Abstract
Aside from HPLC and GC, capillary electrophoresis (CE) is one of the most important techniques for high-performance separations in modern analytical chemistry. Its main advantages are the possibility of using different detection techniques, the possibility of in-capillary sample processing for preconcentration or derivatization, and ease of instrumental miniaturization down to the microfluidic scale. Those features are utilized in the separation of macromolecules in biochemistry and in genetic investigations, but they can be also used in determinations of inorganic ions in water analysis. This review, based on about 100 original research works, presents applications of CE methods in water analysis reported in recent decade, mostly regarding conductivity detection or indirect UV detection. The developed applications include analysis of high salinity sea waters, as well as analysis of other surface waters and drinking waters.
1. Introduction
The current common requirement for the quality control of water for municipal and industrial needs, and also for environmental protection, has resulted practically in a permanent search for the development of improved analytical techniques and instrumentation for the analytical techniques used for the determination of inorganic macro- and micro-components, as well as organic compounds usually occurring at trace levels. With very wide needs for such analyses, the development of analytical techniques providing the possibility of simultaneous multicomponent determination is especially valuable. Atomic emission spectroscopy techniques were employed quite early for the purpose of determinations of numerous light elements, but the most significant breakthrough for both inorganic ions and organic analytes was the introduction of liquid chromatography techniques to the arsenal of water analysis techniques. The invention of high-performance ion-chromatography (IC) in the 1980s essentially improved determinations of inorganic ions in waters. Currently those IC methods are commonly accepted and routinely used in water analyses for various needs.
Considering both speed of analysis and the possibility of multicomponent determinations in IC determinations of anions and cations in waters, the only technique competitive with IC seems to be the capillary electrophoresis (CE). CE can be generally considered a technique complementary to IC in the determination of small inorganic ions due to its favorable features, such as short analysis times, relatively low running and instrumentation costs, small sample volume and reagent consumption, excellent separation efficiency, suitability for miniaturization, and portability. CE is also able to tolerate injection of samples with heavy loaded matrices as the capillary can be easily cleaned between injections. On the other hand, IC equipment and columns for ion chromatography are usually more expensive, and may have rather short lifetime.
Since its invention and employment in the first works of Virtanen in 1974 [1], and Jacobson and Lucas in 1981 [2], CE is considered an attractive and complementary separation technique to high-performance liquid chromatography (HPLC). This is very well documented by a very vast literature: nearly 1800 original research papers and review articles are now published annually. See, for instance, a review on current state of development and recent trends in analytical capillary electrophoresis [3]. One can notice, however, that although the main mechanism of CE deals with the separation of ions, in fact only a very small part of research efforts are focused on optimizing the separation of inorganic ionic species, while the majority of applications deal with determinations of organic analytes. This is a consequence of the limitations of commercially available CE detectors other than UV-VIS spectrophotometers. Numerous other detection techniques are laboratory adapted for research purposes, but not for routine use. So, in cases of routine applications for determination of ions which do not absorb UV-VIS radiation, indirect UV-VIS detection or laser-induced fluorescence (LIF) are employed, requiring appropriate derivatization of the analytes used.
Numerous studies have compared the detection limits of CE and IC in determinations of inorganic ions. For instance, in determinations of anions in drinking water, LODs for a single-column ion chromatography with conductivity detection were found in a ppm range, which for CE with indirect UV detection was about 0.3 to 0.5 ppm [4]. Detection of LODs for inorganic ions in a suppressed IC with conventional conductivity are similar to those of CE with indirect UV detection [5]. It has to be admitted, however, that additional in-capillary preconcentration, e.g., by so called stacking procedures, may significantly improve the detection limits of CE methods [6]. Without stacking preconcentration, however, the LODs for anions in IC systems with UV detection are lower than those for CE systems with indirect UV detection.
The application of C4D contactless conductivity detection in CE significantly improves the limits of the detection of inorganic cations and anions, which are only 2.7 times higher than non-suppressed single column ICs with conductivity detection, and 10 times higher than those obtained for suppressed IC with conductivity detection, respectively [7]. It was shown, for instance, that for trace determination of Fe(II) and dissolved inorganic phosphorus in sediment porewater, the LOD values for IC with conductivity detection were insufficient, while the values obtained for CE with C4D detection were satisfactory [8].
Several other advantages of CE in comparison to IC should be also mentioned here, such as a better separation efficiency and resolution, the possibility of simultaneous determination of cations and anions in one run and in one capillary, easy miniaturization of instrumentation, and the possibility of the application of different efficient methods for the in-capillary preconcentration of analytes. The appropriate selection of the background electrolyte (BGE) composition may reduce the interfering effects of matrix components, which can affect the selectivity of the determination.
