Cholesterylbutyrate Solid Lipid Nanoparticles as a Butyric Acid Prodrug

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Theoretical and Experimental Background
2.1 Solid Lipid Nanoparticles
2.1.1 SLNs from warm microemulsions
2.1.2 Some peculiarities of SLNs
2.1.3 Administration routes of SLNs
2.2 Butyrate
2.2.1 Anti-inflammatory properties.
2.2.2 Anti-cancer effects.
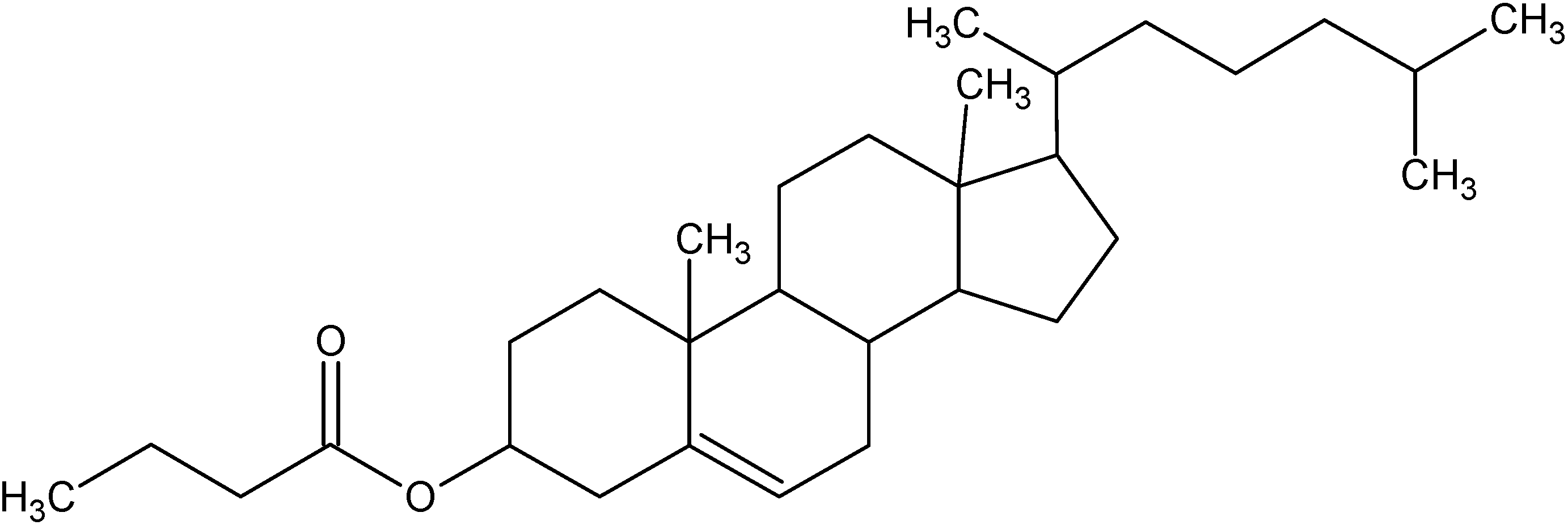
3. Cholesterylbutyrate solid lipid nanoparticles
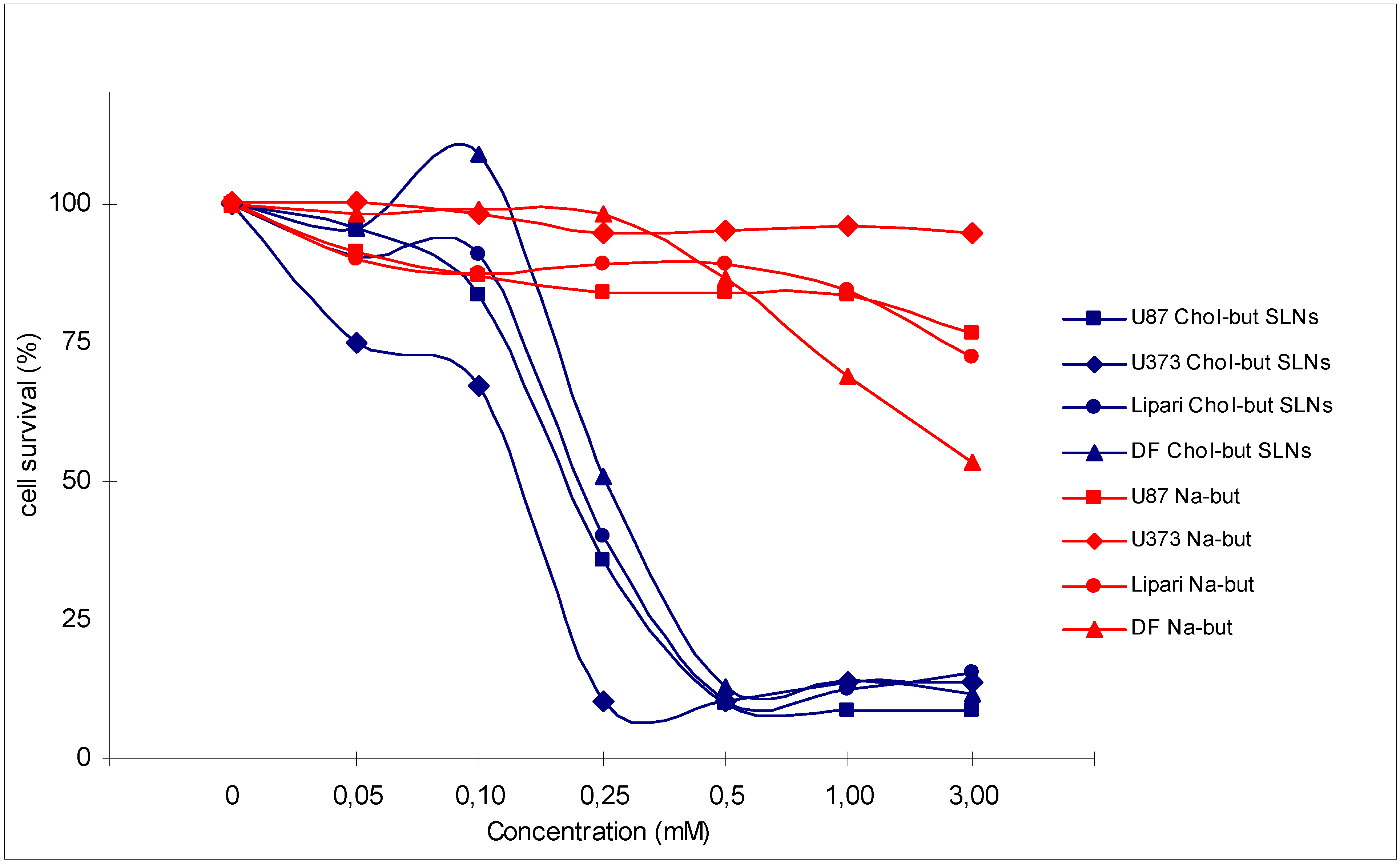
3.1 Anti-inflammatory effects
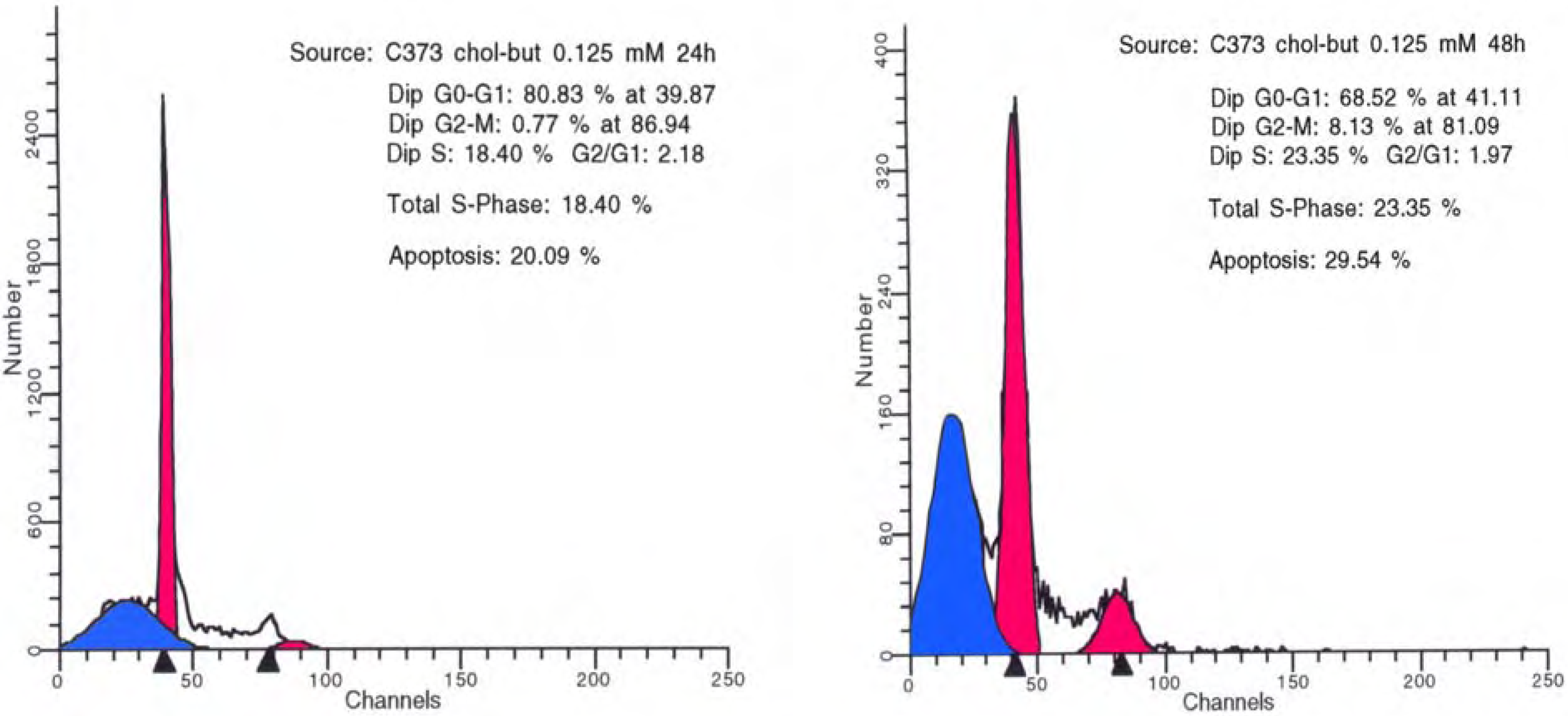
3.2 Antineoplastic effects
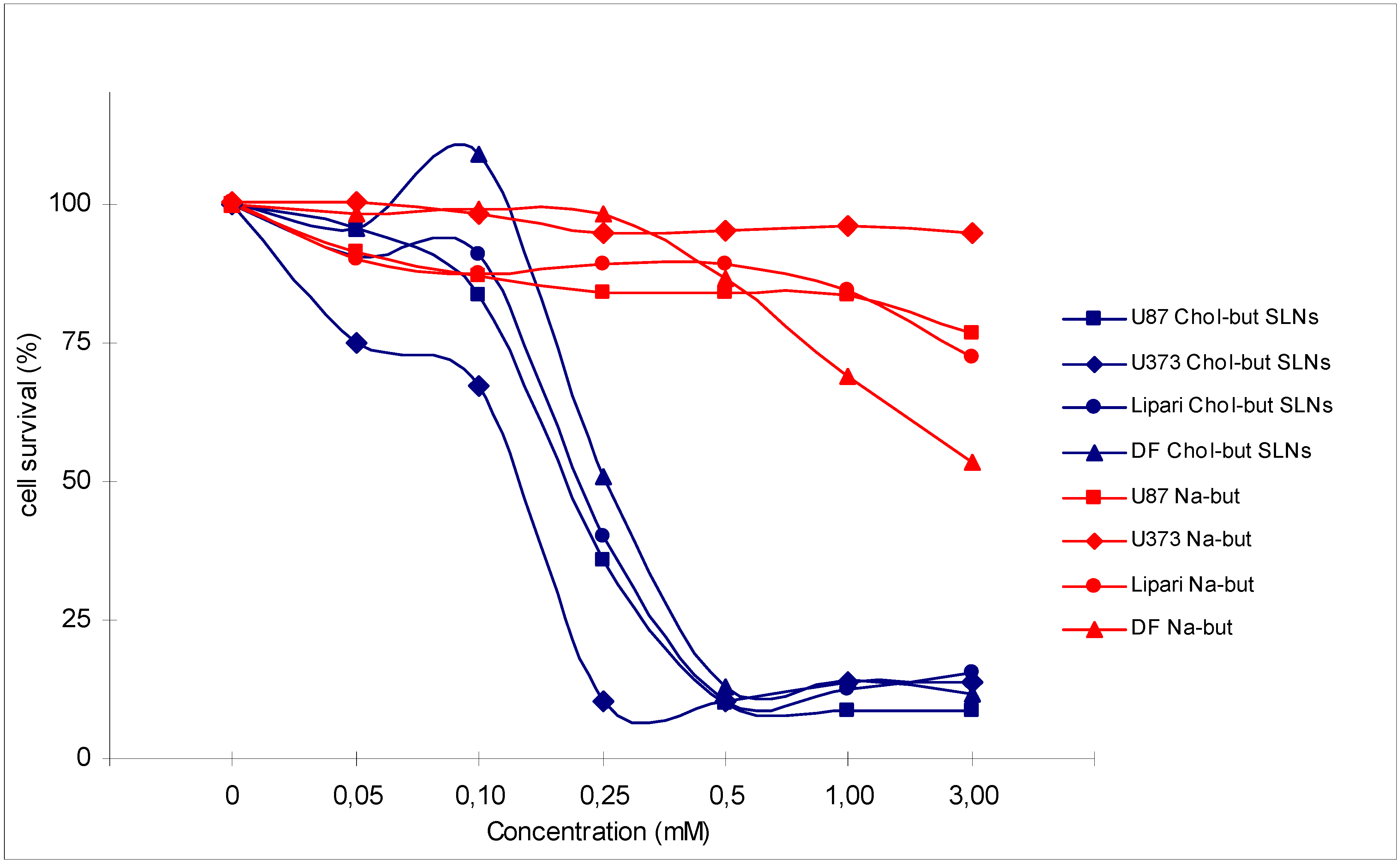

4. Discussion – Conclusions
Acknowledgements
References and Notes
- Gasco, M.R. Lipid nanoparticles: perspectives and challenges. Adv. Drug Deliv. Rev. 2007, 59, 377–378. [Google Scholar] [CrossRef]
- Müller, R.H. Lipid nanoparticles: recent advances. Adv. Drug Deliv. Rev. 2007, 59, 375–376. [Google Scholar] [CrossRef]
- Miller, A.A.; Kurschel, E.; Osieka, R.; Schmidt, C.G. Clinical pharmacology of sodium butyrate in patients with acute leukemia. Eur. J. Cancer Clin. Oncol. 1987, 23, 1283–1287. [Google Scholar] [CrossRef]
- Perrine, S.P.; Ginder, G.D.; Faller, D.V.; Dover, G.H.; Ikuta, T.; Witkowska, H.E.; Cai, S.P.; Vichinsky, E.P.; Olivieri, N.F. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the beta-globin disorders. N. Engl. J. Med. 1993, 328, 81–86. [Google Scholar] [CrossRef]
- Newmark, H.L.; Young, C.W. Butyrate and phenylacetate as differentiating agents: practical problems and opportunities. J. Cell Biochem. Suppl. 1995, 22, 247–253. [Google Scholar] [CrossRef]
- Pouillart, P.R. Role of butyric acid and its derivatives in the treatment of colorectal cancer and hemoglobinopathies. Life Sci. 1998, 63, 1739–1760. [Google Scholar] [CrossRef]
- Egorin, M.J.; Yuan, Z.M.; Sentz, D.L.; Plaisance, K.; Eiseman, J.L. Plasma pharmacokinetics of butyrate after intravenous administration of sodium butyrate or oral administration of tributyrin or sodium butyrate to mice and rats. Cancer Chemother. Pharmacol. 1999, 43, 445–453. [Google Scholar] [CrossRef]
- Santini, V.; Zozzini, A.; Scappini, B.; Grossi, A.; Rossi Ferrini, P. Searching for the magic bullet against cancer: the butyrate saga. Leuk. Lymphoma. 2001, 42, 275–289. [Google Scholar] [CrossRef]
- Chen, J.S.; Faller, D.V.; Spanjaard, R.A. Short-chain fatty acid inhibitors of histone deacetylases: promising anticancer therapeutics? Curr. Cancer Drug Targets 2003, 3, 219–236. [Google Scholar] [CrossRef]
- Miller, S.J. Cellular and physiological effects of short-chain fatty acids. Mini Rev. Med. Chem. 2004, 4, 839–845. [Google Scholar] [CrossRef]
- Serpe, L.; Laurora, S.; Pizzimenti, S.; Ugazio, E.; Ponti, R.; Canapaio, R.; Briatore, F.; Barrera, G.; Gasco, M.R.; Bernengo, M.G.; Eandi, M.; Zara, G.P. Cholesteryl butyrate solid lipid nanoparticles as a butyric acid pro-drug: effects on cell proliferation, cell-cycle distribution and c-myc expression in human leukemic cells. Anticancer Drugs. 2004, 15, 525–536. [Google Scholar] [CrossRef]
- Pellizzaro, C.; Cordini, D.; Morel, S.; Ugazio, E.; Gasco, M.R.; Dandone, M.G. Cholesteryl butyrate in solid lipid nanospheres as an alternative approach for butyric acid delivery. Anticancer Res. 1999, 19, 3921–3925. [Google Scholar]
- Salomone, B.; Ponti, R.; Gasco, M.R.; Ugazio, E.; Quaglino, P.; Osella-Abate, S.; Bernengo, M.G. In vitro effects of cholesteryl butyrate solid lipid nanospheres as a butyric acid pro-drug on melanoma cells: evaluation of antiproliferative activity and apoptosis induction. Clin. Exp. Metastasis 2001, 18, 663–673. [Google Scholar]
- Ugazio, E.; Marengo, E.; Pellizzaro, C.; Coradini, D.; Peira, E.; Daidone, M.G.; Gasco, M.R. The effect of formulation and concentration of cholesteryl butyrate solid lipid nanospheres (SLN) on NIH-H460 cell proliferation. Eur. J. Pharm. Biopharm. 2001, 52, 197–202. [Google Scholar] [CrossRef]
- Dianzani, C.; Cavalli, R.; Zara, G.P.; Gallicchio, M.; Lombardi, G.; Gasco, M.R.; Panzanelli, P.; Fantozzi, R. Cholesteryl butyrate solid lipid nanoparticles inhibit adhesion of human neutrophils to endothelial cells. Br. J. Pharmacol. 2006, 148, 648–656. [Google Scholar] [CrossRef]
- Peira, E.; Marzola, P.; Podio, V.; Aime, S.; Sbarbati, A.; Gasco, M.R. In vitro and in vivo study of solid lipid nanoparticles loaded with superparamagnetic iron oxide. J. Drug Target. 2003, 11, 19–24. [Google Scholar] [CrossRef]
