Bile Acid Scaffolds in Supramolecular Chemistry: The Interplay of Design and Synthesis

{kind=link}
{kind=link}
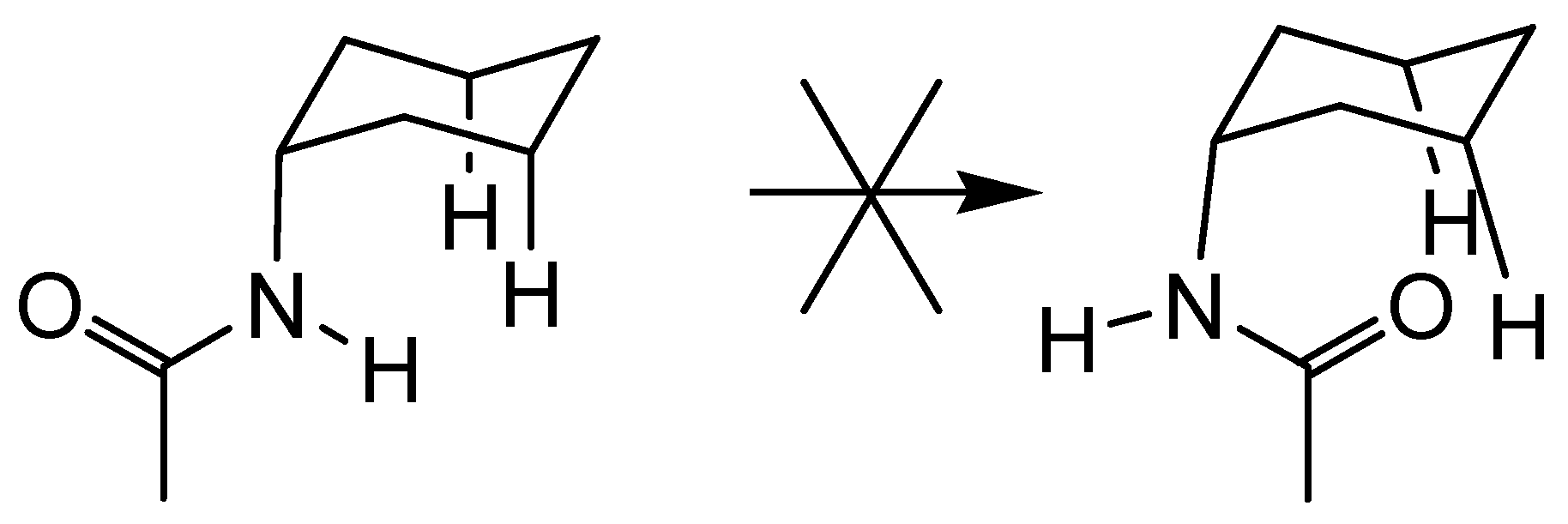{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
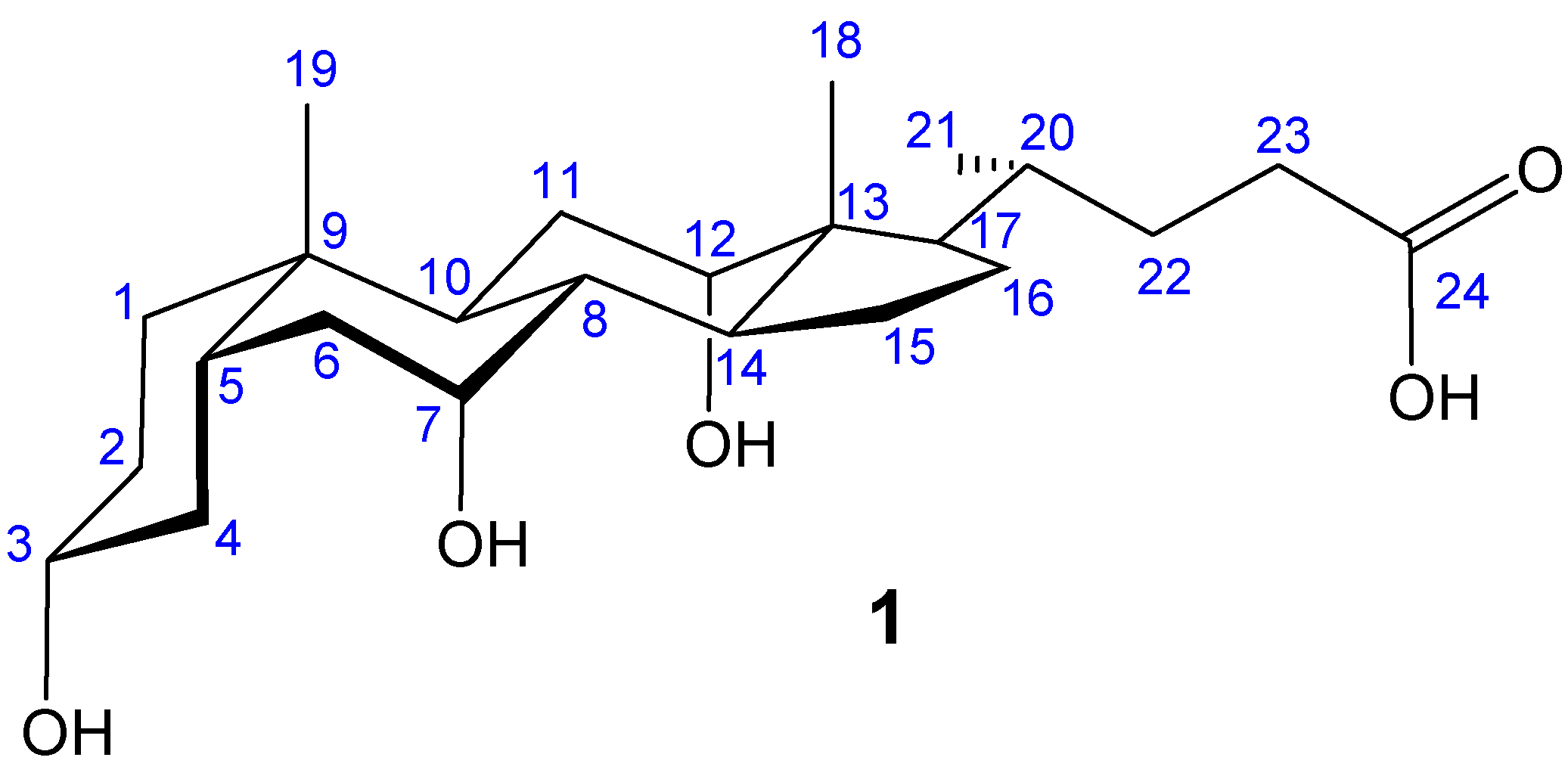
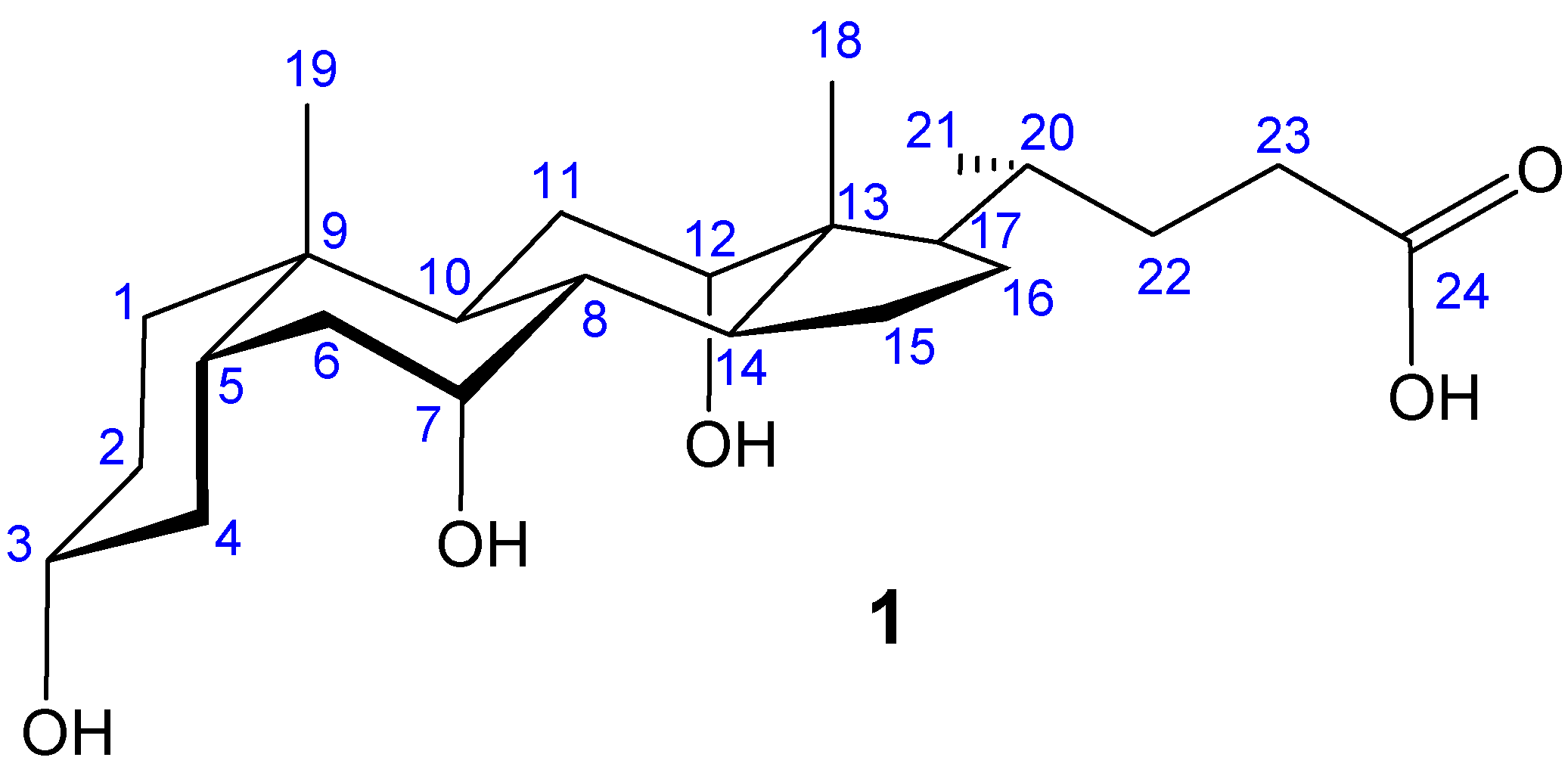
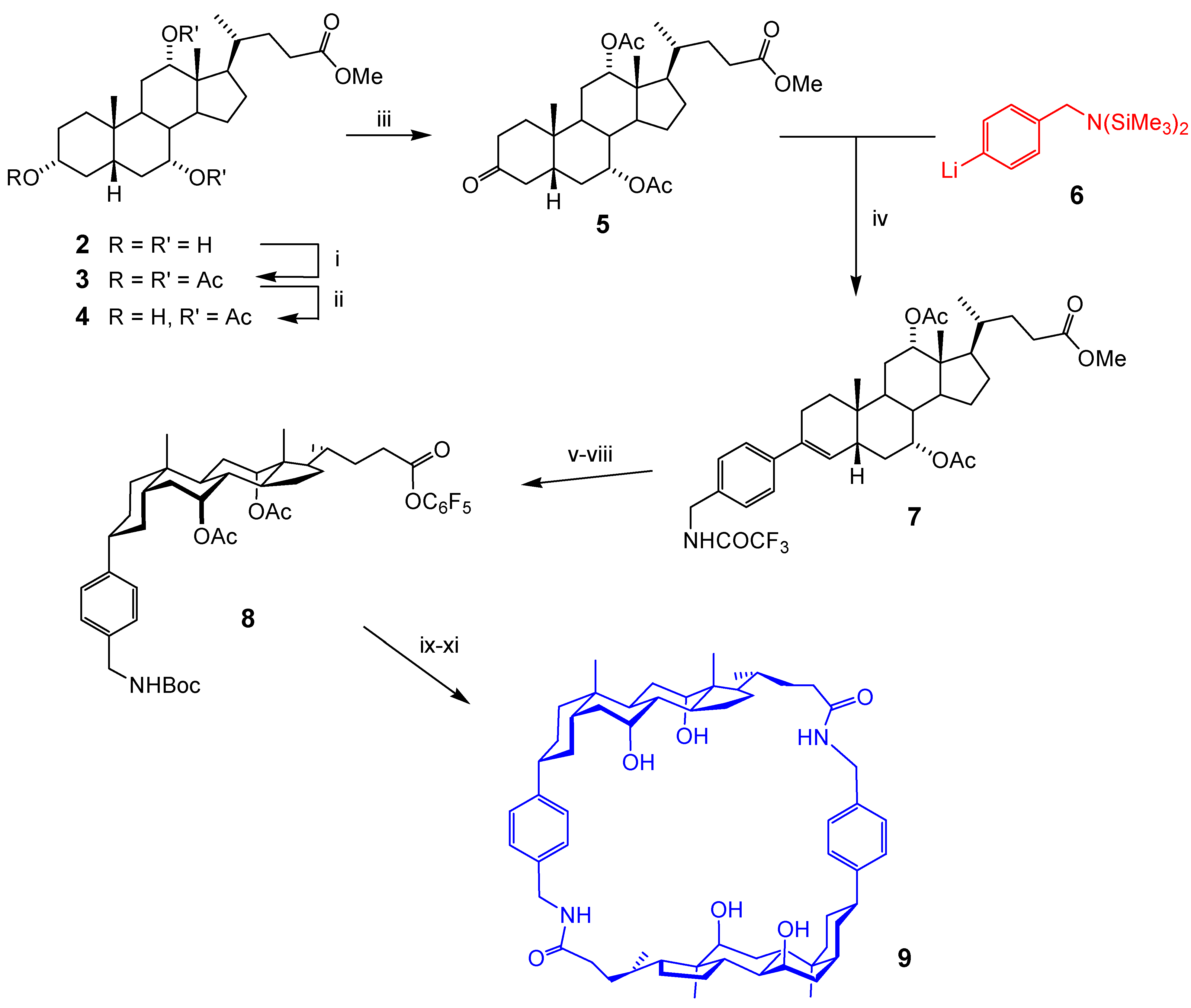
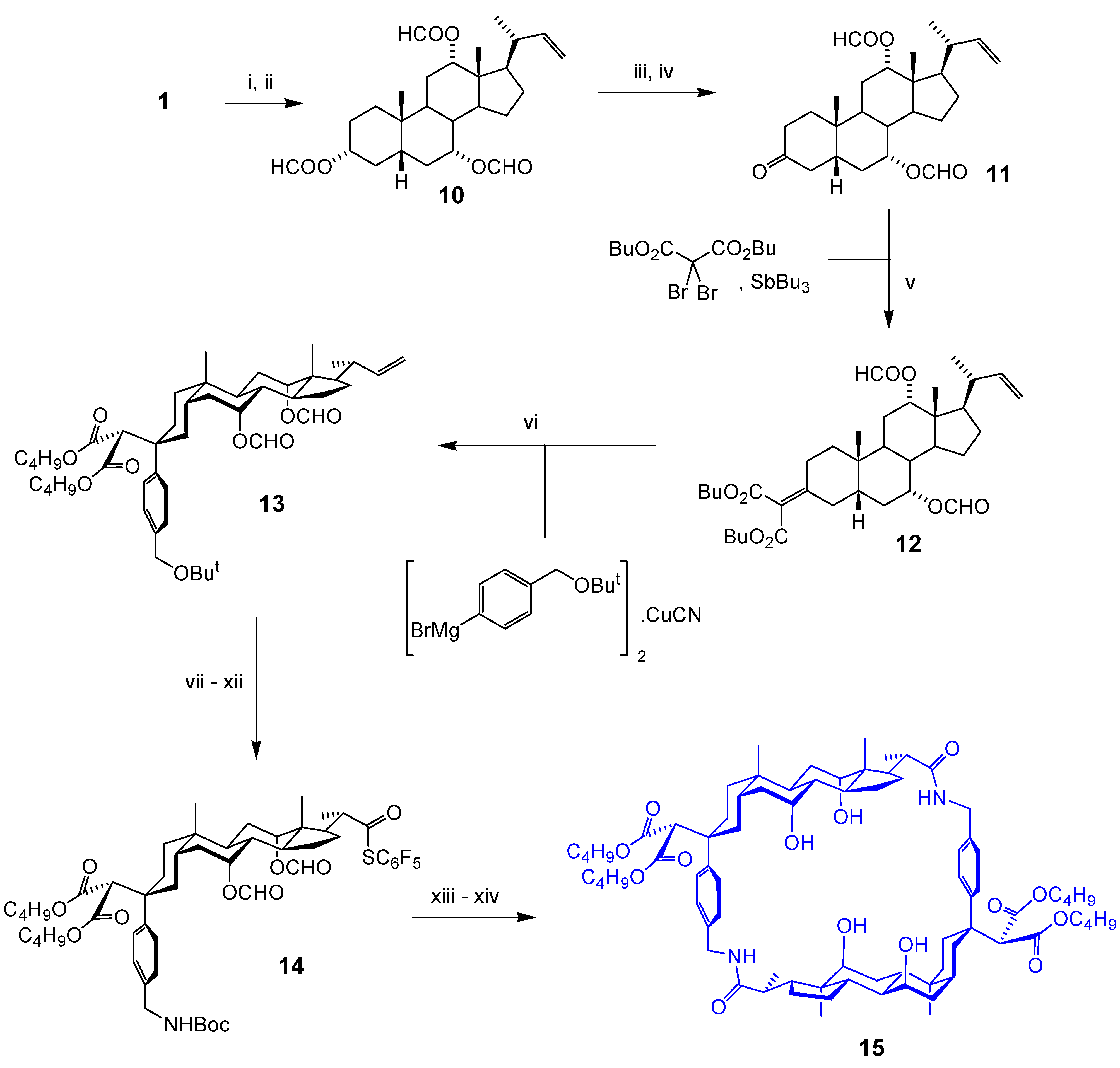