The subject of this review is to present recent achievements in the uses of CE with various detection methods and with different steps of sample processing in the determination of inorganic anions in waters, based on papers published in recent decades. The CE determinations of inorganic ions were already a subject of numerous publications in the past, including numerous reviews, see e.g., [9,10,11], and one on capillary isotachophoresis [12]. Several reviews on the application of CE techniques for the determination of inorganic ions in water analysis [13,14,15] and the use of portable CE systems for applications including water analysis [16] have also been published.
2. Basic Modes of Analytical Capillary Electrophoresis Measurements
Electrophoresis has for more than 200 years been known as the phenomenon of the transport of electrically charged molecules in a liquid or gel phase induced by an electric field. The electrophoretic mobility of ions in a background electrolyte (BGE) filling the capillary is directly proportional to the charge of the ions and inversely proportional to their size. This behavior of ions has been exploited for analytical purposes for many decades. Electrophoretic separations in planar layers of appropriate gels are very commonly used in biochemistry, and also for preparative purposes. In capillary electrophoresis as analytical technique, separation is usually carried out in capillaries of internal diameters of 25 to 75 μm, most often fabricated of fused silica, where the electric field is generated by the application of a high voltage (usually kilovolts) to electrodes which are immersed in solutions, into which two ends of the separation capillary are also immersed. The detection of ions are moving along the capillary is carried out by the detector located at the end of capillary. In CE, the electrophoretic migration of ions is also often associated with the electroosmotic flow (EOF) of the bulk solution within the capillary as the result of the accumulation of electric charge in the solution layer, in the inner capillary surface. The existence, magnitude and direction of EOF under particular conditions, depends on the composition of BGE. For analytical purposes, capillary electrophoretic separations and determinations can be carried out using different methods to provides the efficient separation and determination of electrically charged ions and neutral molecules.
Capillary zone electrophoresis (CZE) is the most commonly employed CE methodology for analytical purposes, where a portion of solution to be analyzed is introduced into the capillary, which is filled with the BGE. The EOF is generated in the whole capillary diameter with the same rate, providing a flat shape of the front of the solution (in contrary to parabolic one in laminar flow). This behavior is crucial to obtain efficient, high-performance separations with chromatographic efficiency close to that of gas chromatography separations. This methodology is the most commonly used for CE determinations of inorganic ions, which is the subject of this review.
Micellar electrokinetic capillary chromatography (MEKC) is based on the use of surface-active additives in BGE, in concentrations sufficient for the formation of micelles. This results in the formation of a so-called pseudostationary phase in the BGE, which creates the possibility of a partition of the analytes between the phase of the micelles and that of the BGE solution. This partition can take place for both charged and non-charged analytes, offering the conditions for capillary chromatographic separation.
Gel capillary electrophoreses (GCE) is carried out in capillaries filled with gels of appropriate pore sizes, and it is mainly employed for the separation of macromolecules. A porous gel phase acts as a molecular sieve, enabling separations according to the sizes of the separated macromolecules.
Capillary isotachophoresis (CITP) is the electrophoretic separation technique carried out with the use of a non-uniform electrolyte filling the separation capillary. The sample segment introduced into the capillary is placed between two zones of different buffer solutions: the leading one containing ions with larger mobility than the analytes, and the terminating one with ions of smaller mobility than the analytes. After the application of a high voltage, the separated ions form a sequence of zones according to their decreasing mobilities, which are detected for analytical determination.
Capillary isoelectric focusing (CIEF) separation is based on differences in the isoelectric point vales (pI) of analytes. In the gradient of pH generated in the capillary, from the lowest value at the capillary inlet to the highest at the outlet is measured. After application of a high voltage, the analytes move in the capillary until they reach an electrolyte zone where their pH are is equal to their pI.
Capillary electrochromatography (CEC) is carried out in a capillary packed with a stationary phase, where mobile phase movement is based on electroosmotic flow. The separation of analytes is based on the combined effect of the partition between their stationary and mobile phases and their electrophoretic mobility.