- Mauro, A.; Miglietta, A.; Cavalli, R.; Bocca, C.; Guido, M.; Di Sapio, A.; Pradotto, L.; Schiffer, D.; Gasco, M.R. Enhanced cytotoxycity of Paclitaxel incorporated in Solid Lipid Nanoparticles against human glioma cells. Proceed. Int’l. Symp. Control. Rel. Bioact. Mater. 2000, 27, 377–378. [Google Scholar]
- Miglietta, A.; Cavalli, R.; Bocca, C.; Gabriel, L.; Gasco, M.R. Cellular uptake and cytotoxicity of solid lipid nanospheres (SLN) incorporating doxorubicin or paclitaxel. Int. J. Pharm. 2000, 210, 61–67. [Google Scholar] [CrossRef]
- Serpe, L.; Guido, M.; Canaparo, R.; Muntoni, E.; Cavalli, R.; Panzanelli, P.; Della Pepal, C.; Bargoni, A.; Mauro, A.; Gasco, M.R.; Eandi, M.; Zara, G.P. Intracellular accumulation and cytotoxicity of doxorubicin with different pharmaceutical formulations in human cancer cell lines. J. Nanosci. Nanotechnol. 2006, 6, 3062–3069. [Google Scholar] [CrossRef]
- Brioschi, A.; Zenga, F.; Zara, G.P.; Gasco, M.R.; Ducati, A.; Mauro, A. Solid lipid nanoparticles: could they help to improve the efficacy of pharmacologic treatments for brain tumors? Neurol. Res. 2007, 29, 324–330. [Google Scholar] [CrossRef]
- Bargoni, A.; Cavalli, R.; Caputo, O.; Fundarò, A.; Gasco, M.R.; Zara, G.P. Solid lipid nanoparticles in lymph and plasma after duodenal administration to rats. Pharm. Res. 1998, 15, 745–750. [Google Scholar] [CrossRef]
- Bocca, C.; Caputo, O.; Cavalli, R.; Gabriel, L.; Miglietta, A.; Gasco, M.R. Phagocytic uptake of fluorescent stealth and non-stealth solid lipid nanoparticles. Int. J. Pharm. 1998, 175, 185–193. [Google Scholar] [CrossRef]
- Podio, V.; Zara, G.P.; Carazzonet, M.; Cavalli, R.; Gasco, M.R. Biodistribution of stealth and non-stealth solid lipid nanospheres after intravenous administration to rats. J. Pharm. Pharmacol. 2000, 52, 1057–1063. [Google Scholar] [CrossRef]
- Zara, G.P.; Cavalli, R.; Bargoni, A.; Fundarò, A.; Vighetto, D.; Gasco, M.R. Intravenous administration to rabbits of non-stealth and stealth doxorubicin-loaded solid lipid nanoparticles at increasing concentrations of stealth agent: pharmacokinetics and distribution of doxorubicin in brain and other tissues. J. Drug Target. 2002, 10, 327–335. [Google Scholar] [CrossRef]
- Cavalli, R.; Bargoni, A.; Podio, V.; Muntoni, E.; Zara, G.P.; Gasco, M.R. Duodenal administration of solid lipid nanoparticles loaded with different percentages of tobramycin. J. Pharm. Sci. 2003, 92, 1085–1094. [Google Scholar] [CrossRef]
- Zara, G.P.; Bargoni, A.; Cavalli, R.; Fundarò, A.; Vighetto, D.; Gasco, M.R. Pharmacokinetics and tissue distribution of idarubicin-loaded solid lipid nanoparticles after duodenal administration to rats. J. Pharm. Sci. 2002, 91, 1324–1333. [Google Scholar] [CrossRef]
- Priano, L.; Esposti, D.; Esposti, R.; Castagna, G.; De Medici, C.; Fraschini, F.; Gasco, M.R.; Mauro, A. Solid Lipid Nanoparticles incorporating melatonin as a new model for sustained oral and transdermal delivery systems. J. Nanosci. Nanotechnol. 2007, 7, 1–6. [Google Scholar]
- Cummings, J.H. Short chain fatty acids in the human colon. Gut 1981, 22, 763–779. [Google Scholar] [CrossRef]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef]
- Gupta, N.; Martin, P.M.; Prasad, P.D.; Ganapathy, V. SLC5A8 (SMCT1)-mediated transport of butyrate forms the basis for the tumor suppressive function of the transporter. Life Sci. 2006, 78, 2419–2425. [Google Scholar] [CrossRef]
- Roediger, W.E. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 1980, 21, 793–798. [Google Scholar] [CrossRef]
- Kruh, J. Effects of sodium butyrate, a new pharmacological agent, on cells in culture. Mol. Cell Biochem. 1982, 42, 65–82. [Google Scholar]
- Jaskiewicz, J.; Zhao, Y.; Hawes, J.W.; Shimomura, Y.; Crabb, D.W.; Harris, R.A. Catabolism of isobutyrate by colonocytes. Arch. Biochem. Biophys. 1996, 327, 265–270. [Google Scholar] [CrossRef]
- Bach Knudsen, K.E.; Serena, A.; Canibe, N.; Juntunen, K.S. New insight into butyrate metabolism. Proc. Nutr. Soc. 2003, 62, 81–86. [Google Scholar] [CrossRef]
- Sartor, R.B. Pathogenesis and immune mechanism of chronic inflammatory bowel diseases. Am. J. Gastroenterol. 1997, 92 (12 Suppl.), 5S–11S. [Google Scholar]
- Brynskov, J.; Nielsen, O.H.; Ahnfelt-Rønne, I.; Bendtzen, K. Cytokines (immunoinflammatory hormones) and their natural regulation in inflammatory bowel disease (Crohn’s disease and ulcerative colitis): a review. Dig. Dis. 1994, 12, 290–304. [Google Scholar] [CrossRef]
- Fuss, I.J. Cytokines network in inflammatory bowel disease. Curr. Drug Targets Inflamm. Allergy 2003, 2, 101–112. [Google Scholar] [CrossRef]
- Venkatraman, A.; Ramakrishna, B.S.; Shaji, R.V.; Kumar, N.S.; Pulimood, A.; Patra, S. Amelioration of dextran sulfate colitis by butyrate: role of heat shock protein 70 and NF-kappaB. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G177–G184. [Google Scholar]
