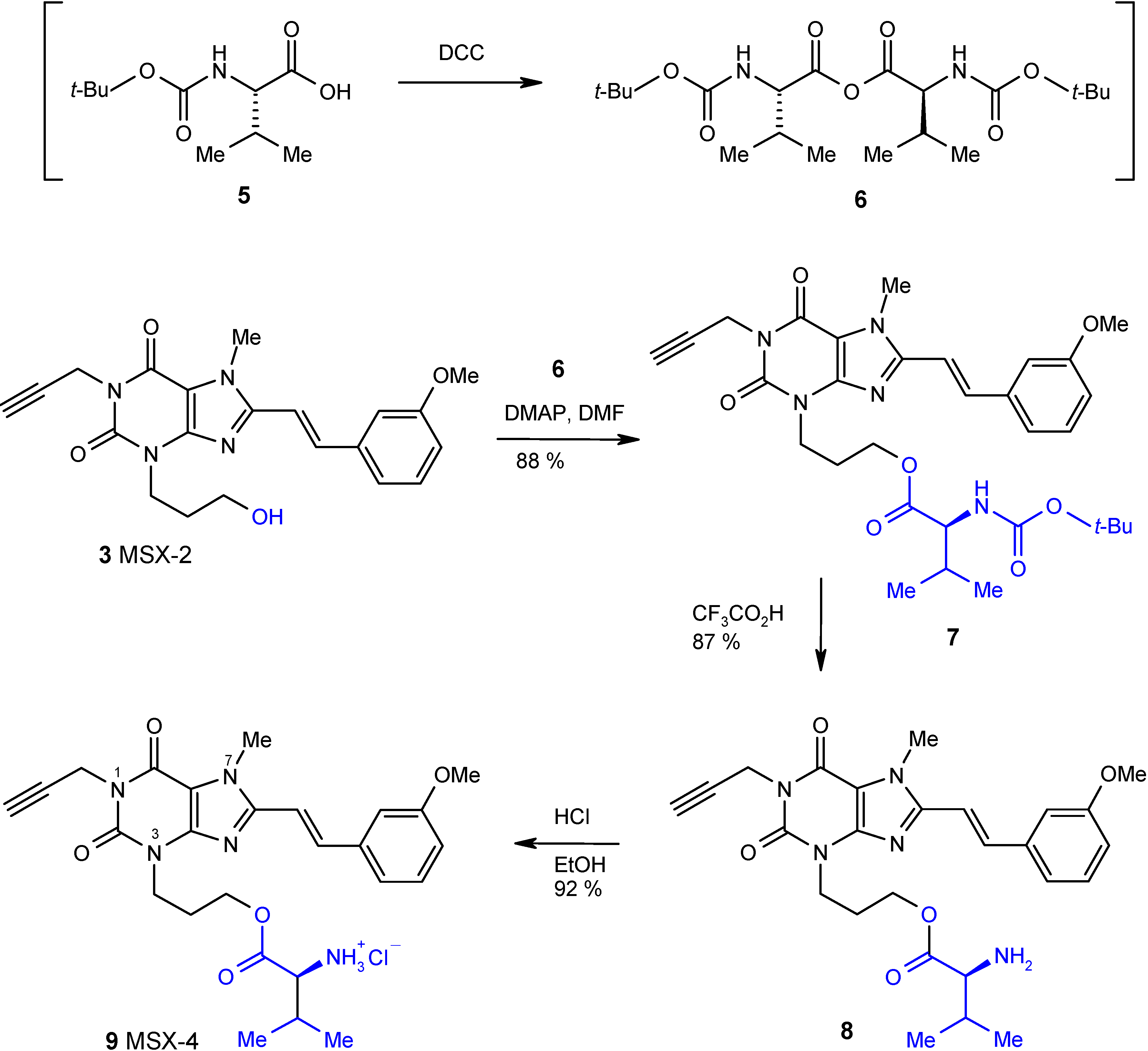
Results and Discussion
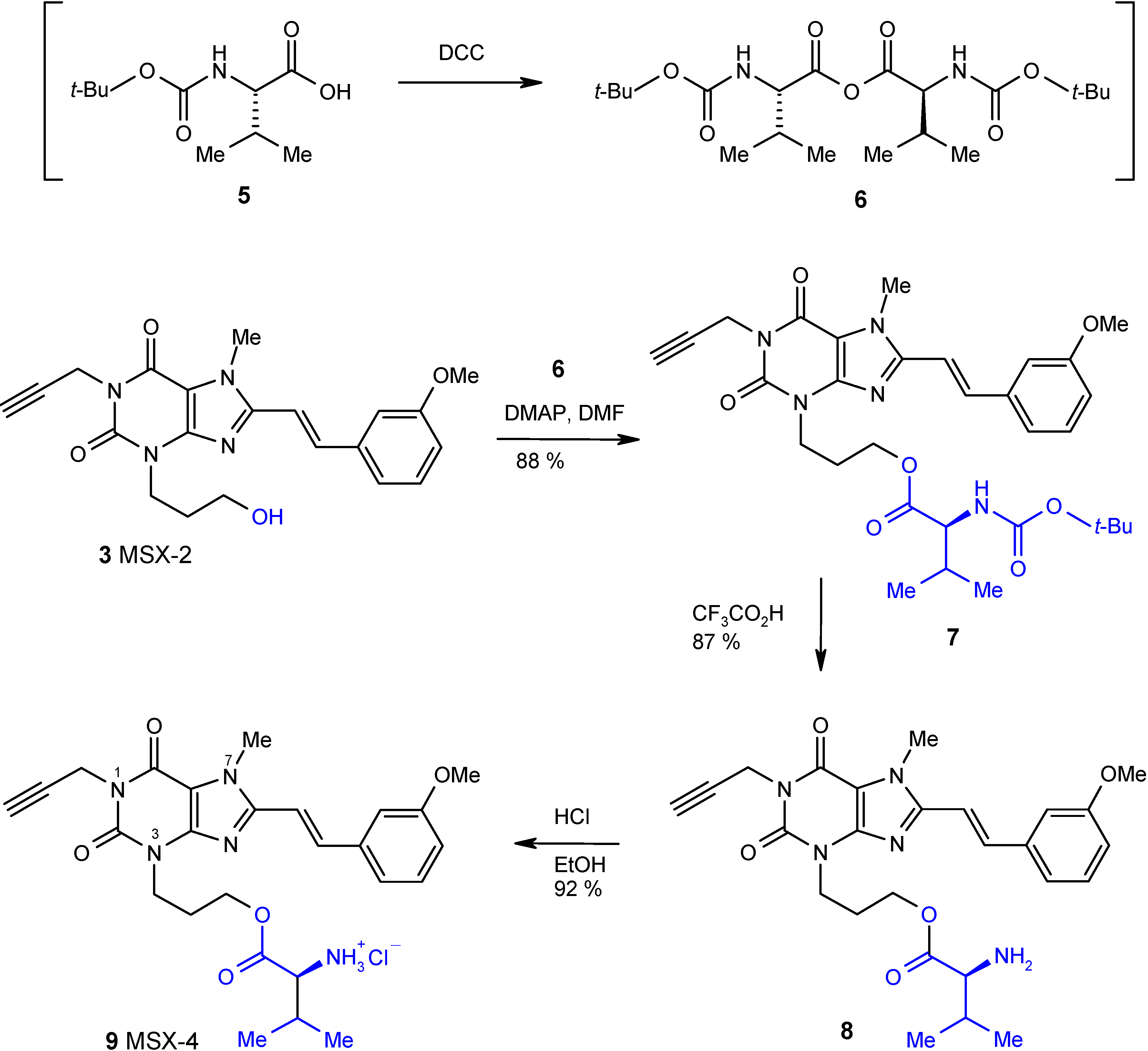
The synthesis of the target compound MSX-4 (
9) was carried out as depicted in
Scheme 1. The N-Boc-protected valine (compound
5) was converted to the corresponding anhydride
6 by the addition of dicyclohexylcarbodiimide (DCC) as a condensing agent.
Scheme 1.
Synthesis of valine ester prodrug MSX-4 (9).
Scheme 1.
Synthesis of valine ester prodrug MSX-4 (9).
Compound
6 was not isolated, but rather reacted immediately with MSX-2 (
3) [
7,
8] in dimethylformamide (DMF) in the presence of dimethylaminopyridine (DMAP) as a coupling reagent to yield the N-Boc-protected ester
7 in high yield. Deprotection was achieved by treatment with trifluoroacetic acid in dichloromethane at low temperature (0°C) yielding
8. Finally, the ester