3. Application of Different Detection Methods
The analytical technique which is applied in CE systems for detection after separation step has a crucial role in the limits of detection of separated analytes obtained when no additional on-line or off-line preconcentration operations are used. The use of different analytical techniques depends on the technical conditions of carrying the separation process in a strong electric field and on the design of the detection cell with small dead volume, in order to avoid disturbances of the quality of the separation process occurring in the capillary. In the abovementioned pioneering works, potentiometric [1] and laser-induced fluorescence [2] detection were employed. The latter is simple to realize by the use of a miniature window in the separation capillary, and this detection is the most commonly offered in commercial instrumentation. Quite early in CE separations, LIF detection was applied as an especially sensitive method, and kits for such techniques are also offered by several manufacturers. Applications for electroanalytical techniques requiring the electroactivity of analytes are reported more rarely, although such detectors also can be found on the instrumental market. The use of a mass spectrometry detection in CE, which allows the identification of separated analytes, is especially powerful, and such hyphenation is also offered by highly specialized producers of CE instrumentation. Applications of other detections, such as those using atomic spectroscopy techniques, are only found practically in the laboratory. The use of each mentioned detection in a direct mode depends on the particular properties of the analyte or the possibility of the suitable derivatization or in-capillary transformation of the analyte into other product, which can be detected by a given detector.
3.1. Application of UV Detection
Detection based on absorption of UV radiation is the most commonly used in routine CE analytical determinations. For analytes, non-absorbing UV radiation an indirect UV detection can be used, based on the addition of the UV absorbing co-ion to BGE, which is locally replaced in the flowing stream by the analyte, resulting in a quantifiable decrease in the background absorbance. This displacement depends on the charges and mobilities of the probe and the analytes. In principle, indirect UV detection is universal, but the optimization of measuring conditions can be quite complex. The limit of detection in that mode of detection depends on the absorption coefficient of the absorbing probe in the BGE and its concentration, and too large a concentration of the probe in the BGE results in an increase of BGE conductivity. This is essential because the BGE conductivity should be smaller or comparable to the conductivity of sample in order to obtain a satisfactory efficiency of separation and prevent electro-dispersion.
The majority of inorganic ions occurring in natural waters do not absorb UV radiation. Thus, they can most commonly be determined by CE through indirect detection or in direct mode after derivatization to form products absorbing UV radiation. A variety of probes can be employed for indirect UV detection. For the detection of anions, chromate, benzoate, and phthalate are most frequently employed, while chromate and 1,2,4,5-benzene tetracarboxylic acid (pyromellitate) are the most popular choices as they give optimal peak shape and sensitivity for small anions. Pyromellitate is preferred to chromate, as chromate is potentially toxic and requires more handling care. For the CE separation of cations with indirect UV detection, imidazole and pyridine are used as additives to the BGE. The limit of detection of such methods depends on difference in absorbances between the co-ion in the BGE and the analyte, on the transfer ratio of the analyte, which is the number of equivalents of the carrier electrolyte co-ion displaced by an equivalent of the analyte ions, the ratio of charges, and the ratio of electrophoretic mobilities of the analyte and the co-ion used as a UV probe. The obtained LOD values at the mg L−1 level are sufficient for use in water analysis without additional preconcentration steps.
Several applications have been reported for inorganic cations with indirect UV detection in low ionic strength samples, such as tap water [17,18,19], wastewater [19], mineral water [18] and diluted high ionic strength sea water samples [20]. In indirect determinations of cations, pyridine [17], 2,4-trimethylpyridine [20], and benzimidazole [18] were recently reported as satisfactory UV probes. For the separation of inorganic cations, in order to selectively modify their similar mobility for a better resolution, the addition of complexing ligands to BGE can be effective, which was reported e.g., with the use of 18-crown-6-ether [17,18,19] or lactate [20]. For instance, CE determination with indirect UV detection was reported for the determination of NH4+, Na+, K+, Ca2+, Mg2+ and Cu2+ cations and Cl−, NO3− and SO42− anions at mg L−1 levels in cold and hot tap waters supplied to households by installations made of copper, plastic and steel water supply pipes [17]. Due to insufficient detectability, however, the determination of ammonium, copper and sulfate was not possible.
Due to its very high salinity, and large differences in the concentration levels of different inorganic ions, the CE analysis of sea water is a much more challenging task. The development of a BGE applicable to the determination of inorganic cations in high ionic strength samples by CE with indirect UV-absorption detection has been reported very recently [20]. Several small-molecule carboxylic acids, as well as several toluidines and pyridines were tested as complexing agents and chromophoric probes, respectively. The application of a BGE containing 200 mM 2,4,6-trimethylpyridine as the chromophoric probe, 250 mM lactic acid as the complexing agent and 5% v/v methanol as an organic solvent modifier allowed for the determination of the main cations in diluted sea water. All analytes were resolved in less than 4 min with obtained concentrations of 270, 8700, 360 and 880 mg L−1 for K+, Na+, Ca2+, Mg2+, respectively.