- Song, M.; Xia, B.; Li, J. Effects of topical treatment of sodium butyrate and 5-aminosalicylic acid on expression of trefoil factor 3, interleukin 1beta, and nuclear factor kappaB in trinitrobenzene sulphonic acid induced colitis in rats. Postgrad. Med. J. 2006, 82, 130–135. [Google Scholar] [CrossRef]
- Vernia, P.; Marcheggiano, A.; Caprilli, R.; Frieri, G.; Corrao, G.; Valpiani, D.; Di Paolo, M.C.; Paoluzi, P.; Torsoli, A. Short-chain fatty acid topical treatment in distal ulcerative colitis. Aliment. Pharmacol. Ther. 1995, 9, 309–313. [Google Scholar]
- Vernia, P.; Annese, V.; Bresci, G.; d’Albasio, G.; D'Incà, R.; Giaccari, S.; Ingrosso, M.; Mansi, C.; Riegler, G.; Valpiani, D.; Caprilli, R.; Gruppo Italiano per lo Studio del Colon and del Retto. Topical butyrate improves efficacy of 5-ASA in refractory distal ulcerative colitis: results of a multicentre trial. Eur. J. Clin. Invest. 2003, 33, 244–248. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Morera, R.; Ciccocioppo, R.; Cazzola, P.; Gotti, S.; Tinozzi, F.P.; Tinozzi, S.; Corazza, G.R. Oral butyrate for mildly to moderately active Crohn's disease. Aliment. Pharmacol. Ther. 2005, 22, 789–794. [Google Scholar] [CrossRef]
- Segain, J.P.; Raingeard de la Blétière, D.; Bourreille, A.; Leray, V.; Gervois, N.; Rosales, C.; Ferrier, L.; Bonnet, C.; Blottière, H.M.; Galmiche, J.P. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut 2000, 47, 397–403. [Google Scholar] [CrossRef]
- Cavaglieri, C.R.; Nishiyama, A.; Fernandes, L.C.; Curi, R.; Miles, E.A.; Calder, P.C. Differential effects of short-chain fatty acids on proliferation and production of pro- and anti-inflammatory cytokines by cultured lymphocytes. Life Sci. 2003, 73, 1683–1690. [Google Scholar] [CrossRef]
- Nancey, S.; Bienvenu, J.; Coffin, B.; Andre, F.; Descos, L.; Flourié, B. Butyrate strongly inhibits in vitro stimulated release of cytokines in blood. Dig. Dis. Sci. 2002, 47, 921–928. [Google Scholar] [CrossRef]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: a study with relevance to inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 2826–2832. [Google Scholar]
- Menzel, T.; Lührs, H.; Zirlik, S.; Schauber, J.; Kudlich, T.; Gerke, T.; Gostner, A.; Neumann, M.; Melcher, R.; Scheppach, W. Butyrate inhibits leukocyte adhesion to endothelial cells via modulation of VCAM-1. Inflamm. Bowel Dis. 2004, 10, 122–128. [Google Scholar] [CrossRef]
- Zapolska-Downar, D.; Siennicka, A.; Kaczmarczyk, M.; Kołodziej, B.; Naruszewicz, M. Butyrate inhibits cytokine-induced VCAM-1 and ICAM-1 expression in cultured endothelial cells: the role of NF-kappaB and PPARalpha. J. Nutr. Biochem. 2004, 15, 220–228. [Google Scholar] [CrossRef]
- Balakin, K.V.; Ivanenkov, Y.A.; Kiselyov, A.S.; Tkachenko, S.E. Histone deacetylase inhibitors in cancer therapy: latest developments, trends and medicinal chemistry perspective. Anticancer Agents Med. Chem. 2007, 7, 576–592. [Google Scholar] [CrossRef]
- Mehnert, J.M.; Kelly, W.K. Histone deacetylase inhibitors: biology and mechanism of action. Cancer J. 2007, 13, 23–29. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Bhalla, K.N. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J. Clin. Oncol. 2005, 23, 3971–3993. [Google Scholar] [CrossRef]
- Sekhavat, A.; Sun, J.M.; Davie, J.R. Competitive inhibition of histone deacetylase activity by trichostatin A and butyrate. Biochem. Cell. Biol. 2007, 85, 751–758. [Google Scholar] [CrossRef]
- Gray, S.G.; Ekström, T.J. The human histone deacetylase family. Exp. Cell Res. 2001, 262, 75–83. [Google Scholar] [CrossRef]
- Miller, T.A.; Witter, D.J.; Belvedere, S. Histone deacetylase inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [Google Scholar] [CrossRef]
- Zhu, W.G.; Otterson, G.A. The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Curr. Med. Chem. Anticancer Agents 2003, 3, 187–199. [Google Scholar] [CrossRef]
- Marks, P.A.; Richon, V.M.; Miller, T.; Kelly, W.K. Histone deacetylase inhibitors. Adv. Cancer Res. 2004, 91, 137–168. [Google Scholar] [CrossRef]
- Marks, P.A.; Richon, V.M.; Rifkind, R.A. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 2000, 92, 1210–1216. [Google Scholar] [CrossRef]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef]
- Drummond, D.C.; Noble, C.O.; Guo, Z.; Hong, K.; Park, J.W.; Kirpotin, D.B. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006, 66, 3271–3277. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Licht, J.D. Histone deacetylase inhibitors in cancer therapy: is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef]