8 was converted to its hydrochloride salt
9 by reaction with a saturated solution of gaseous hydrogen chloride in ethanol.
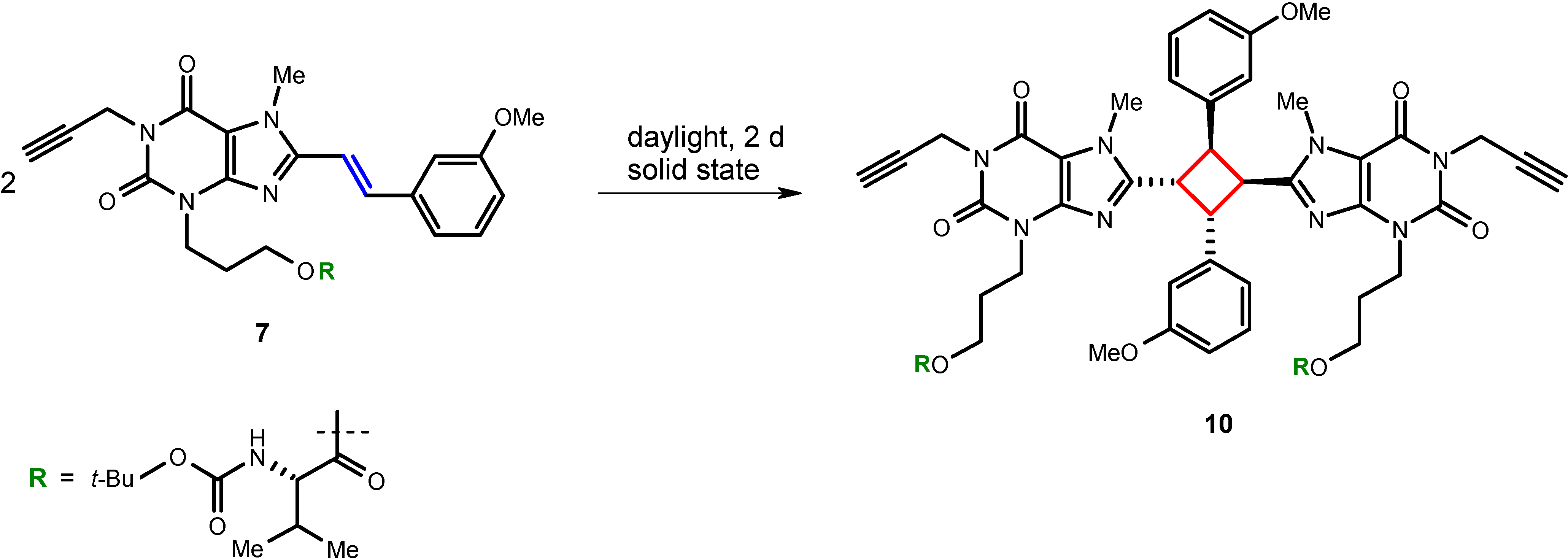
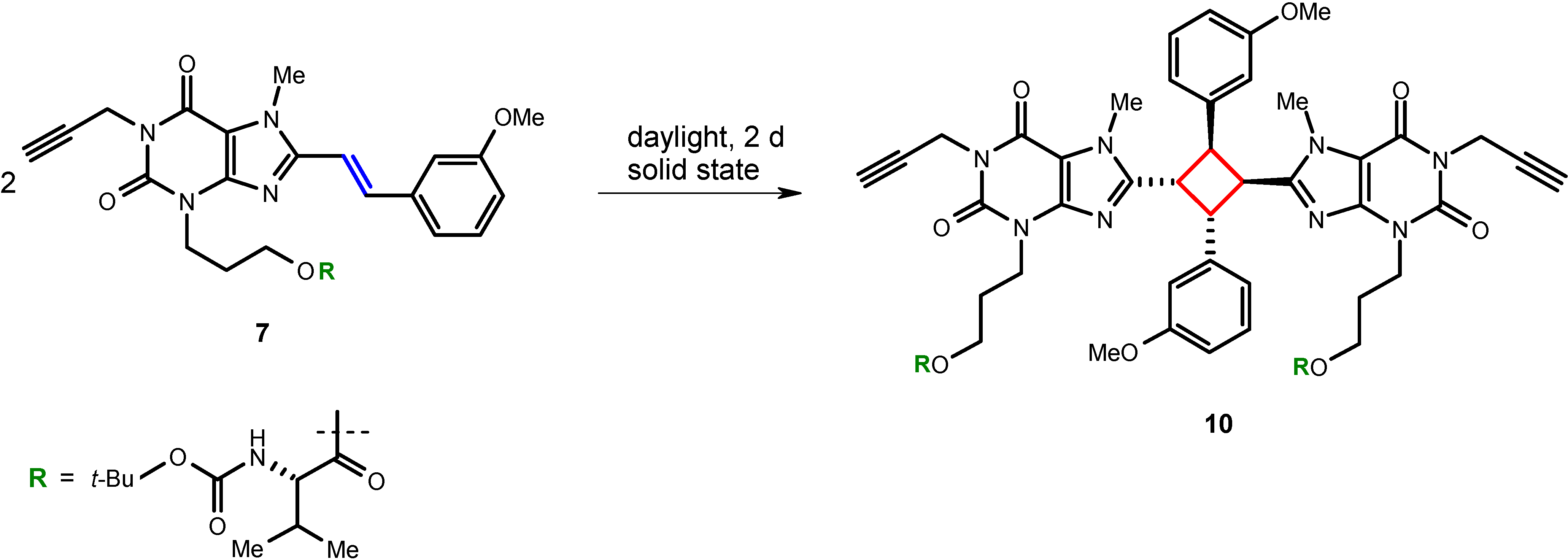
8-Styrylxanthine derivatives have previously been found to undergo light-induced E/Z-isomerization in dilute solutions [
17,
22]. Furthermore, in the solid state, photodimerization upon exposure to light has been observed [
8]. In fact, we found that the intermediate compound
7 also undergoes dimerization when stored unprotected from light in the laboratory yielding cyclobutane derivative
10. With regard to previous findings [
8], a head-to-tail
syn-configuration for the isolated compound can be expected. Therefore, styrylxanthine derivatives have to be strictly protected from light, in the solid state, as well as in solution.
Scheme 2.
Photodimerization of styrylxanthine derivative 7.
Scheme 2.
Photodimerization of styrylxanthine derivative 7.
The water-solubility of product
7 (MSX-4) was determined by UV spectroscopy and found to be 7.3 mg/mL (13.8 mM), which is extraordinarily high compared to most if not all high affinity adenosine receptor A
2A antagonists [
17]. It was in the same range as the solubility of the disodium salt of the phosphate prodrug MSX-3 (
4), which was determined to be 9 mg/mL (17 mM) [
7].
The chemical and enzymatic stability of
9 was investigated by capillary electrophoresis with UV detection. The amino acid ester prodrug was absolutely stable in aqueous solution at 25°C for at least 4 days (data not shown). It was subsequently incubated (i) with simulated gastric acid, and (ii) in the presence of pig liver carboxyl esterase.