Usually, the aforementioned separations are conducted in non-modified fused-silica capillaries, but in order to obtain better reproducibility of CE runs, a double capillary coating with polybrene and polyanion solutions was also employed [18]. The presented method has been successfully applied for the determination of 12 metal cations, including alkali, alkaline earth, transition metals, and ammonium in tap and mineral water samples with base-line resolution, in time periods up to 8 min. CE with indirect UV detection and PMA as UV probe were employed for the determination of sulfur-containing anions in pond water [21], while using chromium trioxide (CrO3) as a UV probe capillary coated with polybrene and SDS for determinations of chloride and sulfate in saline oil-field water [22]. In the determinations of phosphate and calcium, Ca2+ ions were complexed with 2,6-pyridine-dicarboxylic acid (2,6-PDCA) in BGE to form Ca[PDCA]22− complex, and then HPO42− and Ca[PDCA]22− were separated with the direct UV detection used for calcium complex detection, while indirect UV detection was employed for phosphate determination. [23].
In the process of drinking water disinfection with ozone, the formation of toxic bromate as a byproduct takes place, and it can be determined in CE systems. In one such system, isotachophoresis was combined with capillary zone electrophoresis in an automated electrophoretic analyzer with the column-coupling technique, operating in hydrodynamically closed separation system for the determination of bromate in drinking water samples [24]. The detection was carried out at 200 nm, which is selective for the determination of bromate, but not nitrate and nitrite, but other ions do not provide significant absorbance at this detection wavelength. In another example, an open-tubular capillary electrochromatography (OT-CEC) setup with UV detection was designed, where a capillary coated with trimethylamine amination polychloromethyl styrene nanolatex (TMAPL) was successfully applied for the determination of bromate in tap water [25].
CE with indirect UV detection and 2,6-pyridinedicarboxylic acid (PDC) as the UV probe was applied for fluoride determination in sea water. In optimized separation and detection conditions, an LOD of 24 μg·L−1 was obtained [26].
CE with UV detection was also used for the speciation analysis of various trace elements, such as mercury [27,28,29,30], selenium [31] and arsenic [32,33]. Because mercury does not exhibit UV absorption, compounds containing thiol (–SH) groups were used as chelating reagents, including l-cysteine (l-Cys) [27,29,30] and 3-mercapto-1-propane-sulfonic acid [28]. By employing UV detection, determination in real water samples became possible with the use of various pretreatment and preconcentration steps.
3.2. Luminescence-Based Detection Methods
Fluorescence is a very sensitive detection technique, especially when appropriate laser radiation is used as the excitation source. Laser radiation is widely used for the CE determination of numerous organic analytes, but only a few examples can be found in the literature on the determination of inorganic ions. For instance, fluorescent probes have been developed for the ultra-trace determination of heavy metal ions by CE with direct laser-induced fluorescence detection (LIF) [34]. The probes were synthetized from a macrocyclic tetraazacyclododecane-tetraacetic acid (DOTA) or diethylenetriaminepentaacetic acid (DTPA), a spacer 1-(4-aminobenzyl) and fluorescein as a fluorophore. The satisfactory separation of Ca2+, Mg2+, Cu2+, Zn2+, Ni2+, Co2+, Mn2+, Cd2+ and Pb2+ after derivatization was obtained using borate buffer with polybrene as the BGE. Detectability at lower ng L−1 levels, comparable to that obtained by CE-ICP-MS, was achieved. The application of the method for the determination in river water samples was demonstrated.
Indirect fluorescence detection using LED as a light source has been reported for the simultaneous determination of bromide, chloride, nitrate and sulfate [35]. For the fluorescent probe, 8-hydroxy-pyrene-1,3,6-trisulfonic acid (HPTS) was used in the BGE, and limits of detection of 0.4 mg L−1 for sulfate and 1.4 mg L−1 for chloride were obtained.
3.3. Electrochemical Detection Methods
In the search for the most reliable and universal detection methods employing CE determination of inorganic ions, electrochemical methods of detection [36] and conductivity detections play an important role, with a dominant role being played by contactless conductivity detection where the employed electrodes do not contact the BGE in the on-capillary design. Capacitively coupled contactless conductivity detection (C4D), in its currently used form, was invented in 1998 [37,38], and is currently commercially available. Different aspects of its use in conventional capillaries and on microchip electrophoresis devices, have been extensively reviewed by Kuban and Hauser [39,40]. With the axial and contactless configuration of electrodes positioned on the outside surface of the capillary, many advantages can be obtained, such as avoidance of the corrosion of the electrodes, prevention of electrode fouling, inherent decoupling from the electric field applied for separation, simple construction of the detector cell and the possibility of miniaturization. As the detection sensitivity is strongly dependent on background conductivity, it is recommended to select BGEs with low conductivities. In order to obtain the best limits of detection, a difference between the BGE conductivities of the analyte zones should be high, but too high a BGE conductivity can be a source of a large detector noise, while too low a BGE conductivity may cause an unwanted electro-dispersion. Conductivity detection favors the determination of poor- or non-UV absorbing charged species of relatively high specific conductivity, such as inorganic ions. The detection limits achieved for the determination of alkali and alkaline earth cations and ammonium ions with C4D are generally one to two orders of magnitude better than those for the indirect UV detection.