- Entin-Meer, M.; Rephaeli, A.; Yang, X.; Nudelman, A.; VandenBerg, S.R.; Haas-Kogan, D.A. Butyric acid prodrugs are histone deacetylase inhibitors that show antineoplastic activity and radiosensitizing capacity in the treatment of malignant gliomas. Mol. Cancer Ther. 2005, 4, 1952–1961. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef]
- Jiang, Z.; Sharfstein, S.T. Sodium butyrate stimulates monoclonal antibody over-expression in CHO cells by improving gene accessibility. Biotechnol. Bioeng. 2007. [Google Scholar]
- Litvak, D.A.; Evers, B.M.; Hwang, K.O.; Hellmich, M.R.; Ko, T.C.; Townsend, C.M., Jr. Butyrate-induced differentiation of Caco-2 cells is associated with apoptosis and early induction of p21Waf1/Cip1 and p27Kip1. Surgery 1998, 124, 161–169. [Google Scholar] [CrossRef]
- Lallemand, F.; Courilleau, D.; Buquet-Fagot, C.; Atfi, A.; Montagne, M.N.; Mester, J. Sodium butyrate induces G2 arrest in the human breast cancer cells MDA-MB-231 and renders them competent for DNA rereplication. Exp. Cell Res. 1999, 247, 432–440. [Google Scholar] [CrossRef]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 10014–10019. [Google Scholar] [CrossRef]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagosklonny, M.V.; Bates, S.E. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef]
- Kuefer, R.; Hofer, M.D.; Altug, V.; Zorn, C.; Genze, F.; Kunzi-Rapp, K.; Hautmann, R.E.; Gschwend, J.E. Sodium butyrate and tributyrin induce in vivo growth inhibition and apoptosis in human prostate cancer. Br. J. Cancer 2004, 90, 535–541. [Google Scholar] [CrossRef]
- Kim, J.; Park, H.; Im, J.Y.; Choi, W.S.; Kim, H.S. Sodium butyrate regulates androgen receptor expression and cell cycle arrest in human prostate cancer cells. Anticancer Res. 2007, 27, 3285–3292. [Google Scholar]
- el-Deiry, W.S.; Harper, J.W.; O'Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y.; Wiman, K.G.; Mercer, W.E.; Kastan, M.B.; Kohn, K.W.; Elledge, S.J.; Kinzler, K.W.; Vogelstein, B. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994, 54, 1169–1174. [Google Scholar]
- Qiu, L.; Burgess, A.; Fairlie, D.P.; Leonard, H.; Parsons, P.G.; Gabrielli, B.G. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell 2000, 11, 2069–2083. [Google Scholar] [CrossRef]
- Burgess, A.J.; Pavey, S.; Warrener, R.; Hunter, L.J.; Piva, T.J.; Musgrove, E.A.; Saunders, N.; Parsons, P.G.; Gabrielli, B.G. Up-regulation of p21(WAF1/CIP1) by histone deacetylase inhibitors reduces their cytotoxicity. Mol. Pharmacol. 2001, 60, 828–837. [Google Scholar]
- Kurita-Ochiai, T.; Hashizume, T.; Yonezawa, H.; Ochiai, K.; Yamamoto, M. Characterization of the effects of butyric acid on cell proliferation, cell cycle distribution and apoptosis. FEMS Immunol. Med. Microbiol. 2006, 47, 67–74. [Google Scholar] [CrossRef]
- Nudelman, A.; Ruse, M.; Aviram, A.; Rabizadeh, E.; Shaklai, M.; Zimrah, Y.; Rephaeli, A. Novel anticancer prodrugs of butyric acid. 2. J. Med. Chem. 1992, 35, 687–694. [Google Scholar] [CrossRef]
- Vecchia, M.G.; Carnelós Filho, M.; Fellipe, C.R.; Curi, R.; Newsholme, E.A. Acetate and propionate potentiate the antiproliferative effect of butyrate on RBL-2H3 growth. Gen. Pharmacol. 1997, 29, 725–728. [Google Scholar] [CrossRef]
- Siu, L.L.; Von Hoff, D.D.; Rephaeli, A.; Izbicka, E.; Cerna, C.; Gomez, L.; Rowinsky, E.K.; Eckhardt, S.G. Activity of pivaloyloxymethyl butyrate, a novel anticancer agent, on primary human tumor colony-forming units. Invest. New Drugs 1998, 16, 113–119. [Google Scholar] [CrossRef]
- Madigan, M.C.; Chaudhri, G.; Penfold, P.L.; Conway, R.M. Sodium butyrate modulates p53 and Bcl-2 expression in human retinoblastoma cell lines. Oncol. Res. 1999, 11, 331–337. [Google Scholar]
- Hara, I.; Miyake, H.; Hara, S.; Arakawa, S.; Kamidono, S. Sodium butyrate induces apoptosis in human renal cell carcinoma cells and synergistically enhances their sensitivity to anti-Fas-mediated cytotoxicity. Int. J. Oncol. 2000, 17, 1213–1218. [Google Scholar]
- Giermasz, A.; Makowski, M.; Kozłowska, E.; Nowis, D.; Maj, M.; Jalili, A.; Feleszko, W.; Wójcik, C.; Dabrowska, A.; Jakóbisiak, M.; Gołab, J. Potentiating antitumor effects of a combination therapy with lovastatin and butyrate in the Lewis lung carcinoma model in mice. Int. J. Cancer 2002, 97, 746–750. [Google Scholar] [CrossRef]
- Hague, A.; Manning, A.M.; Hanlon, K.A.; Huschtscha, L.I.; Hart, D.; Paraskeva, C. Sodium butyrate induces apoptosis in human colonic tumour cell lines in a p53-independent pathway: implications for the possible role of dietary fibre in the prevention of large-bowel cancer. Int. J. Cancer 1993, 55, 498–505. [Google Scholar] [CrossRef]