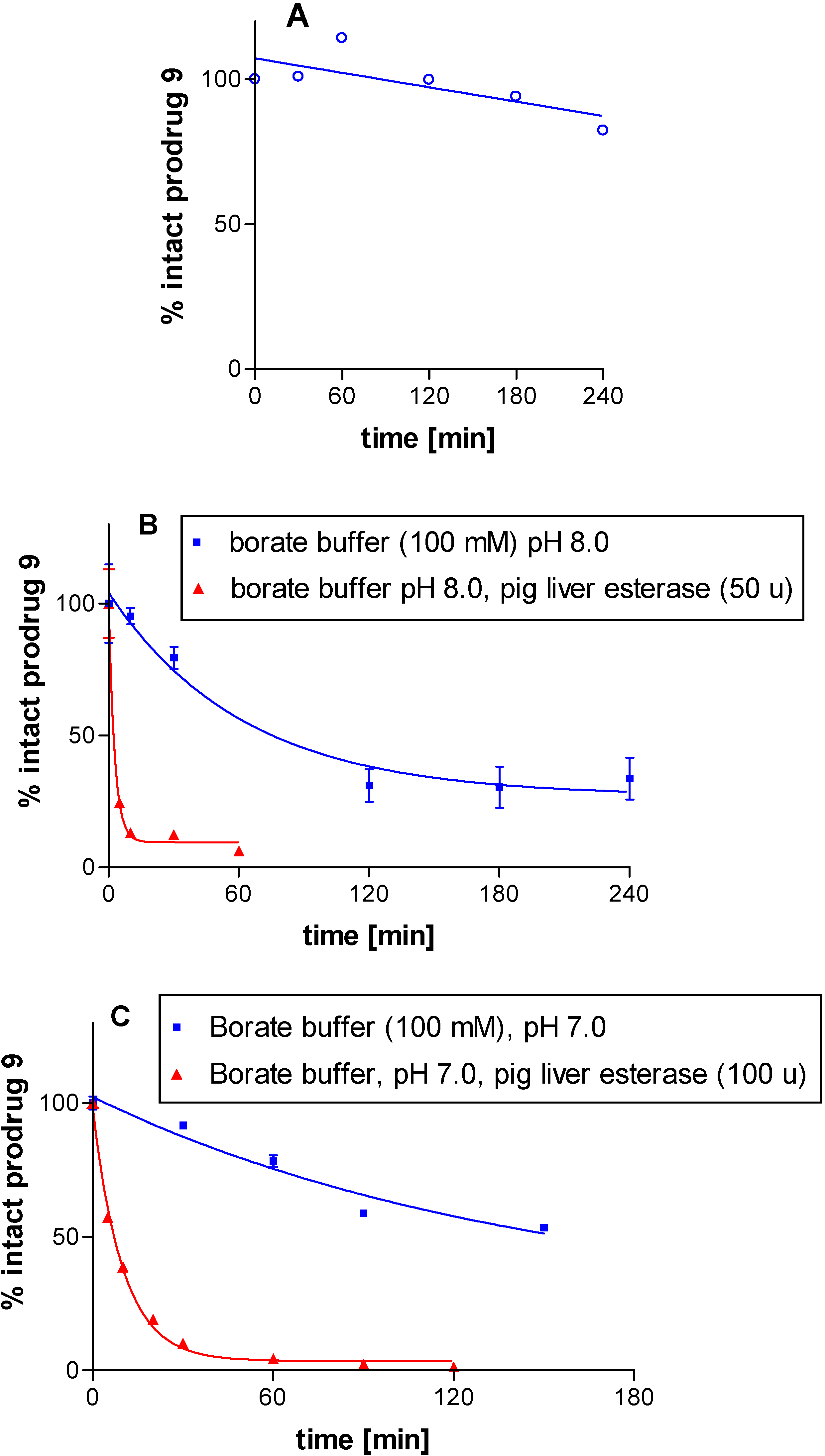
Figure 3A shows the time course for the degradation of prodrug
9 in simulated gastric acid (aqueous HCl solution pH 2 containing pepsin). The incubation was performed for 4 hours since drugs usually do not stay in the stomach any longer. Prodrug
9 was found to be relatively stable under these conditions: after 4 hours of incubation, 82% of the intact ester
9 could still be detected. The antiviral prodrug valaciclovir had also recently been shown to be stable at acidic pH values below 4, but to be slowly cleaved under basic conditions [
23].
Figure 3.
Stability of prodrug 9 (A) in artificial gastric acid (aq. HCl, pH 2, pepsin), (B) in borate buffer pH 8 in the presence and in the absence of pig liver esterase, and (C) in borate buffer pH 7 in the presence and in the absence of pig liver esterase. Data points represent results from 3 independent experiments ± SEM. In some cases the SEM value was smaller than the symbol.
Figure 3.
Stability of prodrug 9 (A) in artificial gastric acid (aq. HCl, pH 2, pepsin), (B) in borate buffer pH 8 in the presence and in the absence of pig liver esterase, and (C) in borate buffer pH 7 in the presence and in the absence of pig liver esterase. Data points represent results from 3 independent experiments ± SEM. In some cases the SEM value was smaller than the symbol.
