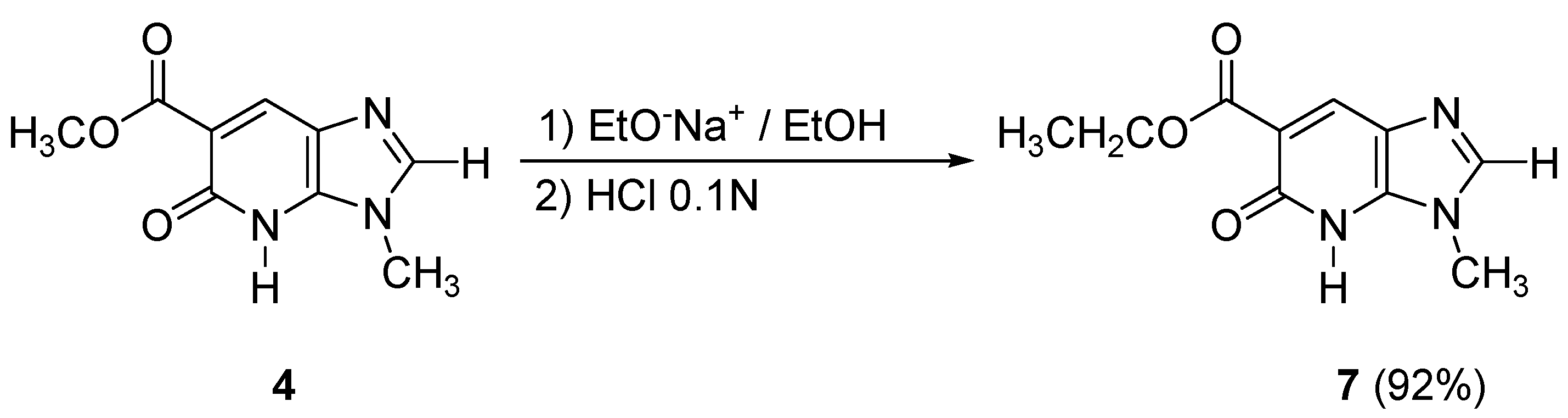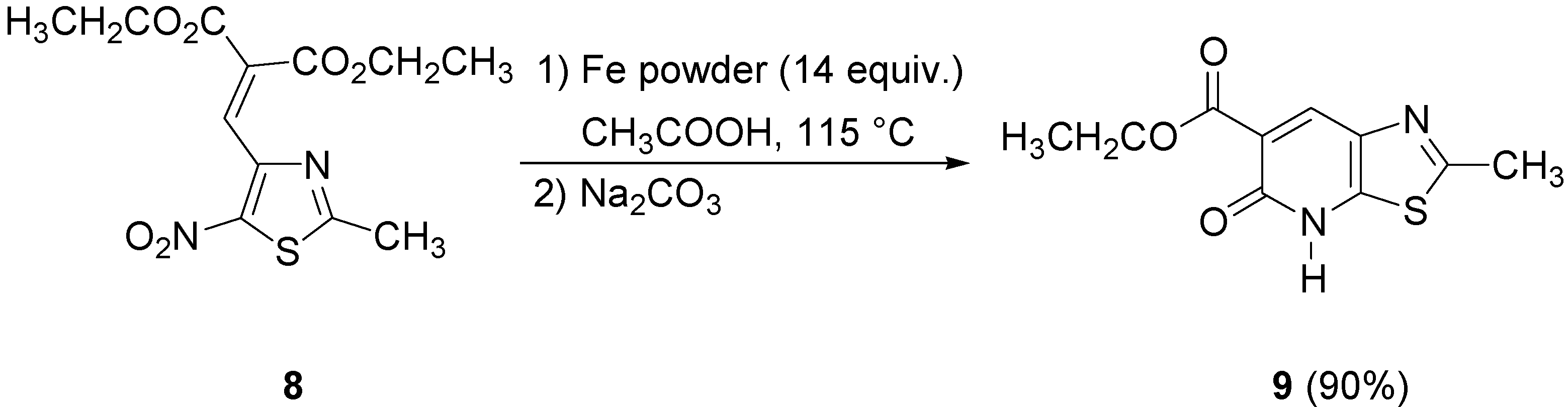Results and Discussion
Due to the high functionalization of the precursors of the pyridones (malonic acid diethyl ester, conjugated double bond, nitro group, heterocyclic ring with a nucleophilic heteroatom), we had to study the reduction reaction in order to selectively reduce the nitro group. Moreover, we studied the reactivity of the amino group obtained from the nitro group reduction and determined if the cyclization reaction could be made in a one-pot reduction-cyclization.