- Mandal, M.; Kumar, R. Bcl-2 expression regulates sodium butyrate-induced apoptosis in human MCF-7 breast cancer cells. Cell Growth Differ. 1996, 7, 311–318. [Google Scholar]
- Janson, W.; Brandner, G.; Siegel, J. Butyrate modulates DNA-damage-induced p53 response by induction of p53-independent differentiation and apoptosis. Oncogene 1997, 15, 1395–1406. [Google Scholar]
- Fan, Y.Y.; Zhang, J.; Barhoumi, R.; Burghardt, R.C.; Turner, N.D.; Lupton, J.R.; Chapkin, R.S. Antagonism of CD95 signaling blocks butyrate induction of apoptosis in young adult mouse colonic cells. Am. J. Physiol. 1999, 277, C310–319. [Google Scholar]
- Terui, T.; Murakami, K.; Takimoto, R.; Takahashi, M.; Takada, K.; Murakami, T.; Minami, S.; Matsunaga, T.; Takayama, T.; Kato, J.; Niitsu, Y. Induction of PIG3 and NOXA through acetylation of p53 at 320 and 373 lysine residues as a mechanism for apoptotic cell death by histone deacetylase inhibitors. Cancer Res. 2003, 63, 8948–8954. [Google Scholar]
- Chopin, V.; Slomianny, C.; Hondermarck, H.; Le Bourhis, X. Synergistic induction of apoptosis in breast cancer cells by cotreatment with butyrate and TNF-alpha, TRAIL, or anti-Fas agonist antibody involves enhancement of death receptors’ signaling and requires P21(waf1). Exp. Cell Res. 2004, 298, 560–573. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, J.W.; Lee, J.Y.; Kwon, T.K. Sodium butyrate sensitizes TRAIL-mediated apoptosis by induction of transcription from the DR5 gene promoter through Sp1 sites in colon cancer cells. Carcinogenesis 2004, 25, 1813–1820. [Google Scholar] [CrossRef]
- Nakata, S.; Yoshida, T.; Horinaka, M.; Shiraishi, T.; Wakada, M.; Sakai, T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene 2004, 23, 6261–6271. [Google Scholar] [CrossRef]
- Joseph, J.; Wajapeyee, N.; Somasundaram, K. Role of p53 status in chemosensitivity determination of cancer cells against histone deacetylase inhibitor sodium butyrate. Int. J. Cancer 2005, 115, 11–18. [Google Scholar] [CrossRef]
- Takimoto, R.; Kato, J.; Terui, T.; Takada, K.; Kuroiwa, G.; Wu, J.; Ohnuma, H.; Takahari, D.; Kobune, M.; Sato, Y.; Takayama, T.; Matsunaga, T.; Niitsu, Y. Augmentation of antitumor effects of p53 gene therapy by combination with HDAC inhibitor. Cancer Biol. Ther. 2005, 4, 421–428. [Google Scholar] [CrossRef]
- Krupitza, G.; Harant, H.; Dittrich, E.; Szekeres, T.; Huber, H.; Dittrich, C. Sodium butyrate inhibits c-myc splicing and interferes with signal transduction in ovarian carcinoma cells. Carcinogenesis 1995, 16, 1199–1205. [Google Scholar] [CrossRef]
- Velázquez, O.C.; Zhou, D.; Seto, R.W.; Jabbar, A.; Choi, J.; Lederer, H.M.; Rombeau, J.L. In vivo crypt surface hyperproliferation is decreased by butyrate and increased by deoxycholate in normal rat colon: associated in vivo effects on c-Fos and c-Jun expression. JPEN J. Parenter. Enteral. Nutr. 1996, 20, 243–250. [Google Scholar]
- Tang, S.J.; Huang, Y.M.; Wang, F.F. Analysis of c-fos expression in the butyrate-induced F-98 glioma cell differentiation. Biochem. J. 1995, 306 (Pt 1), 47–56. [Google Scholar]
- Bonnotte, B.; Favre, N.; Reveneau, S.; Micheau, O.; Droin, N.; Garrido, C.; Fontana, A.; Chauffert, B.; Solary, E.; Martin, F. Cancer cell sensitization to fas-mediated apoptosis by sodium butyrate. Cell Death Differ. 1998, 5, 480–487. [Google Scholar]
- Ogawa, K.; Yasumura, S.; Atarashi, Y.; Minemura, M.; Miyazaki, T.; Iwamoto, M.; Higuchi, K.; Watanabe, A. Sodium butyrate enhances Fas-mediated apoptosis of human hepatoma cells. J. Hepatol. 2004, 40, 278–284. [Google Scholar] [CrossRef]
- Emenaker, N.J.; Calaf, G.M.; Cox, D.; Basson, M.D.; Qureshi, N. Short-chain fatty acids inhibit invasive human colon cancer by modulating uPA, TIMP-1, TIMP-2, mutant p53, Bcl-2, Bax, p21 and PCNA protein expression in an in vitro cell culture model. J. Nutr. 2011, 131 (11 Suppl.), 3041S–3046S. [Google Scholar]
- Joseph, J.; Mudduluru, G.; Antony, S.; Vashistha, S.; Ajitkumar, P.; Somasundaram, K. Expression profiling of sodium butyrate (NaB)-treated cells: identification of regulation of genes related to cytokine signaling and cancer metastasis by NaB. Oncogene 2004, 23, 6304–6315. [Google Scholar] [CrossRef]
- Tong, X.; Yin, L.; Giardina, C. Butyrate suppresses Cox-2 activation in colon cancer cells through HDAC inhibition. Biochem. Biophys. Res. Commun. 2004, 317, 463–471. [Google Scholar] [CrossRef]