In order to obtain high separation efficiency with low conductivity and appropriate ionic strength, organic acids are used as components of BGEs, including acetic acid [41,42,43,44,45], acetic acid with histidine [8,46,47,48,49,50,51,52,53,54,55,56,57,58], 3-(N-morpholino)propane-sulfonic acid (MOPS) and bis(2-hydroxyethyl)amino tris(hydroxymethyl)methane (bis-tris) [59], 2-(N-morpholino) ethanesulfonic acid (MES) with histidine [51,60,61], pyromellitic acid (PMA) with histidine [62], and lactic acid with histidine [49,63]. For instance, the determination of selected anions has been carried out under conditions with suppressed electroosmotic flow by appropriate pH and polarity switching, with capillary coating [54,62], or by reversing the direction of the EOF by adding an EOF modifier (e.g., cetyltrimethylammonium bromide (CTAB) [61]. A low viscosity and low conductivity BGE composed of N,N-dimethylformamide (DMF) and acetic acid (1:1) was proposed for the CE-C4D determination of the same anions and, using such conditions, the separation of BF4−, ClO4−, PF6− I−, NO3−, Br− and Cl− was successfully performed in 30 min, with LODs in the range of 0.83 to 3.83 μM [64].
3.4. Mass Spectrometry Detection
The hyphenation of CE separation to mass spectrometry (MS) detection allows for a highly sensitive and element-specific determination. This has been shown to be advantageous for a variety of applications and has been a crucial tool in the development of a new methods. While coupling CE to electrospray ionization–MS is routinely performed, for example by using a coaxial sheath-flow interface, the hyphenation of inductively coupled plasma mass spectrometry (ICP-MS) is more technically challenging. Direct detection with element specificity and multi-element measurement properties is offered by ICP-MS. In water analysis, CE-ICP-MS systems were used mostly for the speciation analysis of mercury [65,66], arsenic [67,68] and selenium [68].
The main challenge for the successful application of the CE-ICP-MS technique is the development of a suitable interface, a novel and highly efficient interface to be directly used as the nebulizer was developed for the simultaneous determination of ten arsenic species [67]. This interface was fabricated using a CE-ESI-MS sprayer kit, which was previously used to couple CE with ESI-MS. Using this proposed interface, the liquid from the capillary can be introduced directly into the sprayer chamber of the ICP-MS after nebulization. The use of this interface reduced the dead volume, narrowed the peak width and led to a higher sensitivity and a better electrophoretic resolution. Due to the detection limits in the range of 0.9–3 ng·g−1, the method was successfully applied in the determination of arsenic in ground waters without any sample pretreatment. The modified interface was presented by the same research group for the simultaneous separation and determination of six arsenic and five selenium species [68]. Using the same sprayer and a home-made direct-injection high-efficiency nebulizer chamber, the dead volume of the system and the dilution of analytes were decreased significantly. The method was suitable for the direct speciation of arsenic and selenium in real samples. The obtained LODs ranged from 0.11 to 0.37 μg·L−1 for the As compounds, and from 1.27 to 2.31 μg·L−1 for the Se compounds.
A short capillary electrophoresis coupled with ICP-MS was applied for the separation and determination of methylmercury and inorganic mercury Hg(II) [66]. A commercially available MicroMist nebulizer and a laboratory-made removable interface were used in the system. The developed method provided rapid separation in less than 60 s, high throughput of the samples and detectability at the μg·L−1 level without sample pretreatment. The CE-ICP-MS technique was also used for mercury speciation (MeHg, EtHg and Hg(II)) after complexation with mercaptoacetic acid [65]. A commercial CEi-SP20 interface system was applied to the hyphenation of ICP with CE. A successful separation was obtained in 35 min, but application to the analysis of real tap water samples required sample preconcentration. Recently, CE coupled with ICP-MS detection has been also proposed for the determination of total contents of rhenium and the speciation analysis of Re(VII) and Re(IV) in groundwater [69]. LODs of 0.02 μg·L−1 and 0.01 μg·L−1 were obtained for total rhenium and Re(VII), respectively. The quantification of Re(IV) was calculated as the difference between the total amount and the Re(VII) content.