Many reagents can be used to reduce aromatic nitro compounds, the most common being Zn, Sn or Fe (or sometimes other metals) under acidic conditions, or catalytic hydrogenation. Among other reagents we can also mention are Al-NiCl
2·6H
2O-THF [
2], titanium (II) [
3] and titanium (III) [
4] derivatives, FeCl
3·6H
2O-H
2N-N(CH
3)
2 [
5], AlH
3-AlCl
3, formic acid and Pd-C [
6].
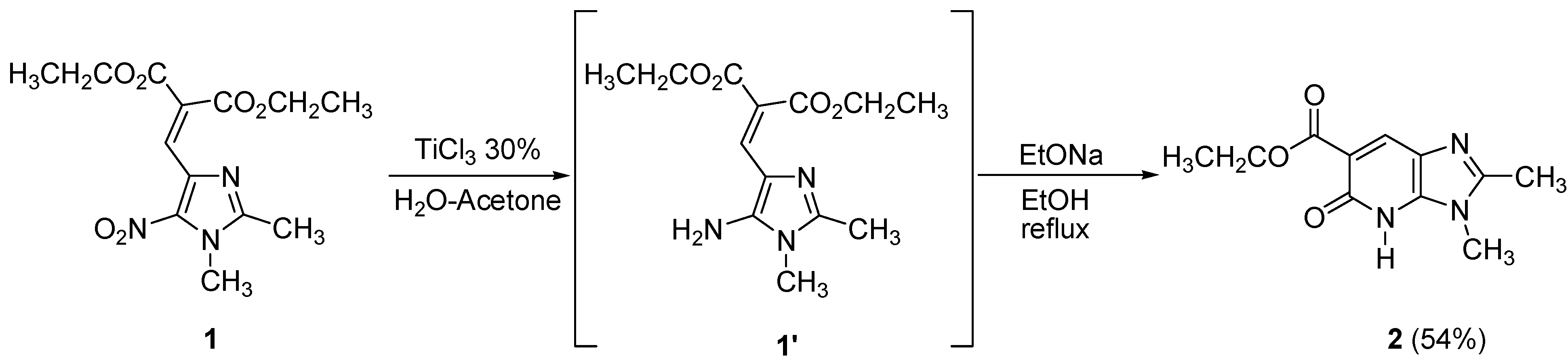
In our preliminary study we focused on imidazoles and our first choice to reduce the nitro group of diethyl 2-[(1,2-dimethyl-5-nitro-1
H-imidazol-4-yl)methylene]malonate (
1) was a weak reducing agent, titanium(III) chloride (30 wt% solution in 2N hydrochloric acid) in a H
2O-acetone mixture at room temperature. The resulting intermediate was then heated in ethanolic sodium ethoxide solution to give the corresponding bicyclic pyridone
2 in 54% yield (
Scheme 1).
Unfortunately, this method was found to not be applicable to all nitroimidazoles. The 5-nitro-imidazoles without alkyl substituents in the 2-position gave poor yields, so we had to find more general conditions and switched to other metal halides. We thus found that SnCl
2 in ethanol gave the best results. Reduction of compound
3 occurred with five equivalents of SnCl
2 in ethanol at 70 °C, then the corresponding amine was treated with two equivalents of magnesium (powder) in methanol at room temperature during 20 h leading to the corresponding bicyclic pyridone
4 in 92% yield (
Scheme 2).