Figure 3 shows the plots of the hydrolysis of
9 at 37°C by esterase (50 u) at pH 8 (
Figure 3B), and by twice the amount of esterase (100 u) at pH 7 (
Figure 3C). The prodrug was readily hydrolyzed in the presence of esterase with a half-life of 6.9 min (100 u, pH 7) and 1.9 min (50 u, pH 8), respectively. As expected, the hydrolysis was faster at the basic pH value of 8, which corresponds to the pH optimum of the enzyme [
24] than at pH 7. At pH 7, the prodrug appeared to be quite stable (half life: 101 min), while it seemed to be somewhat less stable at the more alkaline pH of 8 (half-life: 43 min). However, the disappearance of the prodrug in the absence of esterase may also be at least partly due to deprotonation at neutral to basic pH values and subsequent precipitation of the resulting uncharged molecule
8. Overall we could show that the amino acid ester
9 is an excellent substrate for pig liver esterase and should therefore also be cleaved
in vivo by non-specific esterases releasing the potent drug MSX-2 (
3). In addition to dramatically increasing the water-solubility of MSX-2, the gastrointestinal absorption of the
L-valine ester prodrug
9 may be further increased by active transport via amino acid transporters as previously demonstrated for other
L-amino acid prodrugs, especially
L-valine esters of drugs with a purine scaffold bearing alcoholic functions. Furthermore, CNS bioavailability may also be increased.
Experimental
General
Melting points were determined on a Büchi B-535 apparatus and are uncorrected. TLC was performed on silica gel-coated aluminum sheets Merck 60 F254, and spots were detected by UV light (254 nm). Column chromatography was performed on silica gel 63-200 µm (Merck). Elemental analyses were performed on a VarioEL instrument (Elementar Analysensysteme GmbH). (HR)MS spectra were performed on an MS-50 (Kratos) or a MAT95 (Thermo Quest) instrument. NMR spectra (
1H: 500 MHz,
13C: 126 MHz) were determined on a Bruker 500 spectrometer. The signals of the remaining protons of the deuterated solvents served as internal standard:
1H: δ DMSO-
d6 = 2.49, CDCl
3 = 7.24, MeOH-
d6 = 3.35, 4.78;
13C: DMSO-d
6 = 39.7, CDCl
3 = 77.0, MeOH-
d6 = 49.3. CE measurements were performed on a P/ACE capillary electrophoresis MDQ instrument (Beckman Coulter, Fullerton, CA, USA) equipped with a DAD detector. For incubations an Eppendorf Thermomixer Comfort was used. Optical rotation was measured on a Perkin-Elmer 241 polarimeter (Perkin-Elmer GmbH, Düsseldorf, Germany). DMF was dried over P
2O
5, CH
2Cl
2 was dried over CaCl
2. Ninhydrin solution was prepared as follows: 1 g ninhydrin, 2.5 g cadmium acetate and 10 mL acetic acid were dissolved in ethanol (final volume: 500 mL). The TLC sheets were sprayed with the ninhydrin solution and subsequently heated at 120° for 20 min. All other chemicals were used without further purification. MSX-2 (
3) was prepared as previously described [
7,
8].
[N-tert-Butoxycarbonyl-l-valine]-3-{8-[(E)-2-(3-methoxyphenyl)ethenyl)]-7-methyl-1-propargyl-xanthin-3-yl}propyl ester (7)