The identification and simultaneous quantification of organic and inorganic arsenic (mono-methylarsonate (MMA), dimethylarsinate (DMA), As(III) and As(V)) species at low concentration levels using the CE-ESI-MS setup [70] was recently reported. The coaxial sheath liquid sprayer was used for the CE-MS coupling. Using hexafluoro-2-propanol as the BGE increased the separation efficiency, but the detection of these compounds by ESI-MS was not as sensitive as the detection obtained by ICP-MS. Therefore a preconcentration step was necessary for the analysis of real water samples. Limits of detection between 0.02 and 0.04 μg·L−1 were found using partial evaporation of the water sample (50:1). The proposed method was used in the analysis of groundwater and bottled water (Figure 1).
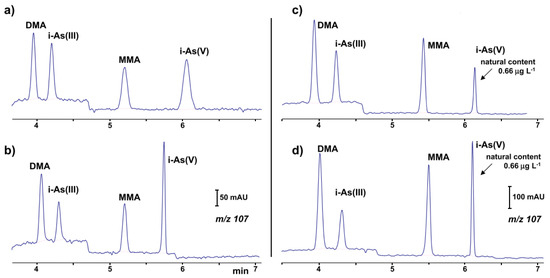
Figure 1.
Ion electropherograms recorded in a CE-MS system for the determination of organic and inorganic arsenic compounds [70]: (a,b) 25 μg·L−1 standard solution (c,d) water sample with a natural content of 0.66 μg·L−1 i-As(V) and spiked with 1.0 μg⋅L−1 of DMA (dimethylarsinate), i-As(III) and MMA (mono-methylarsonate); (b,d) 10% (v/v) of a 11% (v/v) formic acid solution added to sample before injection. CE conditions: capillary—72 cm, 50 μm I.D., BGE—57 mM HFIP (hexafluoro-2-propanol) at pH 10.3, hydrodynamic injection—36 s at 50 mbar, V: +30 kV. (Reproduced under permission from Elsevier. License Number 5173120352687).
In much earlier work, electrospray tandem mass spectroscopy (ESI-MS/MS) with multiple reaction monitoring (MRM) was optimized for the detection of As(III) and As(V), selenium (Se(IV) and Se(VI)) and bromate with on-line enrichment of the target analytes [71]. The LODs obtained were in the range of 1–3 ng·mL−1. ESI-MS detection was also demonstrated for the determination of four chlorine species (ClO3−, ClO2−, ClO4−, Cl−), with LODs at the mg·L−1 level achieved [72].
3.5. Atomic Spectrometry Detection
For the speciation of metal ions, an attractive possibility is CE coupling to atomic absorption spectrometry instruments. The CE separation in a setup using electrothermal atomic absorption spectrometry (ETAAS) has been demonstrated, for instance, for selenium speciation [73]. In a developed interface, the introduction of the CE effluent directly through the end of the graphite tube was used, where the elimination of the upper injection hole of the graphite tube reduced the loss of the analyte and enhanced detection sensitivity. In combination with solid phase extraction (SPE), limits of detection (LOD) values of 0.18, 0.17, 0.54, 0.49 μg·L−1 have been reported for the determination of Se(VI), Se(IV), SeMet, and SeCys2, respectively.
In the case of hydride-forming analytes, the application of a CE–HG–ETAAS setup provides improvement of the detection limits by 1 to 3 orders of magnitude. A designed interface offers good stability, good gas–liquid separation efficiency, a smaller dead volume, good reproducibility and easy operation for As(III) and As(V) determination [74]. The detection limits of As(III) and As(V) were 135 ng·g−1 and 160 ng·g−1, respectively.
Detailed information about applications of CE systems with various detections in water analysis published in recent decades are presented in Table 1.

Table 1.
Applications of capillary electrophoresis methods with various detections in water analysis.
4. Sample Processing in CE Systems for Water Analysis
The determination of numerous environmentally important anions and cations in natural or processed waters which are present in trace concentration levels requires suitable limits of detection and sufficient selectivity of the developed methods. Due to the dimensions of capillaries used, in order to obtain a satisfactory resolution only a very limited sample volume can be introduced, which may usually affect the obtained detectability. An additional factor that has to be considered in case of natural samples in designing the sample processing steps is the matrix of the natural media. Although some water samples do not require preliminary processing besides dilution or filtration, in many cases an insufficient LOD and heavy loaded matrix do require some preliminary processing. In CE techniques, this can be performed using conventional off-line procedures, but in capillary electrophoresis it is quite convenient to carry out them on-line in especially designed CE setups. Particularly attractive are the procedures that can be carried out on-line or, even better, in the in-capillary mode, or directly in the separation capillary. Each of those methods has its limitations and advantages.