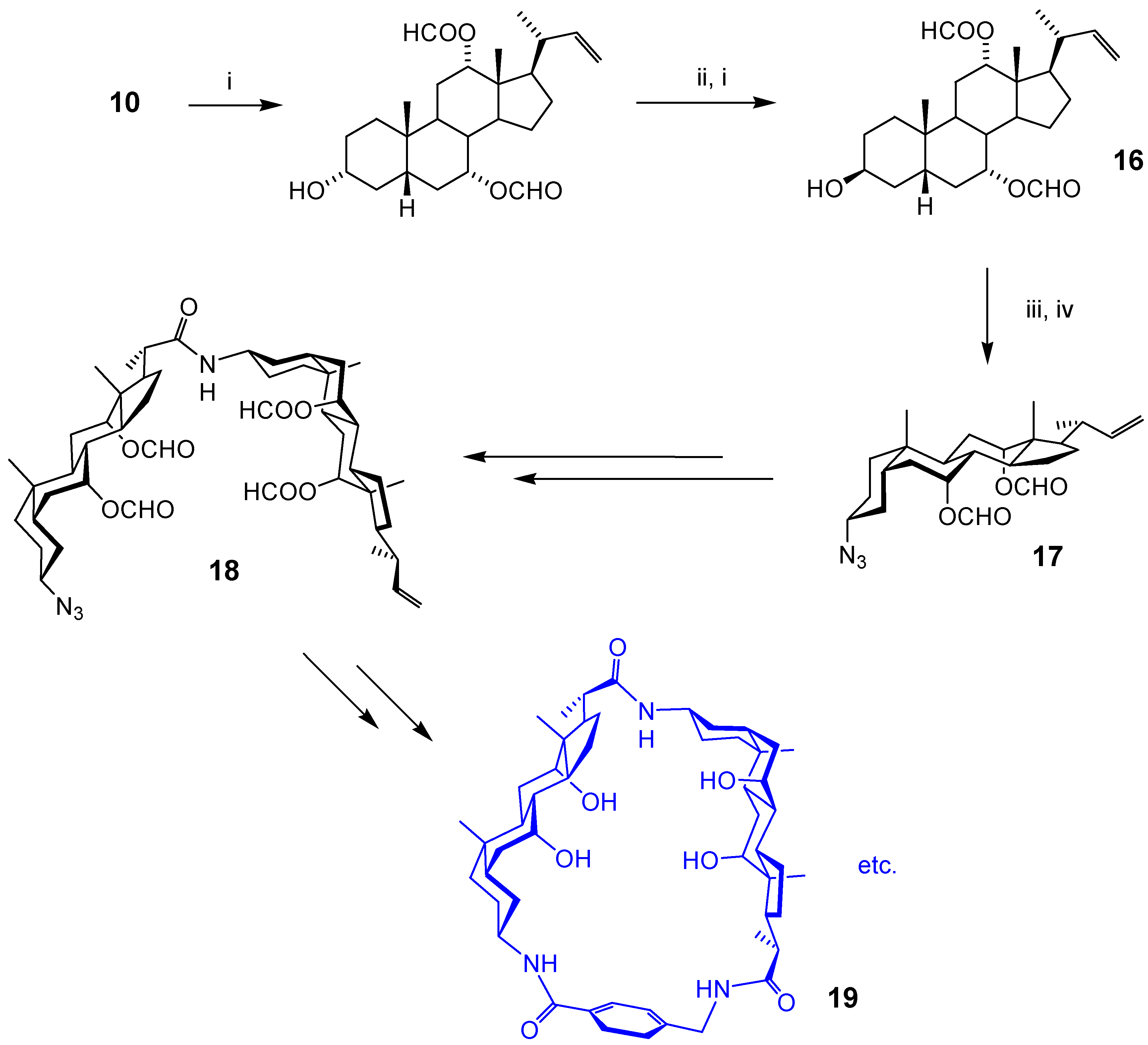
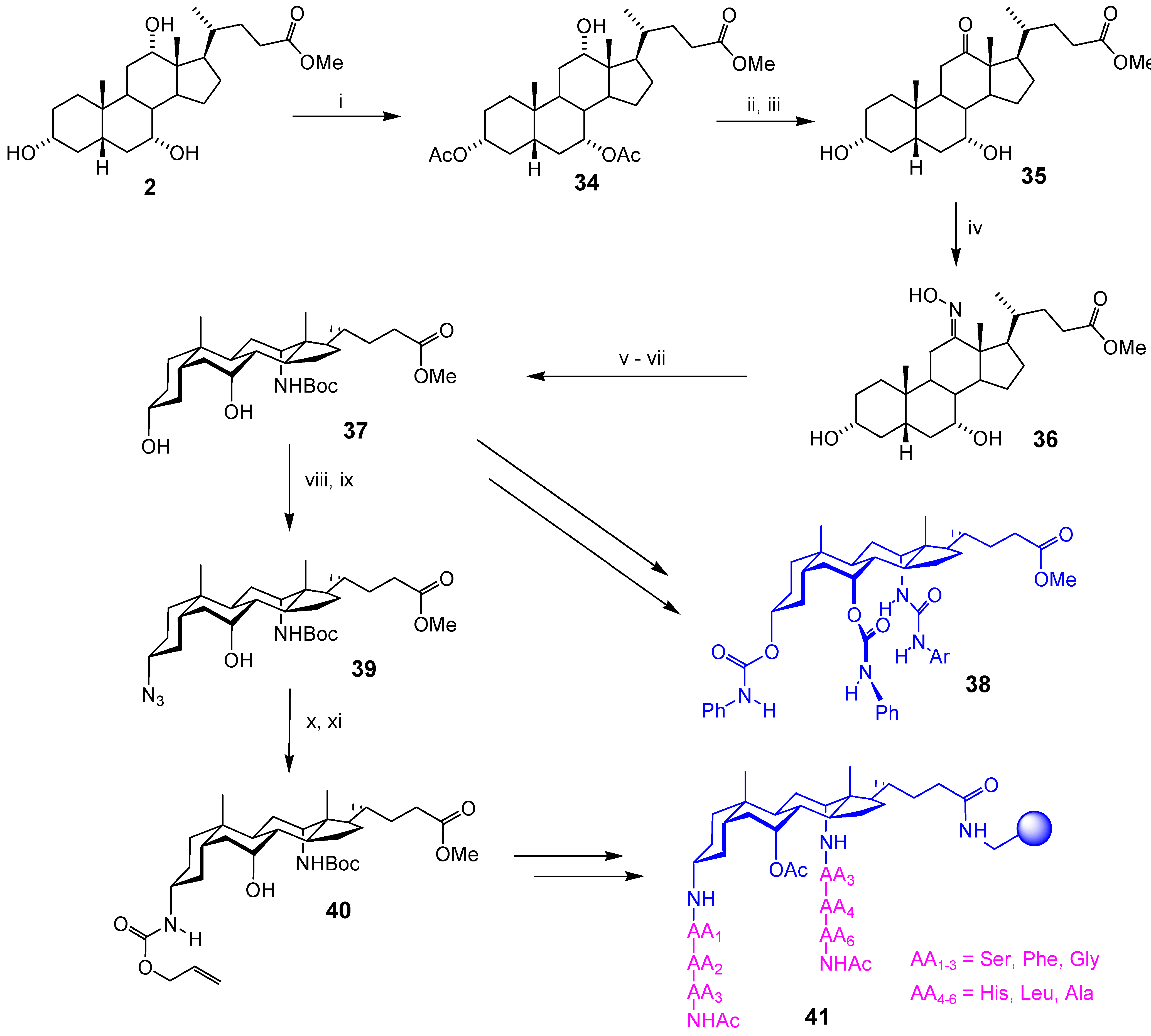
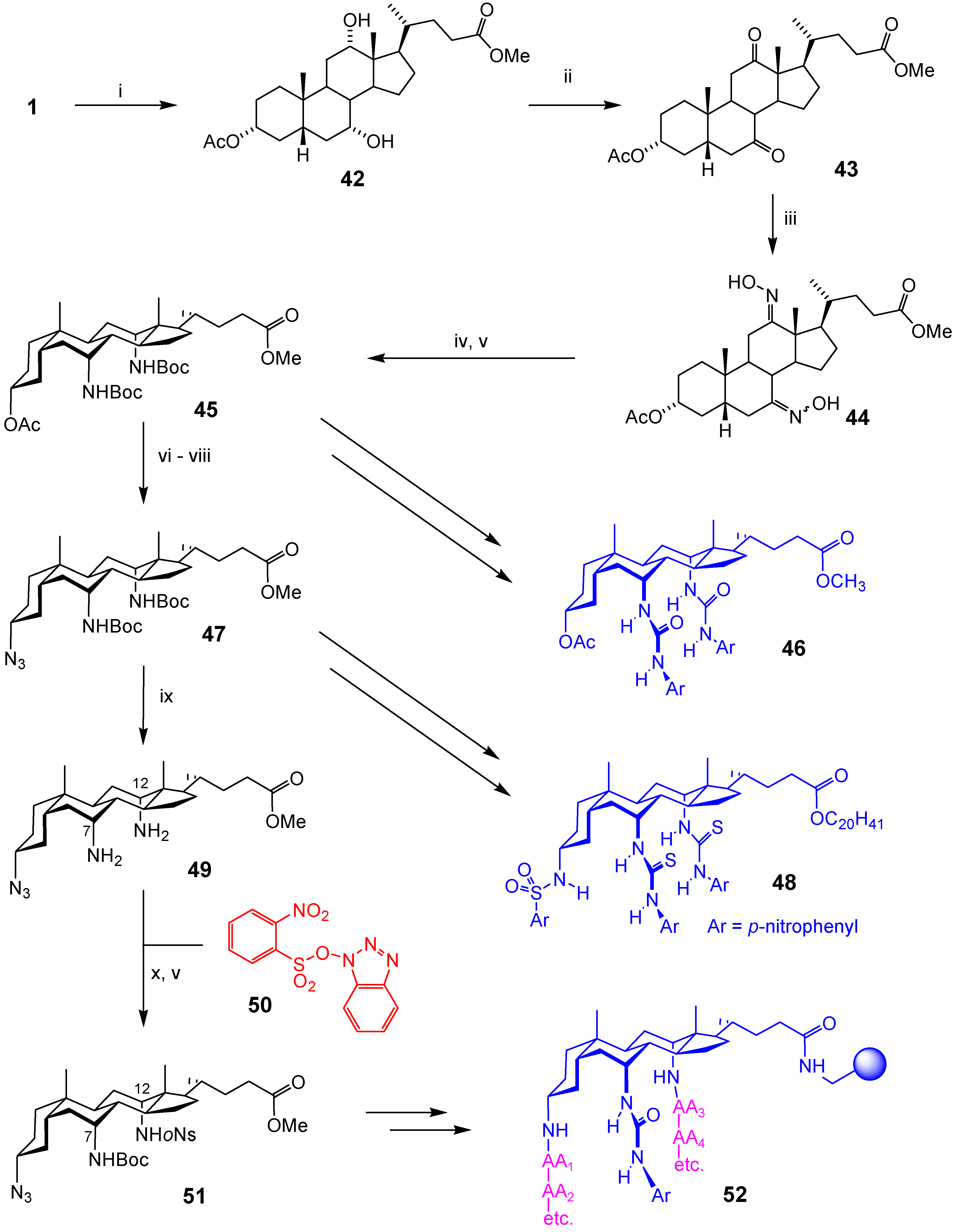
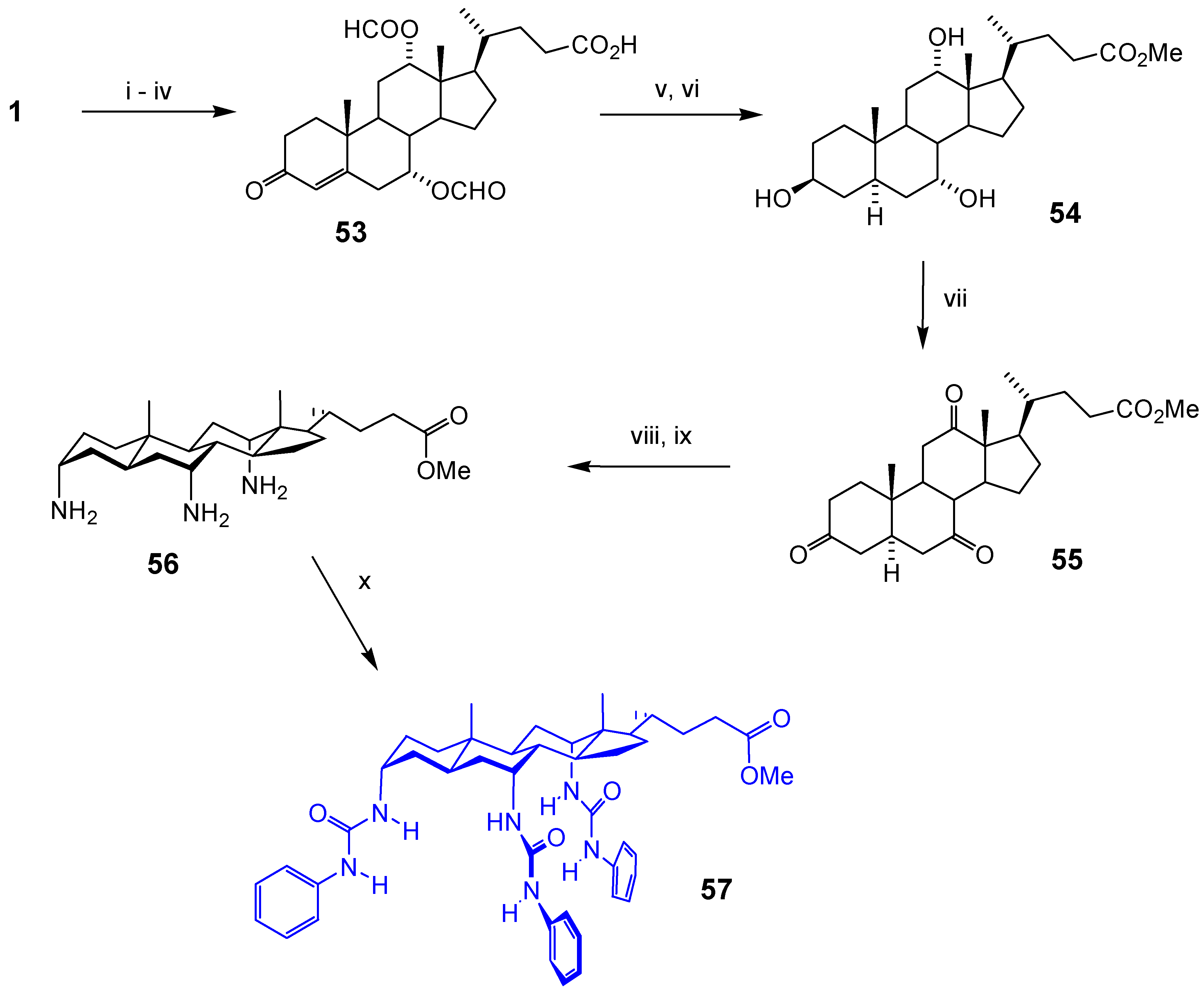
Macrocyclic Architectures - Cholaphanes and Cyclocholamides
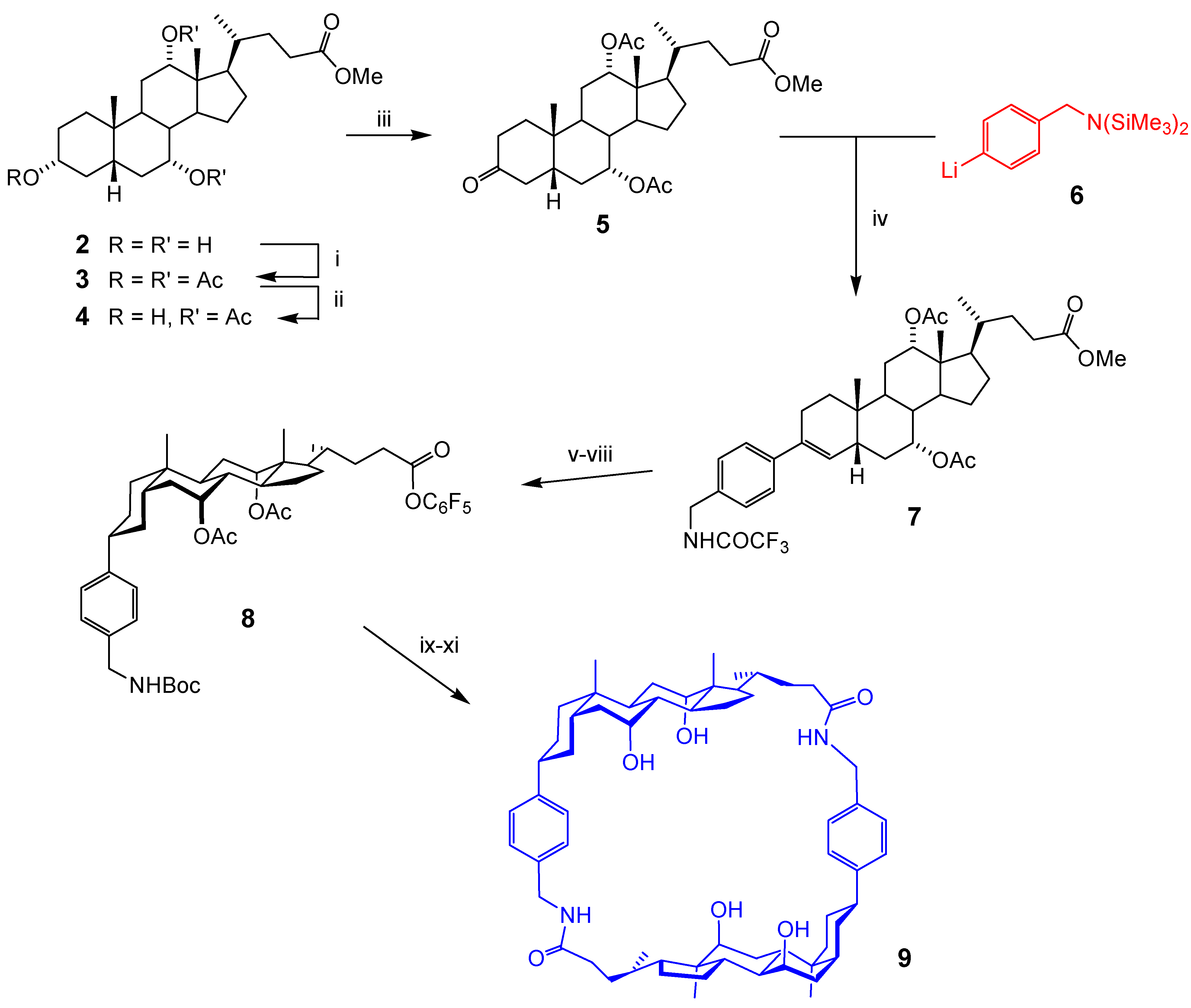
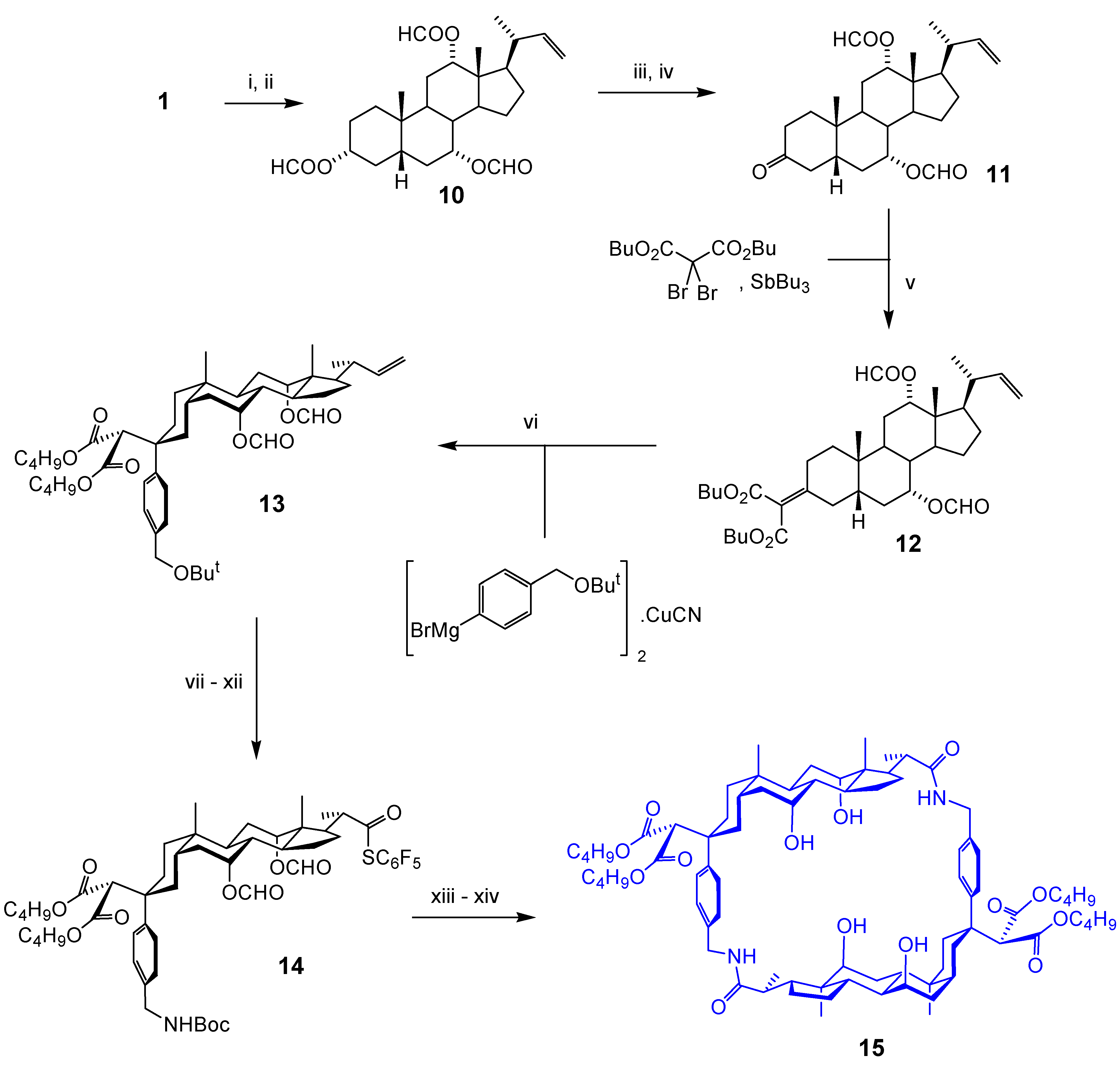
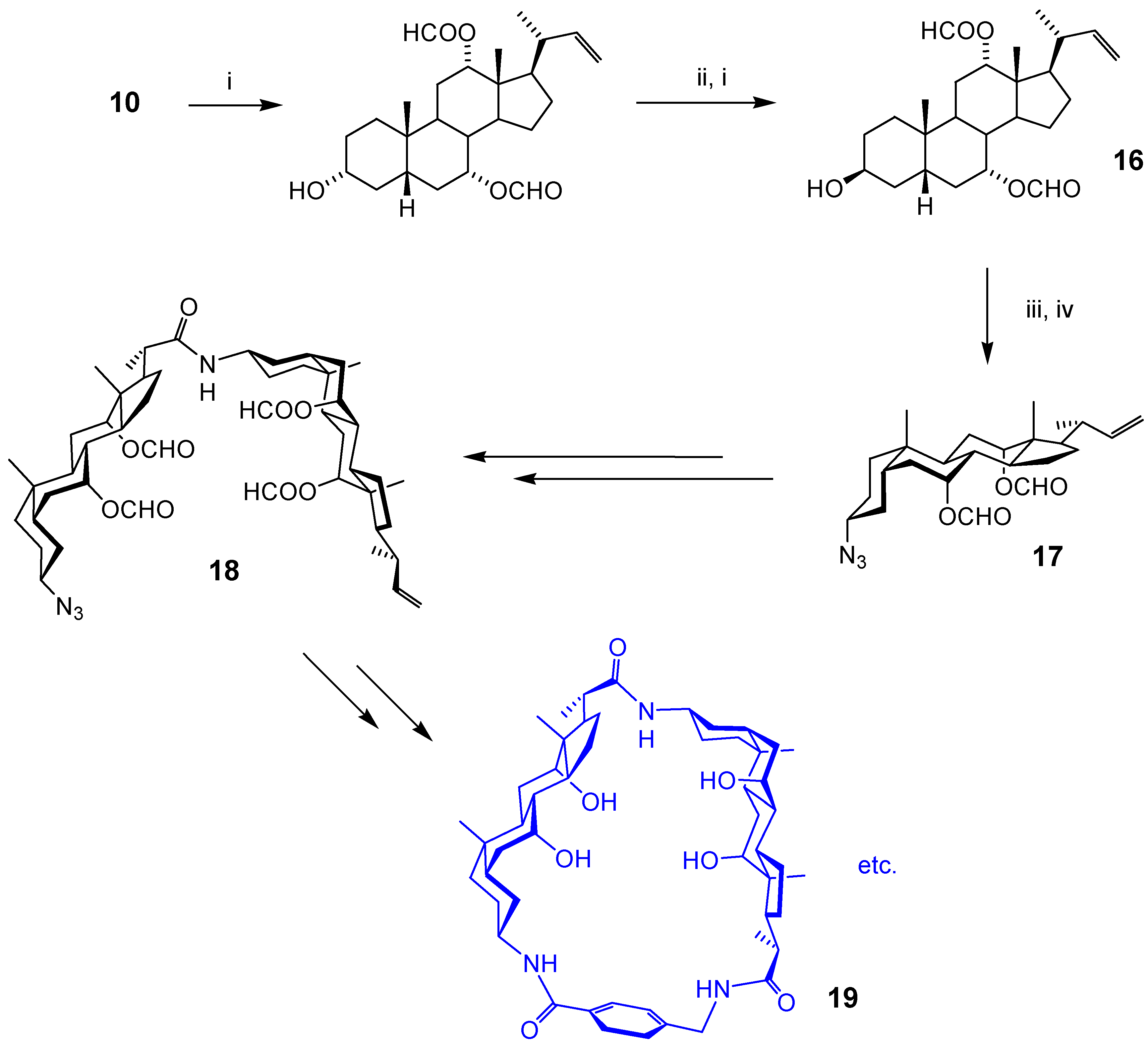