N-Boc-l-valine (1.07 g, 4.94 mmol) was dissolved in CH2Cl2 (20 mL) and dicyclohexyl-carbodiimide (DCC, 602 mg, 2.92 mmol, 0.59 eq) was added. The solution was stirred at rt for 4 h. Then it was filtered under an atmosphere of argon, and the filtrate was evaporated under vacuum. The colorless, oily residue was taken up in DMF (20 mL), and 3 (650 mg, 1.65 mmol) and dimethylaminopyridine (DMAP, 20.1 mg, 0.165 mmol), were added. The mixture was stirred at rt in the dark for ca. 24 h until TLC monitoring indicated complete conversion. The solution was evaporated and the resulting yellow oil was subjected to column chromatography (glass column 45 mm diameter, 46 cm height, 300 g of silica gel, elution with ethyl acetate-CH2Cl2-NH3 conc = 120:80:0.1). Yield: 88 % (863 mg, 1.45 mmol). Rf -value: 0.64 (ethyl acetate-CH2Cl2-NH3 conc = 120:80:0.1), orange-red color after spraying with ninhydrin solution (see above); Rf value (MSX-2): 0.34; 1H-NMR: δ (CDCl3) = 0.86 (d, J = 6.9 Hz, 3H, CHCH3), 0.93 (d, J = 6.9 Hz, 3H, CHCH3), 1.40 (s, 9H, OC(CH3)3), 2.15 (d br, 1H, Me2CH), 2.16 (t, J = 2.4 Hz, 1H, C≡CH), 2.19 (quint, J = 6.8 Hz, 2H, NCH2CH2CH2O), 3.83 (s, 3H, OCH3), 4.03 (s, 3H, NCH3), 4.17 (m br, 1H, NHCH), 4.25 (m, 4H, NCH2, OCH2), 4.76 (d, J = 2.4 Hz, 2H, C≡CCH2), 5.04 (d, J = 8.9 Hz, 1H, NH), 6.86 (d, J = 15.8 Hz, 1H, C=CH), 6.89 (dd, J = 8.1 Hz, 2.3 Hz, 1H, Harom), 7.07 (s, 1H, Harom), 7.17 (d, J = 7.8 Hz, 1H, Harom), 7.30 (dd, J = 8.1 Hz, 7.8 Hz, 1H, Harom), 7.73 (d, J = 15.8 Hz, 1H, C=CH); 13C-NMR: δ (CDCl3) = 17.6 (CH-CH3), 19.1 (CH-CH3), 27.2 (CH-CH3), 28.3 (C(CH3)3), 30.4 (CH2C≡C), 31.2 (NCH2CH2CH2O), 31.6 (NCH3), 40.6 (NCH2), 55.2 (OC(CH3)3), 55.3 (OCH3), 58.5 (NH-CH), 62.8 (OCH2), 70.4 (C≡CH), 78.7 (HC≡C), 107.8 (C5), 111.4 (C=CH), 112.7, 115.2, 120.0, 129.9, 136.6 (C8), 136.8, 138.7 (C=CH), 148.5 (C4), 150.3 (C2), 154.0, 155.7 [OC(O)N], 160.0 (C6), 172.3 (COO); IR (in CH2Cl2): ν (cm-1) = 3440 (N-H), 3300 (≡C-H), 3040 (=C-H), 2950 (C-H), 2920 (C-H), 2860 (C-H), 2450, 1700 (O-CO-NHR, C=O), 1660 (RN-CO-NR), 1590 (CO-NR), 1490 (C=C), 1360, 1160 (C-O), 1090, 1010, 860; [α]D = + 0.22 ° (c = 201.7 mg/mL in CHCl3, 21.5°C); EIMS: 593.3 (M+, 100 %), 519.2 (M+-H-Boc, 26 %), 395.2 (MSX-2); HRMS: calcd., 593.2850; found, 593.2837 (C31H39N5O7); mp 46 - 49 °C.
l-Valine-3-{8-[(E)-2-(3-methoxyphenyl)ethenyl]-7-methyl-1-propargylxanthin-3-yl}propyl ester (8)
Compound 7 (863 mg, 1.45 mmol) was dissolved at 0°C in freshly distilled CF3CO2H (15 mL) and stirred for 50 min in an ice-bath. The trifluoroacetic acid was then distilled off at 0°C in vacuo and the residue was purified by column chromatography (glass column 45 mm diameter, 46 cm height, 300 g of silica gel, ethyl acetate : CH2Cl2-NH3 conc = 200:300:1). Yield: 87 % (623 mg, 1.26 mmol); Rf value: 0.67 (CH2Cl2-MeOH =10:2); 0.83 (starting compound); 1H-NMR: δ (CDCl3) = 1.08 (d, J = 7.0 Hz, 3H, CHCH3), 1.09 (d, J = 7.0 Hz, 3H, CHCH3), 2.19 (t, J = 2.4 Hz, C≡CH), 2.27 (quint, J = 6.4 Hz, 2H, NCH2CH2CH2O), 2.38 [m, 1H, (CH3)2CH], 3.99 (d, J = 4.8 Hz, 1H, H2NCH), 3.84 (s, 3H, OCH3), 4.07 (s, 3H, NCH3), 4.28 (m, 4H, OCH2, NCH2), 4.73 (d, J = 2.2 Hz, 1H, C≡CCH2), 4.74 (d, J = 2.2 Hz, 1H, C≡CCH2), 6.88 (d, J = Hz, 1H, C=CH), 6.93 (dd, J = 8.3 Hz, 2.5 Hz, 1H, Harom), 7.06 (s, 1H, Harom), 7.15 (d, J = 8.0 Hz, 1H, Harom), 7.32 (dd, J = 8.3 Hz, 8.0 Hz, 1H, Harom), 7.73 (d, J = 16.0 Hz, 1H, C=CH); 13C-NMR: δ (CDCl3) = 17.3 (CH-CH3), 17.7 (CH-CH3), 29.6 (CH-CH3), 30.6 (CH2C≡C), 31.7 (NCH2CH2CH2O), 32.3 (NCH3), 40.3 (NCH2), 55.3 (OCH3), 59.0 (NH-CH), 63.7 (OCH2), 70.9 (C≡CH), 78.3 (HC≡C), 106.7 (C5), 110.7, 112.7, 115.5, 120.0, 130.0, 136.4 (C8), 139.3, 139.7, 147.7 (C4), 150.6 (C2), 153.7, 160.0 (C6), 171.5 (COO) (the unassigned signals belong to the eight vinyl and aromatic carbon atoms); Anal. calcd. for C26H31N5O5: C, 63.3; H, 6.33; N, 14, 2; found: C, 63.4; H, 6.60; N, 14.1; EIMS: 493.3 (M+, 59 %), 450.2 (M+-C3H7, 34 %), 395.2 (MSX-2, 43 %), 378.2 (M+-C5H8NO-OH, 100 %), 174.2 ([C11H12NO]+, 63 %), 72.2 ([C4H10N]+, 88 %); HRMS: calcd., 493.2325; found, 493.2329; mp 141 °C.