- Ammanamanchi, S.; Brattain, M.G. Restoration of transforming growth factor-beta signaling through receptor RI induction by histone deacetylase activity inhibition in breast cancer cells. J. Biol. Chem. 2004, 279, 32620–32625. [Google Scholar] [CrossRef]
- Perrin, P.; Cassagnau, E.; Burg, C.; Patry, Y.; Vavasseur, F.; Harb, J.; Le Pendu, J.; Douillard, J.Y.; Galmiche, J.P.; Bornet, F. An interleukin 2/sodium butyrate combination as immunotherapy for rat colon cancer peritoneal carcinomatosis. Gastroenterology 1994, 107, 1697–1708. [Google Scholar]
- Armstrong, F.; Mathers, J.C. Kill and cure: dietary augmentation of immune defences against colon cancer. Proc. Nutr. Soc. 2000, 59, 215–220. [Google Scholar] [CrossRef]
- Miller, S.J.; Zaloga, G.P.; Hoggatt, A.M.; Labarrere, C.; Faulk, W.P. Short-chain fatty acids modulate gene expression for vascular endothelial cell adhesion molecules. Nutrition 2005, 21, 740–748. [Google Scholar] [CrossRef]
- Yee, J.C.; de Leon Gatti, M.; Philp, R.J.; Yap, M.; Hu, W.S. Genomic and proteome exploration of CHO and hybridoma cells under sodium butyrate treatment. Biotechnol. Bioeng. 2007. [Google Scholar]
- Sawa, H.; Murakami, H.; Ohshima, Y.; Murakami, M.; Yamazaki, I.; Tamura, Y.; Mima, T.; Satone, A.; Ide, W.; Hashimoto, I.; Kamada, H. Histone deacetylase inhibitors such as sodium butyrate and trichostatin A inhibit vascular endothelial growth factor (VEGF) secretion from human glioblastoma cells. Brain Tumor Pathol. 2002, 19, 77–81. [Google Scholar] [CrossRef]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.M.; Nusgens, B.V.; Castronovo, V. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef]
- Sawa, H.; Murakami, H.; Kumagai, M.; Nakasato, M.; Yamauchi, S.; Matsuyama, N.; Tamura, Y.; Satone, A.; Ide, W.; Hashimoto, I.; Kamada, H. Histone deacetylase inhibitor, FK228, induces apoptosis and suppresses cell proliferation of human glioblastoma cells in vitro and in vivo. Acta Neuropathol. 2004, 107, 523–531. [Google Scholar] [CrossRef]
- Zgouras, D.; Becker, U.; Loitsch, S.; Stein, J. Modulation of angiogenesis-related protein synthesis by valproic acid. Biochem. Biophys. Res. Commun. 2004, 316, 693–697. [Google Scholar] [CrossRef]
- Heider, U.; Kaiser, M.; Sterz, J.; Zavrski, I.; Jakob, C.; Fleissner, C.; Eucker, J.; Possinger, K.; Sezer, O. Histone deacetylase inhibitors reduce VEGF production and induce growth suppression and apoptosis in human mantle cell lymphoma. Eur. J. Haematol. 2006, 76, 42–50. [Google Scholar] [CrossRef]
- Dong, X.F.; Song, Q.; Li, L.Z.; Zhao, C.L.; Wang, L.Q. Histone deacetylase inhibitor valproic acid inhibits proliferation and induces apoptosis in KM3 cells via downregulating VEGF receptor. Neuro Endocrinol. Lett. 2007, 28(6). [E-pub ahead of print]. [Google Scholar]
- Bar-Sela, G.; Jacobs, K.M.; Gius, D. Histone deacetylase inhibitor and demethylating agent chromatin compaction and the radiation response by cancer cells. Cancer J. 2007, 13, 65–69. [Google Scholar] [CrossRef]
- Arundel, C.M.; Glicksman, A.S.; Leith, J.T. Enhancement of radiation injury in human colon tumor cells by the maturational agent sodium butyrate (NaB). Radiat. Res. 1985, 104, 443–448. [Google Scholar] [CrossRef]
- Arundel, C.M.; Kenney, S.M.; Leith, J.T.; Glicksman, A.S. Contrasting effects of the differentiating agent sodium butyrate on recovery processes after x-irradiation in heterogeneous human colon tumor cells. Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 959–968. [Google Scholar] [CrossRef]
- Lopez, C.A.; Feng, F.Y.; Herman, J.M.; Nyati, M.K.; Lawrence, T.S.; Ljungman, M. Phenylbutyrate sensitizes human glioblastoma cells lacking wild-type p53 function to ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 214–220. [Google Scholar] [CrossRef]
- Entin-Meer, M.; Yang, X.; VandenBerg, S.R.; Lamborn, K.R.; Nudelman, A.; Rephaeli, A.; Haas-Kogan, D.A. In vivo efficacy of a novel histone deacetylase inhibitor in combination with radiation for the treatment of gliomas. Neuro Oncol. 2007, 9, 82–88. [Google Scholar] [CrossRef]
- Ueno, M.; Toyota, M.; Akino, K.; Suzuki, H.; Kusano, M.; Satoh, A.; Mita, H.; Sasaki, Y.; Nojima, M.; Yanagihara, K.; Hinoda, Y.; Tokino, T.; Imai, K. Aberrant methylation and histone deacetylation associated with silencing of SLC5A8 in gastric cancer. Tumour Biol. 2004, 25, 134–140. [Google Scholar] [CrossRef]
- Hong, C.; Maunakea, A.; Jun, P.; Bollen, A.W.; Hodgson, J.G.; Goldenberg, D.D.; Weiss, W.A.; Costello, J.F. Shared epigenetic mechanisms in human and mouse gliomas inactivate expression of the growth suppressor SLC5A8. Cancer Res. 2005, 65, 3617–3623. [Google Scholar] [CrossRef]