The imidazopyridone
4 was obtained as the methyl ester due to the transesterification reaction in methanol. These conditions were applied to the
N-ethyl imidazole analogue of
3, but gave the imidazopyridone
6 in poor yield. No transesterification was observed in this case (
Scheme 3).
In order to obtain the imidazopyridone
7 containing a carboxylic acid ethyl ester moiety in the 6-position, transesterification conditions with sodium ethoxide in ethanol (room temperature, 24 h) were used from compound
4, leading to the desired bicycle
7 in excellent yield (92%) (
Scheme 4).
On the other hand, both the procedures previously described in this paper gave very poor yields of thiazolopyridone
9 when applied to the nitrothiazole
8. Consequently, we tried other reaction conditions using iron in glacial acetic acid as reported in our previous publication [
7]. Under these conditions diethyl 2-[(2-methyl-5-nitrothiazol-4-yl)methylene]malonate (
8) afforded the target lactam
9 in 90% yield (
Scheme 5).
Experimental
General
Melting points were determined on a Büchi B-540 apparatus and are uncorrected. Elemental analyses were performed by the Centre de Microanalyses of The Paul Cézanne University (Aix-Marseille 3). Both
1H- and
13C-NMR spectra were determined on a Bruker ARX 200 spectrometer. The
1H chemical shifts were reported as parts per million downfield from chloroform-d (CDCl
3), and
13C chemical shifts were referenced to the CDCl
3solvent peak (76.9 ppm). Silica gel 60 (Merck, 230-400 mesh) was used as adsorbent for column chromatography. Thin layer chromatography was performed with silica gel Merck 60F-254 (0.25 mm layer thickness). Diethyl 2-[(1,2-dimethyl-5-nitro-1
H-imidazol-4-yl)methylene]malonate (
1), diethyl 2-[(1-methyl-5-nitro-1
H-imidazol-4-yl)methylene]-malonate (
3) and diethyl 2-[(2-methyl-5-nitrothiazol-4-yl)methylene]malonate (
8) were previously described [
1].
Preparation of ethyl 2,3-dimethyl-5-oxo-4,5-dihydro-3H-imidazo[4,5-b]pyridine-6-carboxylate (2)
a) Reduction using Titanium(III) chloride
In a two-necked flask equipped with a reflux condenser, titanium(III) chloride (30 wt% solution in 2N hydrochloric acid, 21 mL) were added dropwise on a solution of 1 (1.25 g, 4.02 mmol) in water (42 mL) and acetone (82 mL). The mixture was stirred at room temperature for 20 h. Then, a saturated aqueous solution of Na2CO3 was added. The green cake obtained was filtered and washed with dichloromethane, then, the solvent was dried over MgSO4 and removed under reduced pressure. The resulting amine was used for the cyclization reaction without any further purification.
b) Cyclization using sodium ethoxide in ethanol
In a two-necked flask equipped with a reflux condenser, ethanol (5 mL) and sodium (100 mg) were stirred until complete dissolution. A solution of the previously obtained amine in ethanol (45 mL) was added rapidly. This mixture was stirred at room temperature for 14 h and then at the reflux temperature of the ethanol for 2 additional hours. After cooling, water was added to the mixture, the solution was then extracted with chloroform. The combined organic layers were dried over MgSO4 and removed under vacuum. Purification by chromatography on silica gel eluting with ethyl acetate and recrystallization led to the corresponding product 2 (400 mg, 54%). Yellow solid, mp 159.6 °C (ethyl acetate); 1H-NMR (CDCl3) δ: 1.44 (t, J = 7.3 Hz, 3H, CH2CH3), 2.59 (s, 3H, CH3), 3.73 (s, 3H, NCH3), 4.43 (q, J = 7.3 Hz, 2H, CH2CH3), 8.42 (s, 1H, Ar-H), 11.85 (s, 1H, NH); 13C-NMR (CDCl3) δ: 15.2 (CH3), 15.4 (CH3), 28.6 (CH3), 62.2 (CH2), 102.5 (C), 128.8 (CH), 130.2 (C), 151.0 (C), 154.2 (C), 162.6 (C), 170.2 (C); Anal. Calcd for C11H13N3O3 : C, 56.16; H, 5.57; N, 17.86. Found : C, 56.13; H, 5.53; N, 17.63.