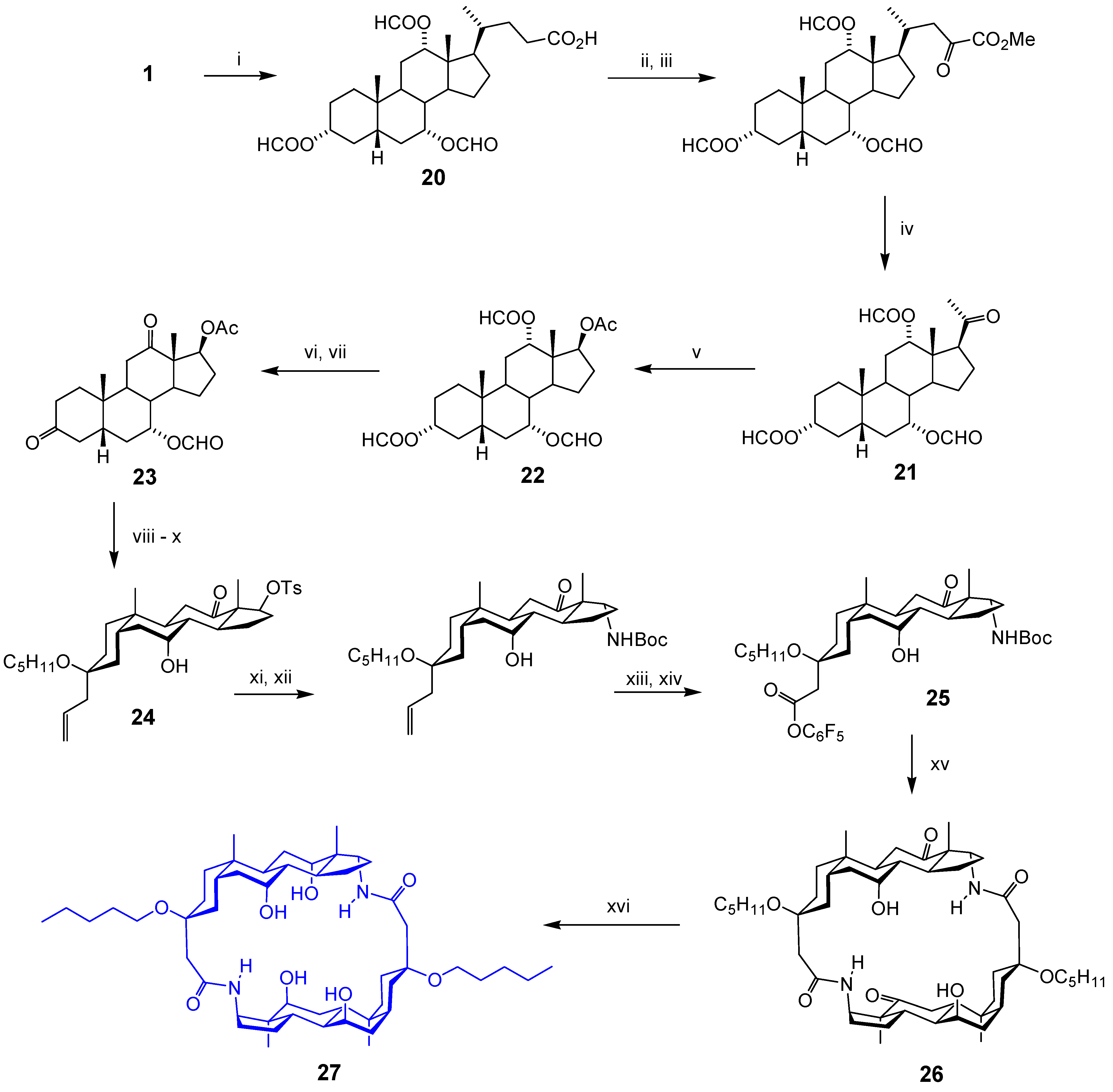
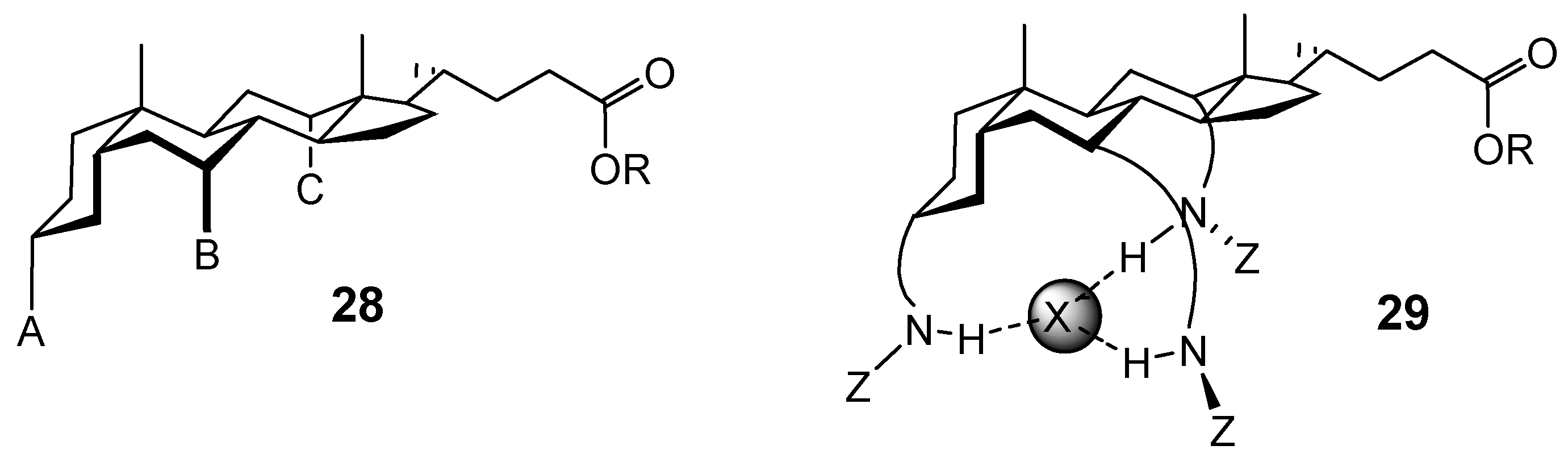
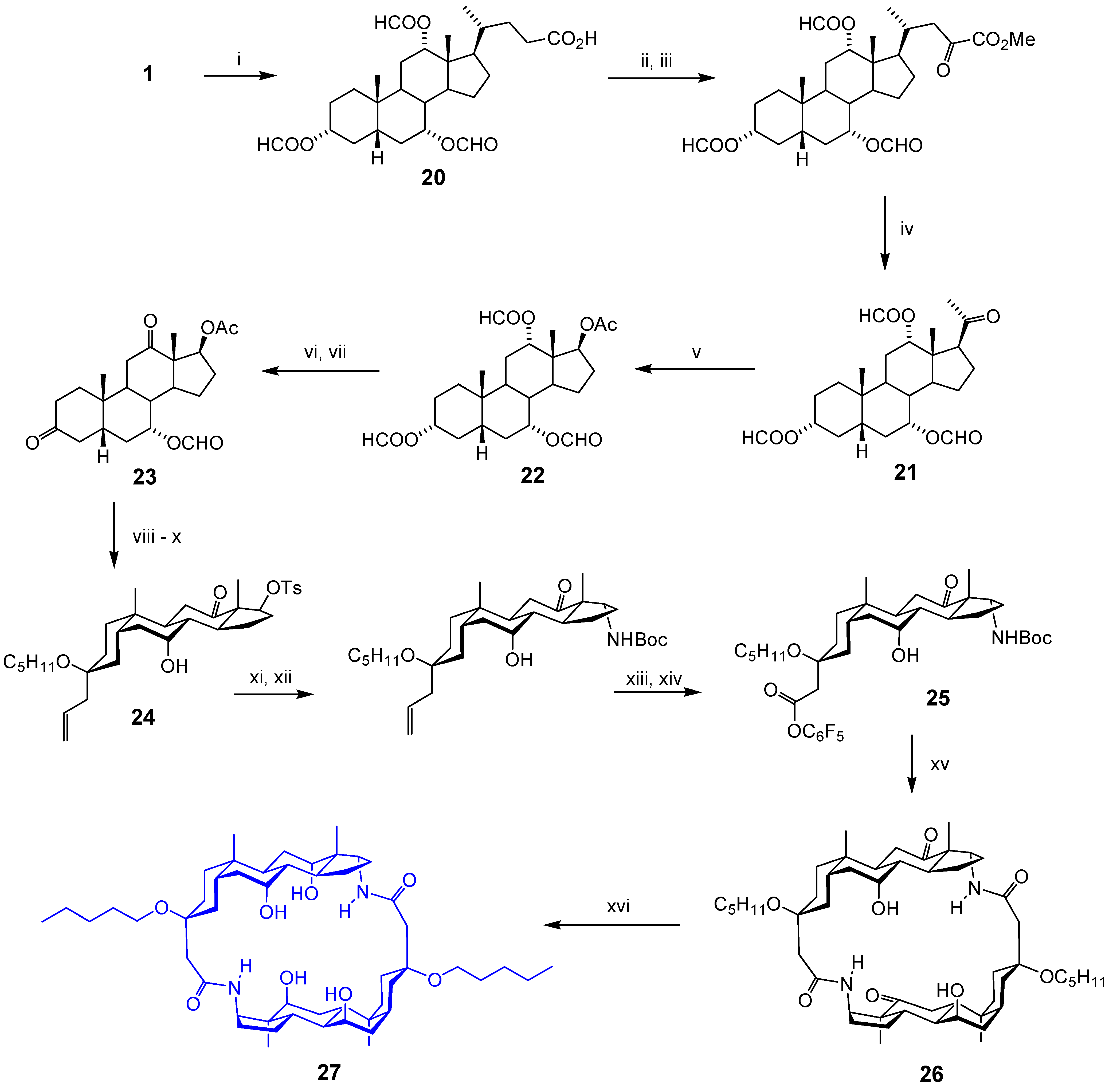
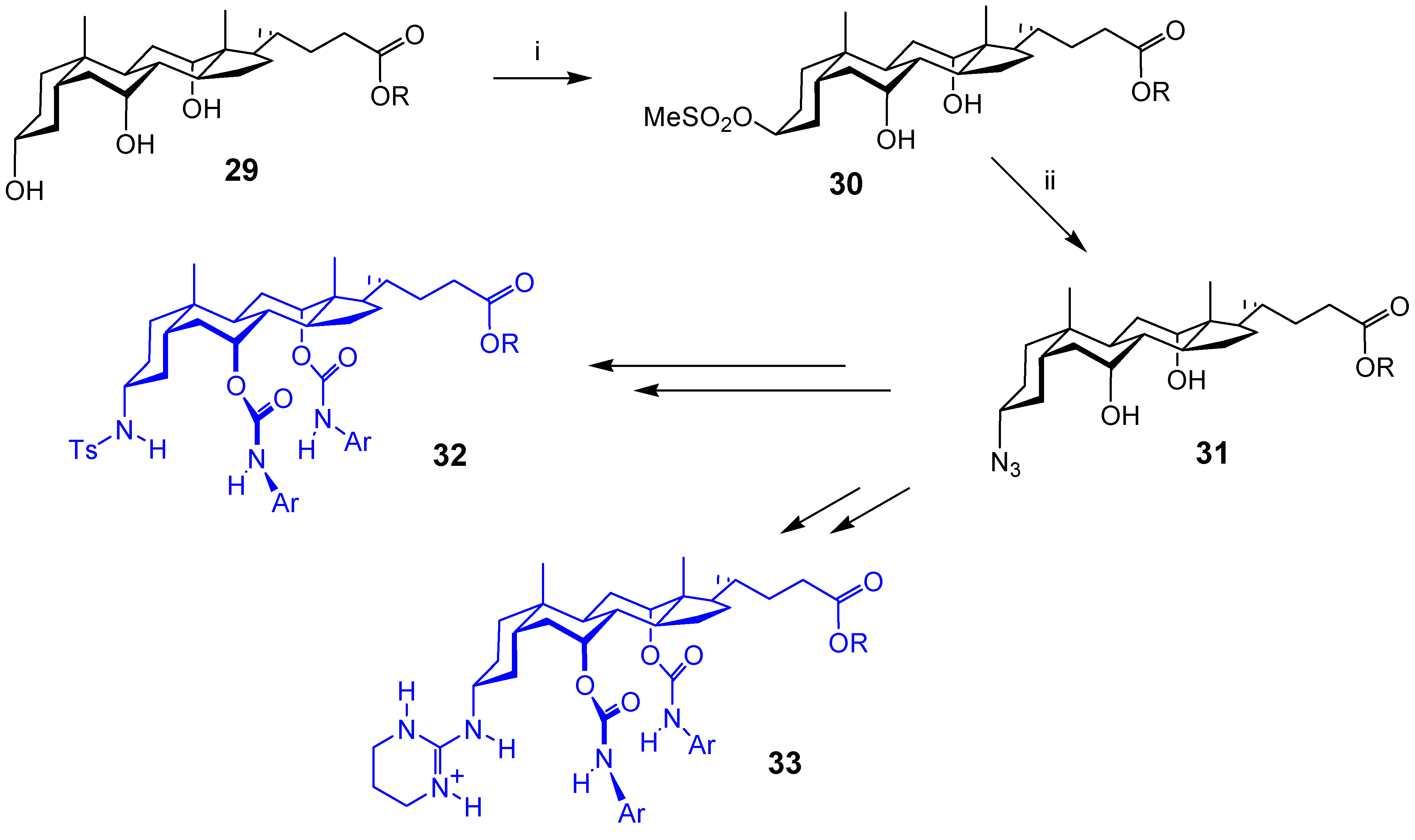
Acyclic Scaffolds - the Cholapod Architecture
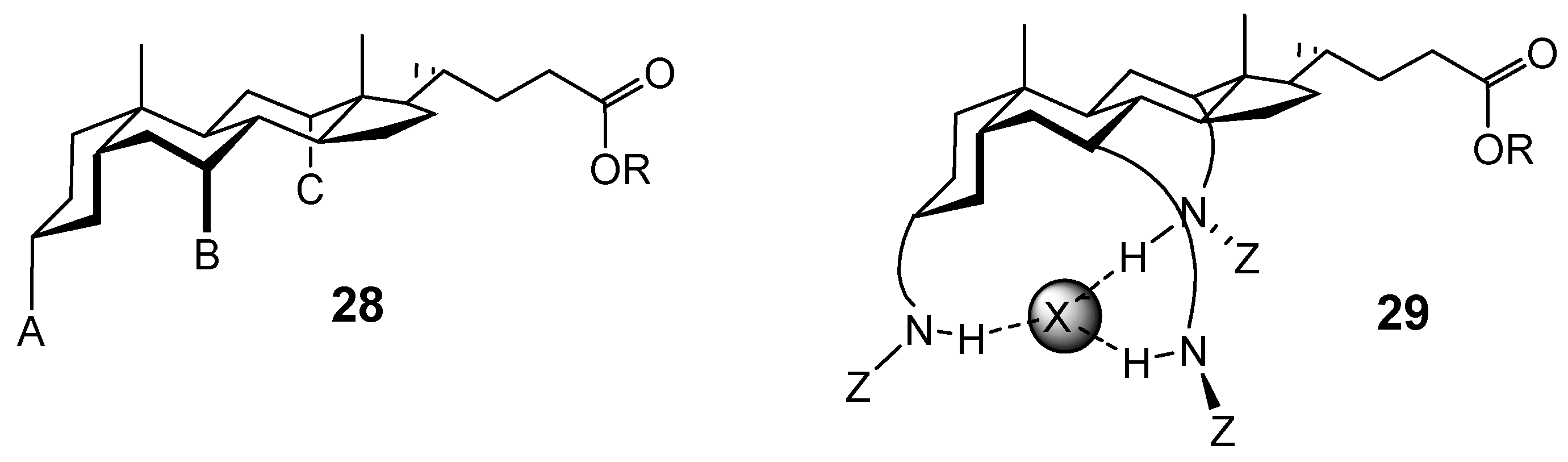
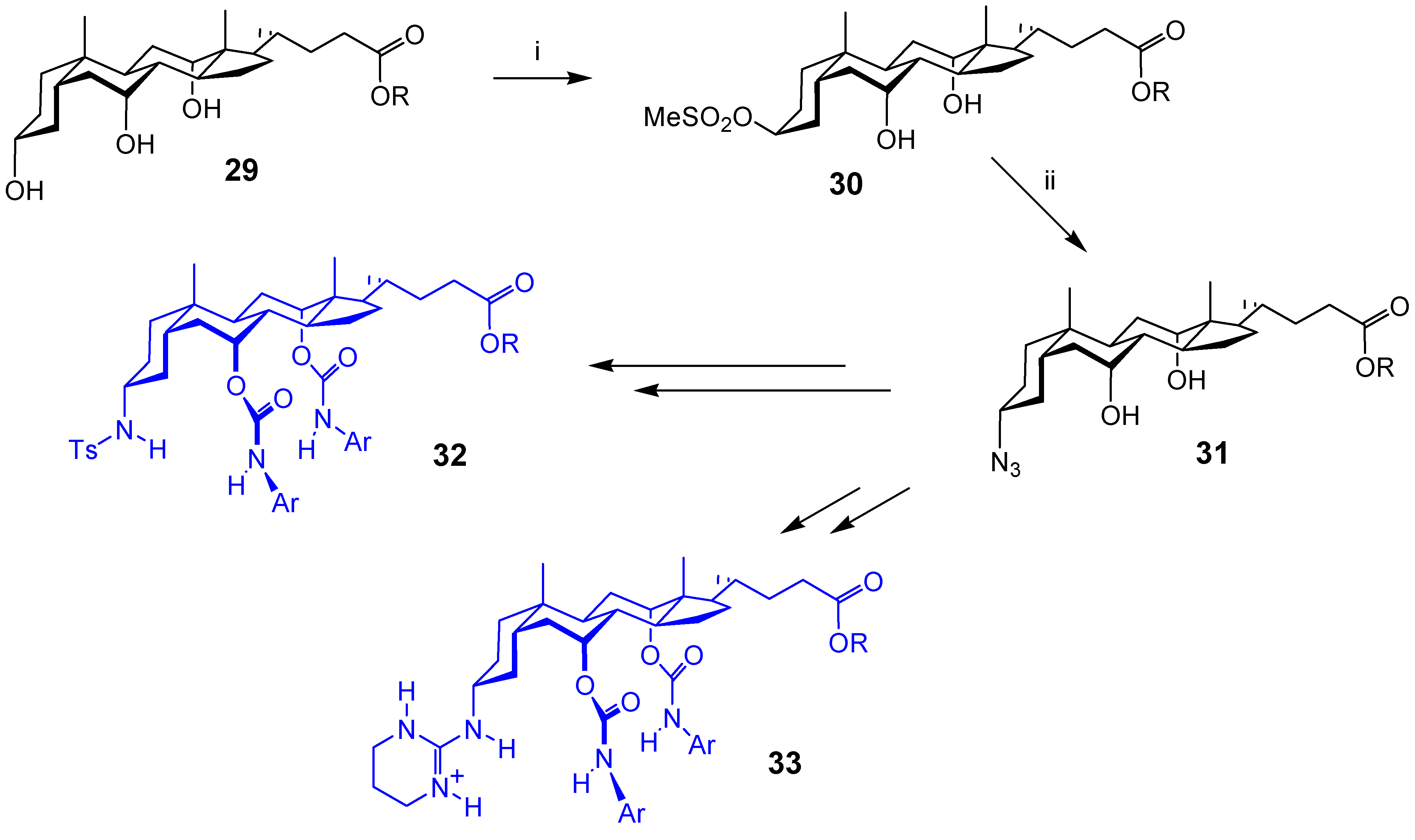
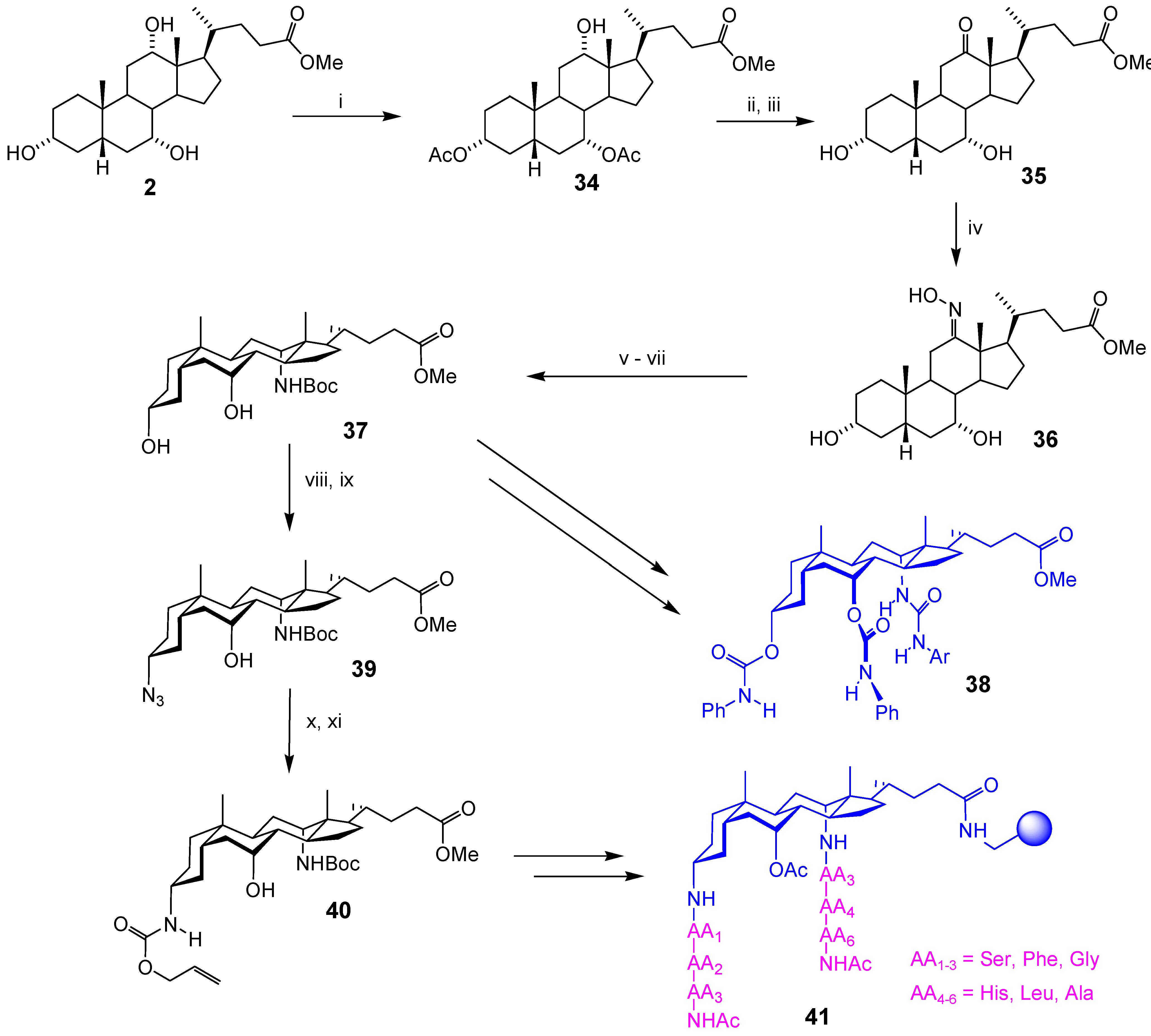