4.1. Off-Line Sample Processing
There are numerous developed sample pretreatment procedures and methods employed in trace analytical techniques, which can be used with unlimited sample volume. The most frequently used are liquid–liquid extraction (LLE) and solid–phase extraction (SPE) methods in their different technical modifications. The usual drawbacks of such procedures are the time needed to perform them, limited precision and also the possibility of the contamination. For instance, SPE with 5-sulfosalicylic acid (SSA)-functionalized silica-coated magnetic nanoparticles (SMNPs), was used for the extraction of selenium species [73]. The enrichment factors for Se(VI), Se(IV), SeMet, and SeCys2 were 21, 29, 18, and 12, respectively. In another example of off-line pretreatment for arsenic speciation, the possibility of carrying out a preconcentration by partial evaporation of the solvent 50:1 from the water sample at 55 °C was reported [70]. The limits of detection were found to be between 0.02 and 0.04 μg·L−1 in a CE-ESI-MS system. The recovery of the analytes during the evaporation was in the range of 76 to 108%.
Both LLE and SPE can be carried in a microscale, as microextraction techniques seems to be more suitable due to a use of significantly lower volume of the organic phase. In the liquid-phase microextraction (LPME) procedure, the analytes from an aqueous sample solution are extracted into a small volume (μL) of organic solvent, which usually eliminates the interfering matrix components. Especially for CE, the use of these techniques is beneficial, but conversion of organic acceptor into an aqueous solution is usually required because aqueous solutions are more suitable for direct injection to CE. For solid-phase micro-extraction (SPME) porous fibers are used for the adsorption of analytes.
One recently-accepted technique is extraction into supported liquid membranes (SLMs), which are usually made of a porous inert material with an immobilized organic solvent; analytes are transferred across the SLM from donor solution into the acceptor due to diffusion or the electric field. In additional electromembrane extraction (EME) carried out in the electric field, a shorter extraction time than that of SLM extraction can be used, and whole process can be more selective [75]. In order to develop an EME procedure for particular determination, the SLM composition, the extraction voltage, pH of the sample and acceptor solutions, extraction time, agitation and temperature must be optimized. Numerous EME procedures have been developed for applications with the LC, GC and CE methods [76].
As far as CE systems are concerned, EME preconcentration was used in the CE determination of metal ions (Mn2+, Cd2+, Zn2+, Co2+, Pb2+, Cu2+, Ni2+), to name one example [46]. The limits of detection obtained in the EME-CE-C4D determination ranged from 25 to 200 nM, which is one to two orders of magnitude improved, compared to CE-C4D without sample treatment. A similar set-up was used for metal determination in diluted saline samples, however due to substantial matrix effects, the obtained sensitivity was worse (500 nM) [41]. An EME with a porous polypropylene hollow fiber impregnated with 1-heptanol was applied for perchlorate extraction from drinking water [53]. Sub-ppb concentrations of perchlorate can be detected in natural water samples, demonstrating that such a method is suitable for the determination of perchlorate below the United States Environmental Protection Agency (US EPA) limit. In the CE-C4D system, EME was also employed for the purification and enrichment of bromate in drinking water [45]. Bromate could be preconcentrated up to 267-fold. For the simultaneous extraction of inorganic and organic mercury species with high efficiency prior to speciation analysis by capillary electrophoresis, the hollow fiber-supported liquid–liquid–liquid membrane microextraction method was reported [30]. For a 50 mL sample solution, the obtained enrichment factor values were 103, 265, 511 and 683 for Hg2+, MeHg, EtHg and PhHg, respectively.
In a micro-electromembrane extraction system, free liquid membranes (FLMs) were reported as being possible to use, being formed in a narrow bore tube as the plug of a water-immiscible organic solvent sandwiched between two plugs of aqueous solutions (donor and acceptor) [43]. This technique was demonstrated in the determination of trace concentrations of perchlorate, where an enrichment factor of up to 30 for perchlorate was achieved, and it made use of the CE-C4D system possible. Organic solvent consumption in extractions across FLMs is reduced compared to SLMs.
The use of pretreatment systems mechanically integrated with CE setups is usually carried out in prototype, laboratory-made devices, which are not commercially available. For instance, a promising device for the efficient preconcentration of a single-drop micro-extraction (SDME) coupled with in-line CE commercial instruments has been presented [77,78]. Analytes are extracted from a sample donor solution into a drop of acceptor solution hanging at the inlet tip of a capillary. The enriched drop is then introduced into the capillary for CE analysis. The acceptor solution and next organic phase are sequentially injected into capillary filled with BGE, then using a backpressure, a drop of the acceptor phase covered with a thin layer of the organic phase is created on the capillary inlet and immersed in sample solution. Finally, the enriched acceptor phase is injected into the capillary. Due to the large volume ratio between the sample donor phase and the acceptor drop as well as the thin organic layer, high enrichment factors can be obtained with SDME in a short time. SDME-CE-UV methods were applied, for instance in arsenic speciation [32]. The preconcentration procedure is schematically shown in Figure 2. The enrichment factors obtained with extraction times of 15 min were 390, 340, 1100, and 1300 for As(III), DMA, MMA, and As(V), respectively. The limits of detection with UV detection were 0.2, 0.7, 0.1, and 0.2 μM for As(III), DMA, MMA, and As(V), respectively.