Preparation of methyl 3-methyl-5-oxo-4,5-dihydro-3H-imidazo[4,5-b]pyridine-6-carboxylate (4)
In a two-necked flask equipped with a reflux condenser, ethanol (32 mL), 3 (940 mg, 3.17 mmol) and tin(II) chloride (3 g, 15.83 mmol) were stirred at 70 °C for 1 h. After cooling, the mixture was poured into an ice bath and made basic to pH=14 with 1N sodium hydroxide solution. The aqueous layer was then extracted with chloroform. The combined organic layer was dried over MgSO4 and evaporated. Methanol (47 mL) was added to the oil previously obtained, then magnesium metal turnings (154 mg, 6.22 mmol) were added. The mixture was stirred at room temperature for 16 h and acidified with 0.1N hydrochloric acid. The aqueous layer was extracted with chloroform. The combined organic layer was dried over MgSO4 and evaporated to give 600 mg (92% yield) of 4 as a brown solid. Mp 345.1 °C (ethanol/ethyl acetate: 2:1); 1H-NMR (CDCl3) δ: 3.84 (s, 3H, CH3), 4.01 (s, 3H, OCH3), 8.01 (s, 1H, Ar-H), 8.60 (s, 1H, Ar-H), 11.81 (s, 1H, NH); 13C-NMR (CDCl3) δ: 29.8 (CH3), 52.9 (CH3), 103.2 (C), 129.9 (C), 132.0 (CH), 145.2 (CH), 149.8 (C), 163.1 (C), 170.5 (C). Anal. Calcd for C9H9N3O3 : C, 52.17; H, 4.38; N, 20.28. Found : C, 52.02; H, 4.38; N, 19.98.
Preparation of ethyl 3-ethyl-5-oxo-4,5-dihydro-3H-imidazo[4,5-b]pyridine-6-carboxylate (6)
Following the procedure described for 4, 200 mg (27% yield) of ethyl 3-ethyl-5-oxo-4,5-dihydro-3H-imidazo[4,5-b]pyridine-6-carboxylate (6) was obtained as a yellow solid, mp 102.5 °C (ethanol); 1H‑NMR (CDCl3) δ: 1.44 (t, J = 7.1 Hz, 3H, CH2CH3), 1.52 (t, J = 7.3 Hz, 3H, CH2CH3), 4.26 (q, J = 7.3 Hz, 2H, CH2CH3), 4.45 (q, J = 7.1 Hz, 2H, CH2CH3), 7.97 (s, 1H, Ar-H), 8.58 (s, 1H, Ar-H), 11.87 (s, 1H, NH); 13C-NMR (CDCl3) δ: 14.1 (CH3), 15.3 (CH3), 38.6 (CH2), 62.1 (CH2), 103.5 (C), 130.0 (C), 131.9 (CH), 144.0 (CH), 149.2 (C), 163.0 (C), 170.1 (C); Anal. Calcd for C11H13N3O3 : C, 56.16; H, 5.57; N, 17.86. Found : C, 56.25; H, 5.57; N, 17.25.