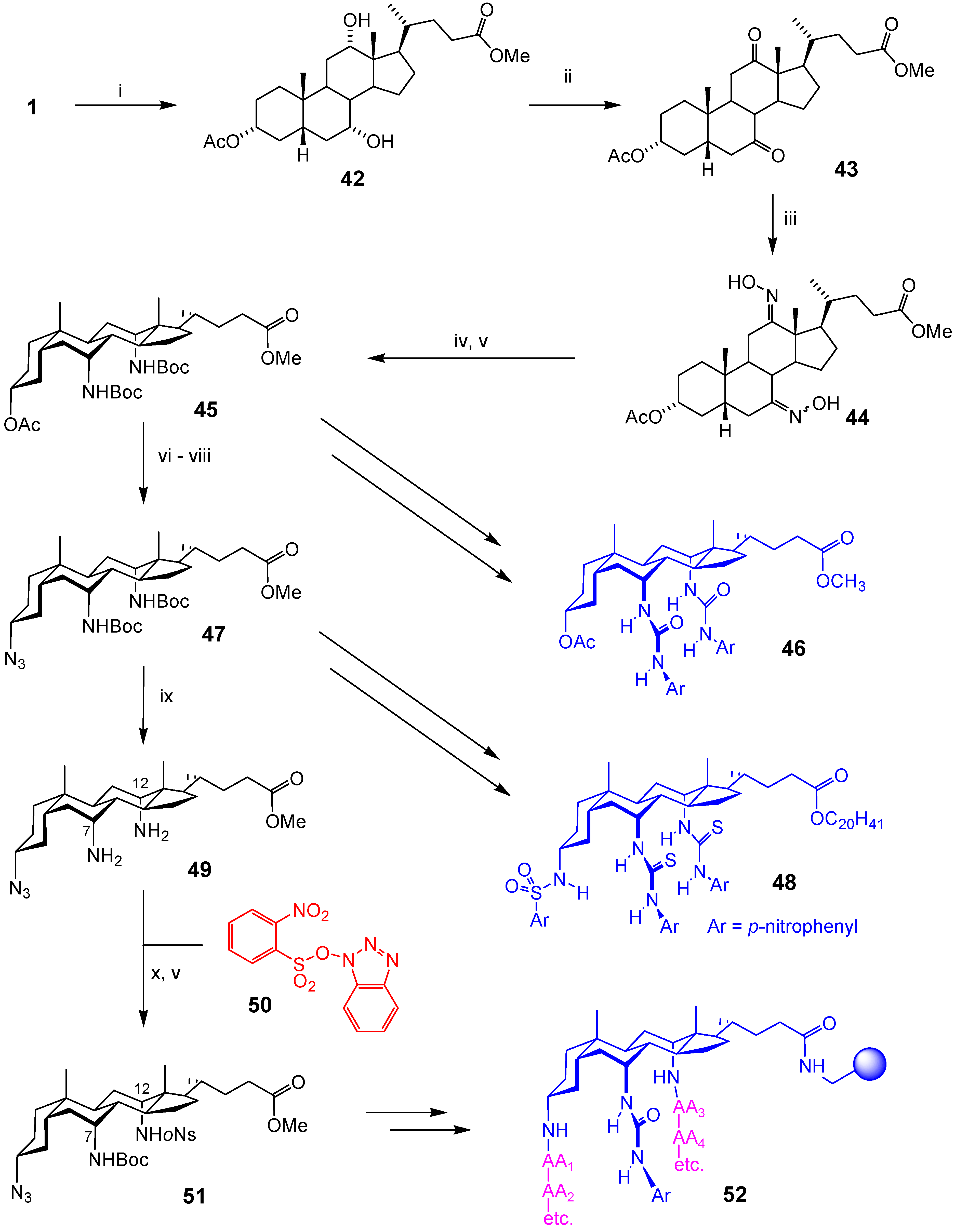
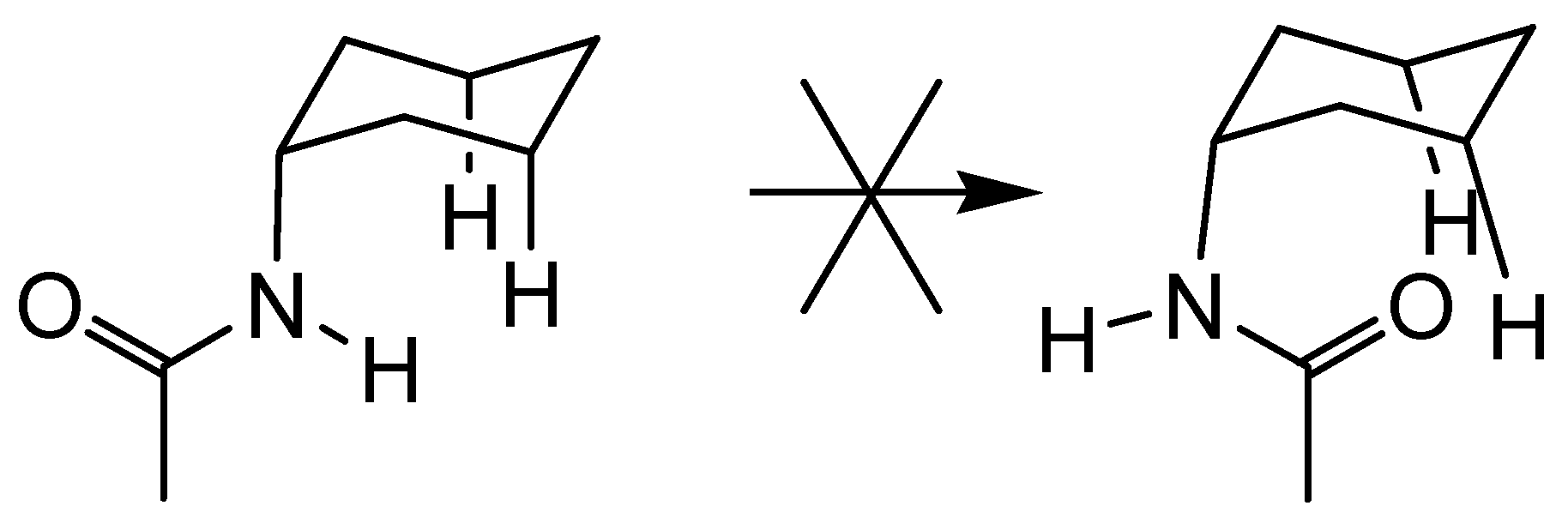
Conclusions
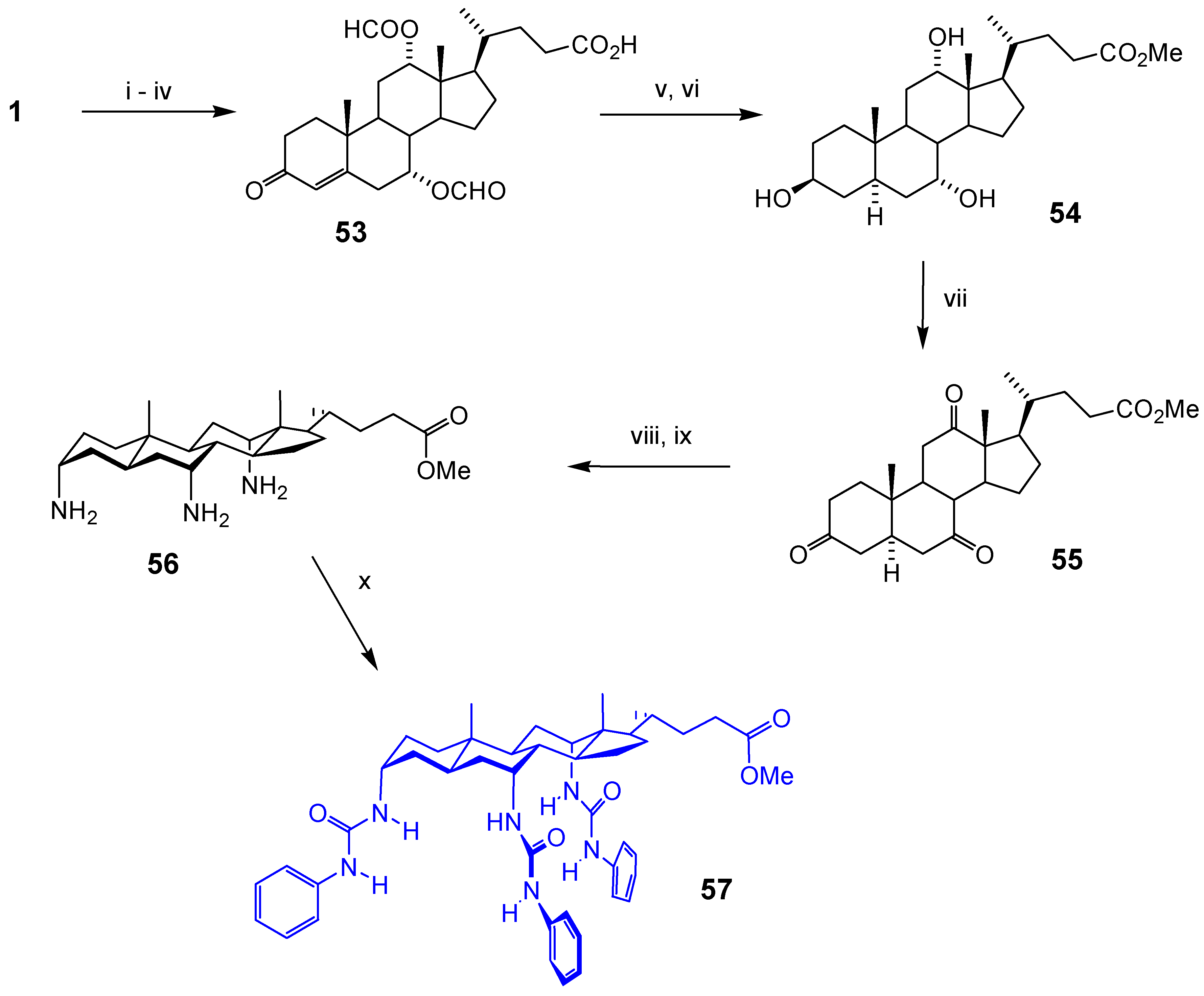
Acknowledgments
References and Notes
- Menger, F. M.; McCreery, M. J. Kinetic characterisation of bile salt micelles. J. Am. Chem. Soc. 1974, 96, 121–126. [Google Scholar] [CrossRef]