l-Valine-3-{8-[(E)-2-(3-ethoxyphenyl)ethenyl]-7-methyl-1-propargylxanthin-3-yl}propyl ester hydro- chloride (9)
Compound 8 (61 mg, 0.123 mmol) was dissolved at 0°C in 3 mL absolute ethanol and 10 mL of a cold saturated solution of HCl gas in ethanol, cooled to 0°C, was added. After 5 min the solvent was partly removed by rotary evaporation, and the precipitated product was filtered off. It is washed twice with 5 mL of CH2Cl2 each. The mother liquor was reduced by rotary evaporation, and the formed precipitate was washed again as described above. Both fractions contained the pure product.Yield: 92 % (60 mg, 0.113 mmol); Rf value: 0.21 (CH2Cl2-MeOH-AcOH = 10:1:0.1); 1H-NMR: δ (methanol-d6) = 1.12 (d, J = 6.9 Hz, 3H, CHCH3), 1.13 (d, J = 6.9 Hz, 3H, CHCH3), 2.28 (quin, J = 6.5 Hz, 2H, NCH2CH2CH2O), 2.35 [m, 1H, (CH3)2CH], 2.62 (t, J = 2.1 Hz, C≡CH), 3.89 (s, 3H, OCH3), 3.99 (d, J = 4.4 Hz, 1H, H3N+CH), 4.11 (s, 3H, NCH3), 4.32 (t, J = 6.8 Hz, 2H, OCH2), 4.43 (t, J = 6.2 Hz, 2H, NCH2), 4.76 (d, J = 2.1 Hz, 2H, C≡CCH2), 6.98 (dd, J = 8.2 Hz, 2.2 Hz, 1H, Harom), 7.25 (m, 3H, C=CH, 2 Harom), 7.35 (dd, J = 7.9 Hz, 7.8 Hz, 1H, ), 7.78 (d, J = 15.8 Hz, C=CH); (NH3+ protons are exchanged in methanol-d6); 13C-NMR: δ (methanol-d6) = 18.6 (CH-CH3), 18.8 (CH-CH3), 28.7 (CH-CH3), 31.2 (CH2C≡C)*, 31.6 (NCH2CH2CH2O), 32.5 (NCH3), 42.0 (NCH2), 56.2 (OCH3), 59.8 (NH-CH), 65.5 (OCH2), 72.1 (C≡CH), 80.1 (HC≡C), 109.4 (C5), 113.1, 114.0, 116.6, 121.6, 131.3, 138.5 (C8), 140.3, 149.6 (C4), 152.3, 152.4, 155.4 (C2), 161.9 (C6), 170.2 (COO); [α]D = -10.0 ° (c = 3.2 mg/mL, H2O, 21.0 °C); Anal. calcd. for C26H32ClN5O5·2.8 CH2Cl2: C, 49.4; H, 5.55; N, 11.1; found, C, 49.5; H, 5.74; N, 11.1; EIMS: 493.3 (M+, 23 %), 450.2 (M+ -C3H7, 13 %), 395.2 (M+-C5H8NO (MSX-2), 24 %), 378.2 (M+-C5H8NO -OH, 47 %), 174.1 ([C11H12NO]+, 42 %),72.1 ([C4H10N]+, 100 %); HRMS: calcd, 529.2092; found, 493.2315; mp 149 - 152 °C.
(1α, 2α, 3β, 4β)-1,3-Bis{3-[(N-tert-butoxycarbonyl-L-valine)-7-methyl-1-propargylxanthin-8-yl]propyl ester}-2,4-bis(3-methoxyphenyl)cyclobutane (10)
A sample solid 7 (90 mg, 0.152 mmol) was left in the laboratory near the window in the sunlight for 2 days and subsequently subjected to column chromatography (glass column, 25 mm diameter, 37 cm height, 70 g of silica gel; CH2Cl2-ethyl acetate = 20:1); Yield: 80 mg (0.0674 mmol). In addition starting compound 7 was recovered (8.0 mg, 0.0135 mmol). Rf value: 0.42 (CH2Cl2-MeOH = 10:1); 0.27 (compound 7); 1H-NMR: δ (CDCl3) = 0.87 (d, J = 7.0 Hz, 3H, CHCH3), 0.89 (d, J = 7.0 Hz, 3H, CHCH3), 0.96 (d, J = 6.7 Hz, 6 H, 2 CHCH3), 1.37 (s, 9H, OC(CH3)3), 1.42 (s, 9H, OC(CH3)3), 2.13 (m, 8 H, 2 Me2CH, 2 C≡CH, 2 NCH2CH2CH2O), 3.65 (s br, 3H, OCH3 or NCH3), 3.67 (s, 6H, NCH3 or OCH3), 3.68 (s, 3H, OCH3 or NCH3), 4.21 (m, 9H, 2 NCH2, 2 OCH2, CH), 4.28 (m, 1H, CH), 4.43 (dd, J = 10.1 Hz, 7.3 Hz, 1H, NHCH), 4.47 (m br, 1H, NHCH), 4.69 (d, J = 2.2 Hz, C≡CCH2), 4.98 (dd, J = 9.8 Hz, 7.3 Hz, 2H, 2 CH), 5.04 (d, J = 9.1 Hz, 2H, NH2), 6.67 (ddd, J = 7.6 Hz, 5.4 Hz, 1.9 Hz, 2H, Harom), 6.75 (m, 2H, Harom), 6.77 (d J = 7.6 Hz, 2H, Harom), 7.09 (m, 2H, Harom); 13C-NMR: δ (CDCl3) = 17.6 (CH-CH3), 19.05 (CH-CH3), 19.08 (CH-CH3), 27.3 (CH3-CH), 27.4 (CH3-CH), 28.26 (C(CH3)3), 28.3 (C(CH3)3), 30.4 (C≡CCH2), 31.2 (NCH2CH2CH2O), 31.3 (NCH2CH2CH2O), 31.7 (NCH3), 40.4 (NCH2), 40.5 (NCH2), 44.3 (CH), 44.4 (CH), 55.10 (OC(CH3)3), 55.13 (OC(CH3)3), 58.56 (NH-CH), 58.6 (NH-CH), 62.7 (OCH2), 62.9 (OCH2), 70.3 (C≡CH), 78.7 (HC≡C), 107.7 (C5), 107.8 (C5), 112.0/112.1, 114.1/114.3, 119.8, 129.6, 139.8/139.9 (2 C8), 147.4/147.5(2 C4), 150.4 (2 C2), 152.6/152.7, 153.9/154.0, 155.6/155.7 (2 OC(O)N), 159.6/159.7 (2 C6), 172.3/172.4 (2 CO2); EIMS (C62H78N10O14, MR = 1187.36 g/mol): 788.5 (M+- 2 C10H16NO3, 5 %), 394.2 (MSX-2, 100 %) (the amino acid esters were cleaved off).