Preparation of ethyl 3-methyl-5-oxo-4,5-dihydro-3H-imidazo[4,5-b]pyridine-6-carboxylate (7)
In a two-necked flask equipped with a reflux condenser, ethanol (10 mL) and sodium (0.03 g, 1.3 mmol) were stirred at room temperature. After formation of the sodium ethoxide, a solution of 4 (0.2 g, 0.97 mmol) in ethanol (10 mL) was added dropwise. The mixture was stirred at room temperature for 48 h, then it was acidified with 0.1N hydrochloric acid to pH=4. The aqueous layer was extracted with chloroform. The combined organic layer was dried over MgSO4 and evaporated to give 200 mg of 7 (92% yield). An analytical sample of 7 (yellow solid, mp 164.0 °C) was obtained by crystallization (ethanol); 1H-NMR (CDCl3) δ: 1.44 (t, J = 7.1 Hz, 3H, CH2CH3), 3.83 (s, 3H, CH3), 4.45 (q, J = 7.1 Hz, 2H, CH2CH3), 7.94 (s, 1H, Ar-H), 8.60 (s, 1H, Ar-H), 11.89 (s, 1H, NH); 13C-NMR (CDCl3) δ: 14.1 (CH3), 29.8 (CH3), 62.2 (CH2), 103.5 (C), 129.3 (C), 129.8 (C), 132.0 (CH), 145.1 (CH), 163.3 (C), 170.1 (C). Anal. Calcd for C10H11N3O3 : C, 54.29; H, 5.01; N, 19.00. Found : C, 54.27; H, 4.96; N, 18.76.
Preparation of ethyl 2-methyl-5-oxo-4,5-dihydrothiazolo[5,4-b]pyridine-6-carboxylate (9)
In a two-necked flask equipped with a reflux condenser, 8 (100 mg, 0.314 mmol) and glacial acetic acid (5 mL) were stirred and heated at reflux. To this solution was added iron powder (250 mg, 4.46 mmol) and the stirred mixture was heated at reflux for 2 h. After cooling, the solution was filtered through Celite and washed with glacial acetic acid. The acetic acid solution was evaporated on a rotary evaporator and the residue made basic with aqueous Na2CO3. The aqueous layer was extracted with chloroform. The combined organic layer was dried over MgSO4 and evaporated to give 70 mg of 9 (90% yield). An analytical sample of 9 was obtained as a beige solid, mp 114.6 °C, by crystallization (isopropanol); 1H-NMR (CDCl3) δ: 1.43 (t, J = 6.8 Hz, 3H, CH2CH3), 2.78 (s, 3H, CH3), 4.46 (q, J = 6.8 Hz, 2H, CH2CH3), 8.61 (s, 1H, Ar-H), 11.63 (s, 1H, NH); 13C-NMR (CDCl3) δ: 14.0 (CH3), 20.8 (CH3), 62.6 (CH2), 106.3 (C), 132.3 (CH), 141.2 (C), 162.4 (C), 163.6 (C), 166.1 (C), 169.3 (C); Anal. Calcd for C10H10N2O3S : C, 50.41; H, 4.23; N, 11.76. Found : C, 50.44; H, 4.20; N, 11.68.