- McKenna, J.; McKenna, J. M.; Thornthwaite, D. W. Bis-steroids as potential enzyme models: Perylene solubilisation and dye spectral changes with aqueous solutions of some derivatives of conessine and cholic acid. J. Chem. Soc., Chem. Commun. 1977, 809–811. [Google Scholar] [CrossRef]
- Davis, A. P. Cholaphanes et al.; Steroids as structural components in molecular engineering. Chem. Soc. Rev. 1993, 22, 243–253. [Google Scholar] Davis, A. P.; Bonar-Law, R. P.; Sanders, J. K. M. Comprehensive Supramolecular Chemistry; Murakami, Y., Ed.; Pergamon: Oxford, 1996; Vol. 4, (Supramolecular Reactivity and Transport: Bioorganic Systems); pp. 257–286. [Google Scholar] Li, Y. X.; Dias, J. R. Dimeric and oligomeric steroids. Chem. Rev. 1997, 97, 283–304. [Google Scholar] Tamminen, J.; Kolehmainen, E. Bile acids as building blocks of supramolecular hosts. Molecules 2001, 6, 21–46. [Google Scholar] Virtanen, E.; Kolehmainen, E. Use of bile acids in pharmacological and supramolecular applications. Eur. J. Org. Chem. 2004, 3385–3399. [Google Scholar]
- We estimate that at least 40 groups have contributed to the area.