Determination of the water solubility of 9 by UV spectroscopy
A saturated solution of 9 was prepared by suspending the compound (15.1 mg, 28.5 μmol) in H2O (1 mL). The suspension was subsequently centrifuged twice at 4000 rpm for 30 min each. The clear supernatant was appropriately diluted in water analyzed by UV spectroscopy. A standard curve was determined using known concentrations of 9: A stock solution was prepared (1 mg/10 mL water corresponding to 189 µmol/L) and further diluted to obtain six different concentrations ranging from 9.45 µmol/L to 94.5 µmol/L. The concentration of the saturated solution was determined to be 13.8 ± 0.76 μmol/mL or 13.8 mmol/L corresponding to 7.3 ± 0.4 mg/mL.
Stability studies using capillary electrophoresis with UV detection
A fused-silica capillary of 60 cm length, internal diameter 75 µm was used; the temperature was kept at 25°C. A 50 mM phosphate (KH2PO4) buffer pH 6.1 was used as running buffer, and a constant current of 90 µA was applied. Samples were injected for 5 s with a pressure of 0.5 p.s.i. Injection was made from the anodic side of the capillary. Compounds were detected by UV absorption at 254 nm. Before each measurement the capillary was washed with 0.1 M NaOH, water, and running buffer, for 1 min each. The stability studies were performed in a volume of 500.0 µL at a concentration of 500 µg/mL in borate buffer (100 mM) at pH values of 7.0 and 8.0, respectively. An aqueous solution of quinidine sulfate (50.0 µg/mL) was used as internal standard. Samples (50.0 µL) were drawn and diluted with the internal standard solution (1:10, 450 µL). From the resulting solution, 50 µL were taken and diluted 1:1 with demineralized water. The final concentration of 9 in the CE measurements was therefore 25.0 µg/mL and for quinidine sulfate 22.5 µg/mL. Samples were taken after 10, 30, 60, and 90 min, or after 1, 2, 3 and 4 d. Each of the three independent experiments was performed with exclusion of light.
Enzymatic hydrolysis
Carboxyl esterase from pig liver (Sigma, E2884, in 2 M (NH4)2SO4 solution pH 8.0) was used for the experiments. The enzyme suspension was thoroughly shaken prior to use. An amount of 0.27 µL of enzyme suspension corresponded to 1 unit (defined as the amount of enzyme which hydrolyzes 1.0 µmol of butyric acid ethyl ester at pH 8.0, 25°C within 1 min). Samples for studying enzymatic hydrolysis were prepared as described above for stability studies. The sample (50 µL) for the time zero was taken before the addition of the enzyme. To the remaining 450 µL of solution 100 u (27.0 µL undiluted), or 50 u (27.0 µL, diluted 1:1 with buffer) of enzyme solution was added, and the mixture was incubated at 37 °C. After addition of the enzyme solution, the resulting substrate concentration was 472 µg/mL, and the reaction volume was 477 µL. After 5, 10, 20, 30, 60, 90 and 120 min samples (50 µl) were taken and diluted 1:10 with 450 µL of a solution of the internal standard and heated at 99°C for 10 min in order to inactivate the enzyme. Heating at 99°C for 10 min did not lead to hydrolysis of 9. After cooling to room temperature, 50.0 µL of the solutions were further diluted with 50.0 µL of demineralized water and subjected to analysis by capillary electrophoresis. The final concentration of quinidine sulfate was 22.5 µg/mL and of 9 23.6 µg/mL (at time zero). In all cases, three independent experiments were performed, with exclusion of light.


{kind=link}
{kind=link}
{kind=link}
{kind=link}