{kind=link}
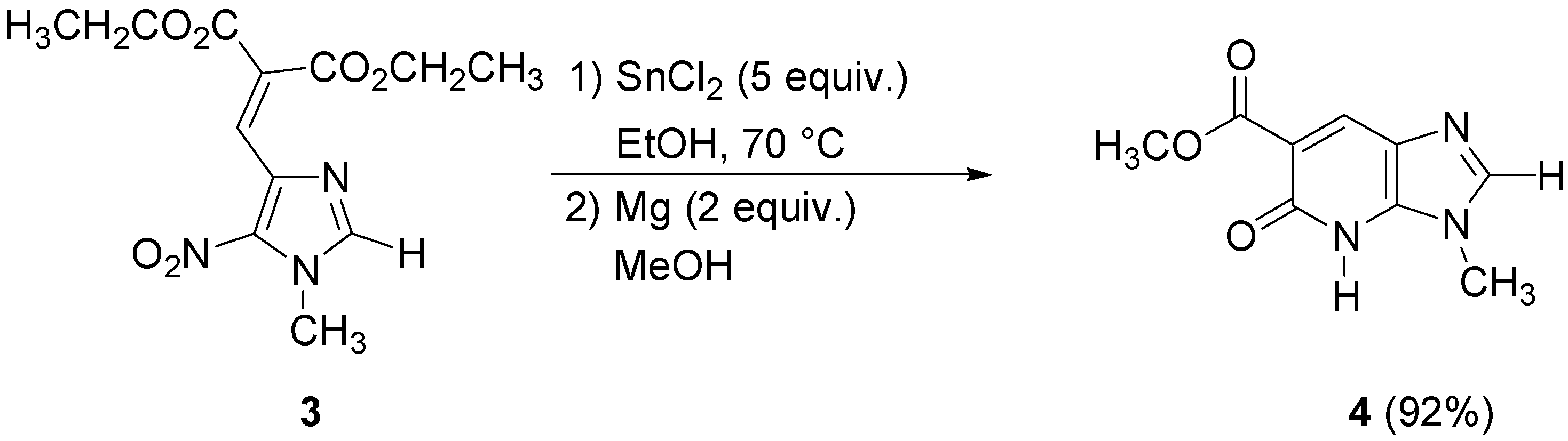{kind=link}
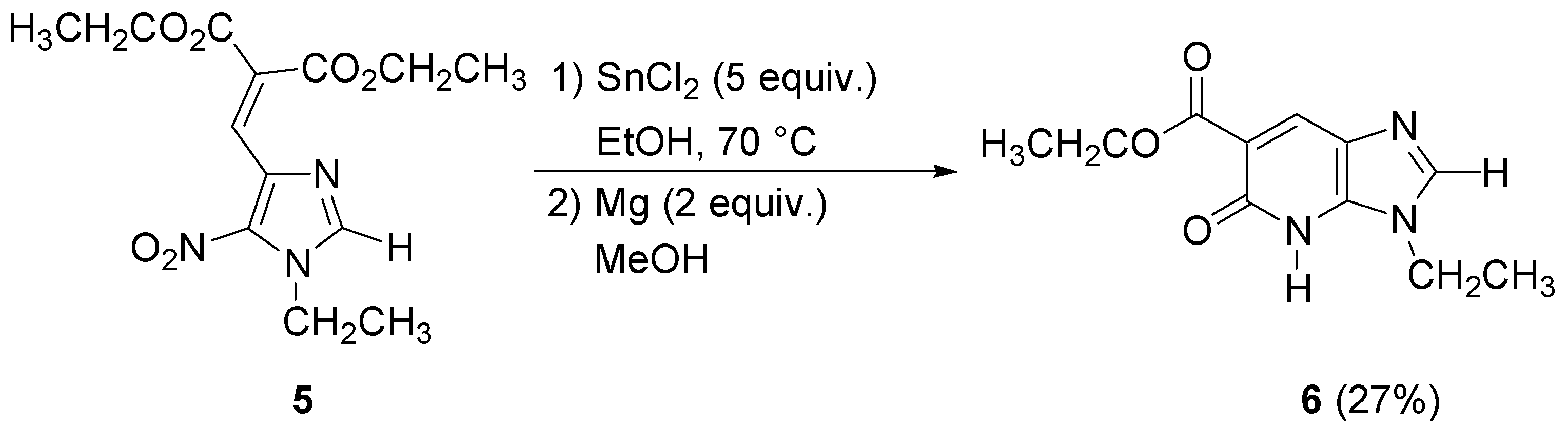{kind=link}