- Davis, A. P.; Wareham, R. S. Carbohydrate recognition through noncovalent interactions: A challenge for biomimetic and supramolecular chemistry. Angew. Chem., Int. Ed. 1999, 38, 2978–2996. [Google Scholar] [CrossRef]
- Davis, A. P.; Joos, J.-B. Steroids as organising elements in anion receptors. Coord. Chem. Rev. 2003, 240, 143–156. [Google Scholar] [CrossRef] Davis, A. P. Anion binding and transport by steroid-based receptors. Coord. Chem. Rev. 2006, 250, 2939–2951. [Google Scholar]
- Bonar-Law, R. P.; Davis, A. P. Synthesis of steroidal cyclodimers from cholic acid; A molecular framework with potential for recognition and catalysis. J. Chem. Soc., Chem. Commun. 1989, 1050–1052. [Google Scholar] [CrossRef] Bonar-Law, R. P.; Davis, A. P. Cholic acid as an architectural component in molecular recognition chemistry. Synthesis of the first cholaphanes. Tetrahedron 1993, 49, 9829–9844. [Google Scholar]
- Höfle, G.; Steglich, W.; Vorbruggen, H. 4-Dialkylaminopyridines as acylation catalysts. 4. 4- Dialkylaminopyridines as highly active acylation catalysts. Angew. Chem., Int. Ed. Engl. 1978, 17, 569–583. [Google Scholar] [CrossRef]
- Dias, J. R.; Ramachandra, R. Studies directed toward synthesis of quassinoids. 3. Selective hydrolysis of 3-alpha-acetate functional-group of cholic-acid derivatives. Synth. Commun. 1977, 7, 293–297. [Google Scholar] [CrossRef]
- Cahiez, G.; Normant, J. F. Reactivity of organomanganese(ii) reagents - reaction with carbonyl- compounds, selective preparation of ketols from keto-aldehydes. Tetrahedron Lett. 1977, 3383–3384. [Google Scholar] [CrossRef]
- Bhattarai, K. M.; Davis, A. P.; Perry, J. J.; Walter, C. J.; Menzer, S.; Williams, D. J. A new generation of "cholaphanes": Steroid-derived macrocyclic hosts with enhanced solubility and controlled flexibility. J. Org. Chem. 1997, 62, 8463–8473. [Google Scholar] [CrossRef]
- Liao, Y.; Huang, Y. Z. A novel olefination of diazo-compounds with carbonyl-compounds mediated by tributylstibine and catalytic amount of Cu(I)I. Tetrahedron Lett. 1990, 31, 5897–5900. [Google Scholar] [CrossRef] Zhou, Z. L.; Shi, L. L.; Huang, Y. Z. The synthetic application of elementoorganic compounds of 15th and 16th groups. 89. A novel olefination of carbonyl-compounds with dibromomalonate promoted by dibutyl telluride. Synth. Commun. 1991, 21, 1027–1037. [Google Scholar]
- Davis, A. P.; Bhattarai, K. M. Antimony-based “forcing Knoevenagel” methodology for the conversion of ketones into alkylidenemalonates. Tetrahedron 1995, 51, 8033–8042. [Google Scholar] [CrossRef]
- Davis, A. P.; Walsh, J. J. Amide bond formation via pentafluorothiophenyl active esters. Tetrahedron Lett. 1994, 35, 4865–4868. [Google Scholar] [CrossRef]
- Davis, A. P.; Walsh, J. J. Steroid-based receptors with tunable cavities; Stepwise and direct syntheses of a C-3-symmetrical prototype. Chem. Commun. 1996, 449–451. [Google Scholar] Davis, A. P.; Menzer, S.; Walsh, J. J.; Williams, D. J. Steroid-based receptors with tunable cavities; A series of polyhydroxylated macrocycles of varying size and flexibility. Chem. Commun. 1996, 453–455. [Google Scholar]
- Davis, A. P.; Gilmer, J. F.; Perry, J. J. A steroid-based cryptand for halide anions. Angew. Chem., Int. Ed. Engl. 1996, 35, 1312–1315. [Google Scholar] [CrossRef]
- Barton, D. H. R.; Wozniak, J.; Zard, S. Z. A short and efficient degradation of the bile-acid side- chain - some novel reactions of sulfines and alpha-ketoesters. Tetrahedron 1989, 45, 3741–3754. [Google Scholar] [CrossRef]
- Dias, J. R.; Ramachandra, R. Studies directed toward synthesis of quassinoids. 4. D-Ring cleavage of cholic-acid derivatives. Org. Prep. Proc. Int. 1977, 9, 109–115. [Google Scholar] [CrossRef]
- Mekhalfia, A.; Markó, I. E. The silyl modified Sakurai (SMS) reaction. An efficient and versatile one-pot synthesis of homoallylic ethers. Tetrahedron Lett. 1991, 32, 4779–4782. [Google Scholar] [CrossRef]
- Cheng, Y.; Ho, D. M.; Gottlieb, C. R.; Kahne, D.; Bruck, M. A. Facial amphiphiles. J. Am. Chem. Soc. 1992, 114, 7319–7320. [Google Scholar] [CrossRef]
- Boyce, R.; Li, G.; Nestler, H. P.; Suenaga, T.; Still, W. C. Peptidosteroidal receptors for opioid peptides. Sequence-selective binding using a synthetic receptor library. J. Am. Chem. Soc. 1994, 116, 7955–7956. [Google Scholar] [CrossRef]
- Davis, A. P.; Dresen, S.; Lawless, L. J. Mitsunobu reactions with methanesulfonic acid; The replacement of equatorial hydroxyl groups by azide with net retention of configuration. Tetrahedron Lett. 1997, 38, 4305–4308. [Google Scholar] [CrossRef]
- del Amo, V.; Siracusa, L.; Markidis, T.; Baragaña, B.; Bhattarai, K. M.; Galobardes, M.; Naredo, G.; Pérez-Payán, M. N.; Davis, A. P. Differentially-protected steroidal triamines; Scaffolds with potential for medicinal, supramolecular, and combinatorial chemistry. Org. Biomol. Chem. 2004, 2, 3320–3328. [Google Scholar] [CrossRef]
- Davis, A. P.; Perry, J. J.; Williams, R. P. Anion recognition by tripodal receptors derived from cholic acid. J. Am. Chem. Soc. 1997, 119, 1793–1794. [Google Scholar] [CrossRef] Ayling, A. J.; Broderick, S.; Clare, J. P.; Davis, A. P.; Pérez-Payán, M. N.; Lahtinen, M.; Nissinen, M. J.; Rissanen, K. An extraction-based assay for neutral anionophores: The measurement of high binding constants to steroidal receptors in a nonpolar solvent. Chem. Eur. J. 2002, 8, 2197–2203. [Google Scholar]
- Lawless, L. J.; Blackburn, A. G.; Ayling, A. J.; Pérez-Payán, M. N.; Davis, A. P. Steroidal guanidines as enantioselective receptors for N-acyl alpha-amino acids. Part 1. 3 alpha-Guanylated carbamates derived from cholic acid. J. Chem. Soc., Perkin Trans. 1 2001, 1329–1341. [Google Scholar] Baragaña, B.; Blackburn, A. G.; Breccia, P.; Davis, A. P.; de Mendoza, J.; Padron-Carrillo, J. M.; Prados, P.; Riedner, J.; de Vries, J. G. Enantioselective transport by a steroidal guanidinium receptor. Chem. Eur. J. 2002, 8, 2931–2936. [Google Scholar]
- For an example, see Hsieh, H.-P.; Muller, J. G.; Burrows, C. J. Structural effects in novel steroidal polyamine-DNA binding. J. Am. Chem. Soc. 1994, 116, 12077–12078. [Google Scholar] [CrossRef]
- Barry, J. F.; Davis, A. P.; Pérez-Payán, M. N.; Elsegood, M. R. J.; Jackson, R. F. W.; Gennari, C.; Piarulli, U.; Gude, M. A trifunctional steroid-based scaffold for combinatorial chemistry. Tetrahedron Lett. 1999, 40, 2849–2852. [Google Scholar] [CrossRef]
- Fieser, L. F.; Rajagopalan, S. Oxidation of Steroids. III. Selective oxidations and acylations in the bile acid series. J. Am. Chem. Soc. 1950, 72, 5530–5536. [Google Scholar] [CrossRef]
- Siracusa, L.; Hurley, F. M.; Dresen, S.; Lawless, L. J.; Pérez-Payán, M. N.; Davis, A. P. Steroidal ureas as enantioselective receptors for an N-acetyl alpha-amino carboxylate. Org. Lett. 2002, 4, 4639–4642. [Google Scholar] [CrossRef]
- De Muynck, H.; Madder, A.; Farcy, N.; De Clercq, P. J.; Pérez-Payán, M. N.; Öhberg, L. M.; Davis, A. P. Application of combinatorial procedures in the search for serine-protease-like activity with focus on the acyl transfer step. Angew. Chem., Int. Ed. 2000, 39, 145–148. [Google Scholar] [CrossRef]
- Kuhajda, K.; Kandrac, J.; Cirin-Novta, V.; Miljkovic, D. One-pot esterification and selective 3 alpha-acetylation of cholic and deoxycholic acid. Coll. Czech. Chem. Comm. 1996, 61, 1073–1076. [Google Scholar] [CrossRef]
- Davis, A. P.; Pérez-Payán, M. N. The "triamino-analogue" of methyl cholate; A practical, large- scale synthesis. Synlett 1999, 991–993. [Google Scholar]
- Koulov, A. V.; Lambert, T. N.; Shukla, R.; Jain, M.; Boon, J. M.; Smith, B. D.; Li, H. Y.; Sheppard, D. N.; Joos, J. B.; Clare, J. P.; Davis, A. P. Chloride transport across vesicle and cell membranes by steroid-based receptors. Angew. Chem., Int. Ed. 2003, 42, 4931–4933. [Google Scholar] [CrossRef]
- For a complementary approach to a triamino steroidal scaffold see Li, C. H.; Rehman, A.; Dalley, N. K.; Savage, P. B. Short syntheses of triamine derivatives of cholic acid. Tetrahedron Lett. 1999, 40, 1861–1864. [Google Scholar] [CrossRef]
- Ayling, A. J.; Pérez-Payán, M. N.; Davis, A. P. New "cholapod" anionophores; High-affinity halide receptors derived from cholic acid. J. Am. Chem. Soc. 2001, 123, 12716–12717. [Google Scholar] Clare, J. P.; Ayling, A. J.; Joos, J. B.; Sisson, A. L.; Magro, G.; Pérez-Payán, M. N.; Lambert, T. N.; Shukla, R.; Smith, B. D.; Davis, A. P. Substrate discrimination by cholapod anion receptors: Geometric effects and the "affinity-selectivity principle". J. Am. Chem. Soc. 2005, 127, 10739–10746. [Google Scholar]
- McNally, B. A.; Koulov, A. V.; Smith, B. D.; Joos, J. B.; Davis, A. P. A fluorescent assay for chloride transport; Iidentification of a synthetic anionophore with improved activity. Chem. Commun. 2005, 1087–1089. [Google Scholar]
- Davis, A. P.; Sheppard, D. N.; Smith, B. D. Development of synthetic membrane transporters for anions. Chem. Soc. Rev. 2007, 36, 348–357. [Google Scholar] [CrossRef]
- Fukuyama, T.; Jow, C.-K.; Cheung, M. 2- nd 4-Nitrobenzenesulfonamides: Exceptionally versatile means for preparation of secondary amines and protection of amines. Tetrahedron Lett. 1995, 36, 6373–6374. [Google Scholar] [CrossRef]
- Bhattarai, K. M.; del Amo, V.; Magro, G.; Sisson, A. L.; Joos, J. B.; Charmant, J. P. H.; Kantacha, A.; Davis, A. P. The "triamino-analogue" of methyl allocholate; A rigid, functionalised scaffold for supramolecular chemistry. Chem. Commun. 2006, 2335–2337. [Google Scholar]
- Kallner, A. A method of synthesis of allocholanoic acids - bile acids and steroids 182. Acta Chem. Scand. 1967, 21, 322–328. [Google Scholar] [CrossRef]
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Davis, A.P. Bile Acid Scaffolds in Supramolecular Chemistry: The Interplay of Design and Synthesis. Molecules 2007, 12, 2106-2122. https://doi.org/10.3390/12082106
Davis AP. Bile Acid Scaffolds in Supramolecular Chemistry: The Interplay of Design and Synthesis. Molecules. 2007; 12(9):2106-2122. https://doi.org/10.3390/12082106
Chicago/Turabian StyleDavis, Anthony P. 2007. "Bile Acid Scaffolds in Supramolecular Chemistry: The Interplay of Design and Synthesis" Molecules 12, no. 9: 2106-2122. https://doi.org/10.3390/12082106
APA StyleDavis, A. P. (2007). Bile Acid Scaffolds in Supramolecular Chemistry: The Interplay of Design and Synthesis. Molecules, 12(9), 2106-2122. https://doi.org/10.3390/12082106