- Conley, B.A.; Egorin, M.J.; Tait, N.; Rosen, D.M.; Sausville, E.A.; Dover, G.; Fram, R.J.; Van Echo, D.A. Phase I study of the orally administered butyrate prodrug, tributyrin, in patients with solid tumors. Clin. Cancer Res. 1998, 4, 629–634. [Google Scholar]
- Patnaik, A.; Rowinsky, E.K.; Villalona, M.A.; Hammond, L.A.; Britten, C.D.; Siu, L.L.; Goetz, A.; Felton, S.A.; Burton, S.; Valone, F.H.; Eckhardt, S.G. A phase I study of pivaloyloxymethyl butyrate, a prodrug of the differentiating agent butyric acid, in patients with advanced solid malignancies. Clin. Cancer Res. 2002, 8, 2142–2148. [Google Scholar]
- Edelman, M.J.; Bauer, K.; Khanwani, S.; Tait, N.; Trepel, J.; Karp, J.; Nemieboka, N.; Chung, E.J.; Van Echo, D. Clinical and pharmacologic study of tributyrin: an oral butyrate prodrug. Cancer Chemother. Pharmacol. 2003, 51, 439–444. [Google Scholar]
- Reid, T.; Valone, F.; Lipera, W.; Irwin, D.; Paroly, W.; Natale, R.; Sreedharan, S.; Keer, H.; Lum, B.; Scappaticci, F.; Bhatnagar, A. Phase II trial of the histone deacetylase inhibitor pivaloyloxymethyl butyrate (Pivanex, AN-9) in advanced non-small cell lung cancer. Lung Cancer 2004, 45, 381–386. [Google Scholar] [CrossRef]
- Cutts, S.M.; Rephaeli, A.; Nudelman, A.; Hmelnitsky, I.; Phillips, D.R. Molecular basis for the synergistic interaction of adriamycin with the formaldehyde-releasing prodrug pivaloyloxymethyl butyrate (AN-9). Cancer Res. 2001, 61, 8194–8202. [Google Scholar]
- Perrine, S.P.; Dover, G.H.; Daftari, P.; Walsh, C.T.; Jin, Y.; Mays, A.; Faller, D.V. Isobutyramide, an orally bioavailable butyrate analogue, stimulates fetal globin gene expression in vitro and in vivo. Br. J. Haematol. 1994, 88, 555–561. [Google Scholar] [CrossRef]
- Nudelman, A.; Gnizi, E.; Katz, Y.; Azulai, R.; Cohen-Ohana, M.; Zhuk, R.; Sampson, S.R.; Langzam, L.; Fibach, E.; Prus, E.; Pugach, V.; Rephaeli, A. Prodrugs of butyric acid. Novel derivatives possessing increased aqueous solubility and potential for treating cancer and blood diseases. Eur. J. Med. Chem. 2001, 36, 63–74. [Google Scholar] [CrossRef]
- Nudelman, A.; Rephaeli, A. Novel mutual prodrug of retinoic and butyric acids with enhanced anticancer activity. J. Med. Chem. 2000, 43, 2962–2966. [Google Scholar] [CrossRef]
- Rephaeli, A.; Rabizadeh, E.; Aviram, A.; Shaklai, M.; Ruse, M.; Nudelman, A. Derivatives of butyric acid as potential anti-neoplastic agents. Int. J. Cancer 1991, 49, 66–72. [Google Scholar] [CrossRef]
- Rephaeli, A.; Entin-Meer, M.; Angel, D.; Tarasenko, N.; Gruss-Fischer, T.; Bruachman, I.; Phillips, D.R.; Cutts, S.M.; Haas-Kogan, D.; Nudelman, A. The selectivty and anti-metastatic activity of oral bioavailable butyric acid prodrugs. Invest. New Drugs 2006, 24, 383–392. [Google Scholar] [CrossRef]
- Zimra, Y.; Nudelman, A.; Zhuk, R.; Rabizadeh, E.; Shaklai, M.; Aviram, A.; Rephaeli, A. Uptake of pivaloyloxymethyl butyrate into leukemic cells and its intracellular esterase-catalyzed hydrolysis. J. Cancer Res. Clin. Oncol. 2000, 126, 693–698. [Google Scholar]
- Nakase, H.; Okazaki, K.; Tabata, Y.; Uose, S.; Ohana, M.; Uchida, K.; Matsushima, Y.; Kawanami, C.; Oshima, C.; Ikada, Y.; Chiba, T. Development of an oral drug delivery system targeting immune-regulating cells in experimental inflammatory bowel disease: a new therapeutic strategy. J. Pharmacol. Exp. Ther. 2000, 292, 15–21. [Google Scholar]
- Lamprecht, A.; Ubrich, N.; Yamamoto, H.; Schäfer, U.; Takeuchi, H.; Maincent, P.; Kawashima, Y.; Lehr, C.M. Biodegradable nanoparticles for targeted drug delivery in treatment of inflammatory bowel disease. J. Pharmacol. Exp. Ther. 2001, 299, 775–781. [Google Scholar]
- Peart, M.J.; Smyth, G.K.; van Laar, R.K.; Bowtell, D.D.; Richon, V.M.; Marks, P.A.; Holloway, A.J.; Johnstone, R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 3697–3702. [Google Scholar] [CrossRef]
- Epping, M.T.; Wang, L.; Plumb, J.A.; Lieb, M.; Gronemeyer, H.; Brown, R.; Bernards, R. A functional genetic screen identifies retinoic acid signaling as a target of histone deacetylase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 17777–17782. [Google Scholar] [CrossRef]
- Lindemann, R.K.; Newbold, A.; Whitecross, K.F.; Cluse, L.A.; Frew, A.J.; Ellis, L.; Williams, S.; Wiegmans, A.P.; Dear, A.E.; Scott, C.L.; Pellegrini, M.; Wei, A.; Richon, V.M.; Marks, P.A.; Lowe, S.W.; Smyth, M.J.; Johnstone, R.W. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 8071–8076. [Google Scholar] [CrossRef]
- Fundarò, A.; Cavalli, R.; Bargoni, A.; Vighetto, D.; Zara, G.P.; Gasco, M.R. Non-stealth and stealth solid lipid nanoparticles (SLN) carrying doxorubicin: pharmacokinetics and tissue distribution after i.v. administration to rats. Pharmacol. Res. 2000, 42, 337–343. [Google Scholar] [CrossRef]