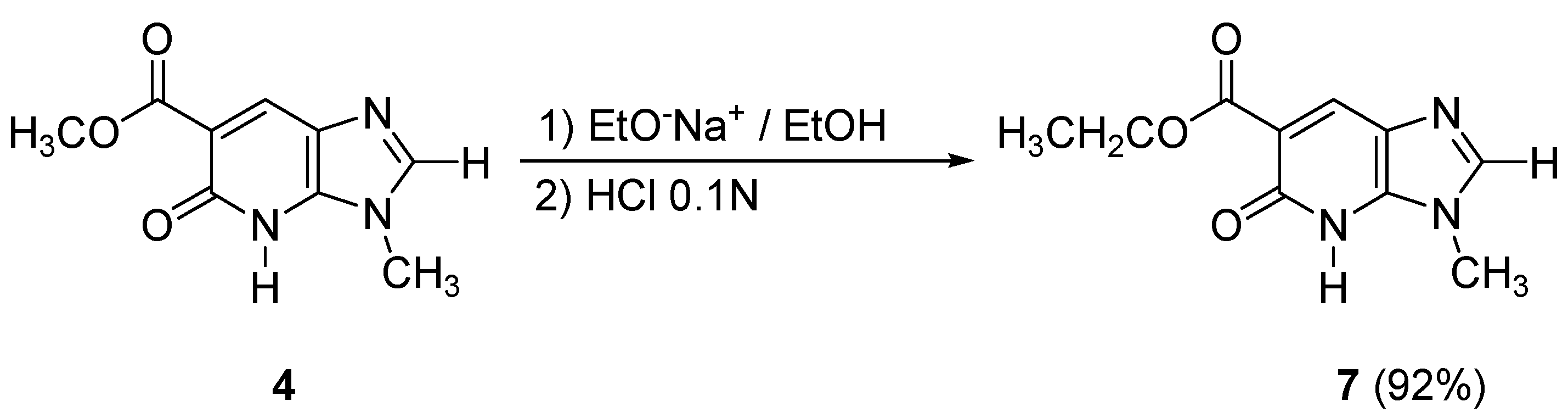{kind=link}
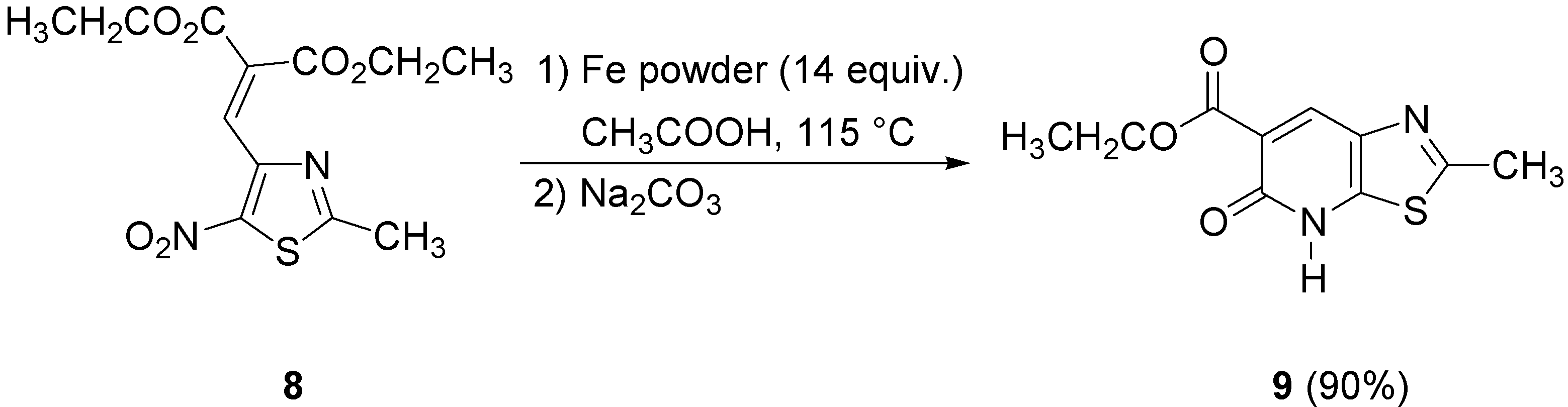{kind=link}