- Blasi, P.; Giovagnoli, S.; Schoubben, A.; Ricci, M.; Rossi, C. Solid lipid nanoparticles for targeted brain drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 454–477. [Google Scholar] [CrossRef]
- Zara, G.P.; Cavalli, R.; Fundarò, A.; Bargoni, A.; Caputo, O.; Gasco, M.R. Pharmacokinetics of doxorubicin incorporated in solid lipid nanospheres (SLN). Pharmacol. Res. 1999, 40, 281–286. [Google Scholar] [CrossRef]
- Rich, J.N.; Bigner, D.D. Development of novel targeted therapies in the treatment of malignant glioma. Nat. Rev. Drug Discov. 2004, 3, 430–446. [Google Scholar] [CrossRef]
- Dehais, C.; Laigle-Donadey, F.; Marie, Y.; Kujas, M.; Lejeune, J.; Benouaich-Amiel, A.; Pedretti, M.; Polivka, M.; Xuan, K.H.; Thillet, J.; Delattre, J.Y.; Sanson, M. Prognostic stratification of patients with anaplastic gliomas according to genetic profile. Cancer 2006, 107, 1891–1897. [Google Scholar] [CrossRef]
- Sanson, M.; Laigle-Donadey, F.; Benouaich-Amiel, A. Molecular changes in brain tumors: prognostic and therapeutic impact. Curr. Opin. Oncol. 2006, 18, 623–630. [Google Scholar] [CrossRef]
- Carpentier, A.F.; Meng, Y. Recent advances in immunotherapy for human glioma. Curr. Opin. Oncol. 2006, 18, 631–636. [Google Scholar] [CrossRef]
- Gray, S.G.; Dangond, F. Rationale for the use of histone deacetylase inhibitors as a dual therapeutic modality in multiple sclerosis. Epigenetics 2006, 1, 67–75. [Google Scholar] [CrossRef]
- Boutillier, A.L.; Rouaux, C.; Pantaleeva, I.; Loeffler, J.Philippe. Chromatin acetylation status in the manifestation of neurodegenerative diseases: HDAC inhibitors as therapeutic tools. Subcell. Biochem. 2007, 41, 263–293. [Google Scholar]
- Morrison, B.E.; Majdzadeh, N.; D'Mello, S.R. Histone deacetylases: focus on the nervous system. Cell. Mol. Life Sci. 2007, 64, 2258–2269. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kubilus, J.K.; Lee, J.; Ryu, H.; Beesen, A.; Zucker, B.; Smith, K.; Kowall, N.W.; Ratan, R.R.; Luthi-Carter, R.; Hersch, S.M. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 2003, 23, 9418–9427. [Google Scholar]
- Oliveira, J.M.; Chen, S.; Almeida, S.; Riley, R.; Gonçalves, J.; Oliveira, C.R.; Hayden, M.R.; Nicholls, D.G.; Ellerby, L.M.; Rego, A.C. Mitochondrial-dependent Ca2+ handling in Huntington's disease striatal cells: effect of histone deacetylase inhibitors. J. Neurosci. 2006, 26, 11174–11186. [Google Scholar] [CrossRef]
- Borovecki, F.; Lovrecic, L.; Zhou, J.; Jeong, H.; Then, F.; Rosas, H.D.; Hersch, S.M.; Hogarth, P.; Bouzou, B.; Jensen, R.V.; Krainc, D. Genome-wide expression profiling of human blood reveals biomarkers for Huntington’s disease. Proc Natl. Acad. Sci. U.S.A. 2005, 102, 11023–11028. [Google Scholar] [CrossRef]
- Chang, J.G.; Hsieh-Li, H.M.; Jong, Y.J.; Wang, N.M.; Tsai, C.H.; Li, H. Treatment of spinal muscular atrophy by sodium butyrate. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 9808–9813. [Google Scholar] [CrossRef]
- Minamiyama, M.; Katsuno, M.; Adachi, H.; Waza, M.; Sang, C.; Kobayashi, Y.; Tanaka, F.; Doyu, M.; Inukai, A.; Sobue, G. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2004, 13, 1183–1192. [Google Scholar] [CrossRef]
- Mercuri, E.; Bertini, E.; Messina, S.; Solari, A.; D'Amico, A.; Angelozzi, C.; Battini, R.; Berardinelli, A.; Boffi, P.; Bruno, C.; Cini, C.; Colitto, F.; Kinali, M.; Minetti, C.; Mongini, T.; Morandi, L.; Neri, G.; Orcesi, S.; Pane, M.; Pelliccioni, M.; Pini, A.; Tiziano, F.D.; Villanova, M.; Vita, G.; Brahe, C. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology 2007, 68, 51–55. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Brioschi, A.; Zara, G.P.; Calderoni, S.; Gasco, M.R.; Mauro, A. Cholesterylbutyrate Solid Lipid Nanoparticles as a Butyric Acid Prodrug. Molecules 2008, 13, 230-254. https://doi.org/10.3390/molecules13020230
Brioschi A, Zara GP, Calderoni S, Gasco MR, Mauro A. Cholesterylbutyrate Solid Lipid Nanoparticles as a Butyric Acid Prodrug. Molecules. 2008; 13(2):230-254. https://doi.org/10.3390/molecules13020230
Chicago/Turabian StyleBrioschi, Andrea, Gian Paolo Zara, Sara Calderoni, Maria Rosa Gasco, and Alessandro Mauro. 2008. "Cholesterylbutyrate Solid Lipid Nanoparticles as a Butyric Acid Prodrug" Molecules 13, no. 2: 230-254. https://doi.org/10.3390/molecules13020230
APA StyleBrioschi, A., Zara, G. P., Calderoni, S., Gasco, M. R., & Mauro, A. (2008). Cholesterylbutyrate Solid Lipid Nanoparticles as a Butyric Acid Prodrug. Molecules, 13(2), 230-254. https://doi.org/10.3390/molecules13020230