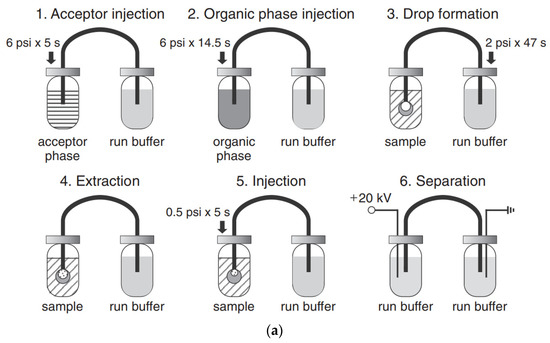
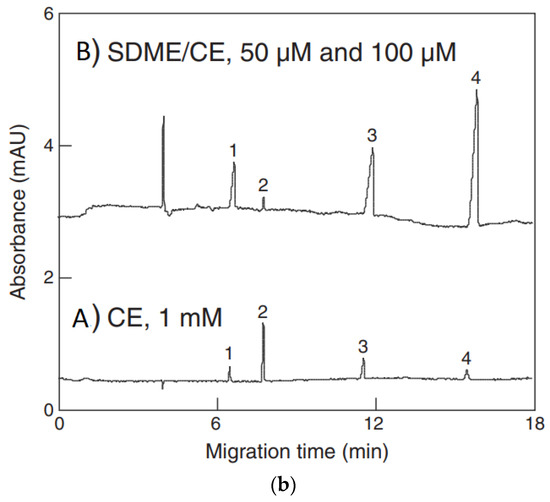
Figure 2.
Application of a single drop microextraction (SDME) procedure (a) for inorganic arsenic species in water by CE and recorded example electropherograms (b) [32]. In (a): (1) acceptor injection, (2) organic injection (trioctylmethylammonium chloride, aliquat 336 in octanol), (3) drop formation, (4) extraction, (5) sample injection, and (6) separation. In (b): (A) CE of 1 mM arsenic compounds, (B) SDME/CE of 100 μM of DMA and As(III) and 50 μM MMA and As(V) in unbuffered water (5 min SDME). Arsenic compounds: DMA (1), As(III) (2), MMA (3), and As(V) (4). CE conditions: capillary—60 cm (50 cm), 25 μm I.D., BGE—15 mM phosphate buffer at pH 10.6, V: +20 kV, UV detection at 200 nm, hydrodynamic injection for 5 s at 0.5 psi. (Reproduced under permission from Elsevier. License Number 5173111119111).
One kind of instrumental integration was demonstrated by the design of an online two-dimensional ion chromatography (IC)-CE system (Figure 3), developed for the determination of major inorganic anions, i.e., bromate and organic acids, in tap water [79]. A six-port, two-position injection valve in the sequential injection (SI)-CE setup and a T-piece interface were used for coupling. A two-dimensional IC effluent was sequentially and electrokinetically injected into the CE with a sampling time of 18.4 s. The system was tested for the determination of anions typical for the drinking water matrix and drinking water disinfection by-products. A 10-fold increase in peak capacity compared to IC was obtained due to the orthogonality of the separation mechanisms of those two methods.
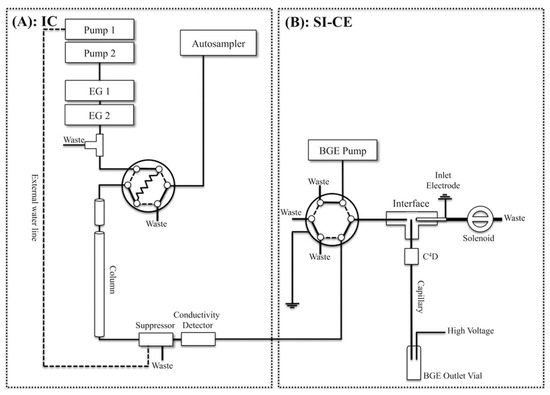
Figure 3.
Schematic diagram of a two-dimensional ion chromatography (IC) (A) and sequential injection (SI)-capillary electrophoresis (CE) system with conductivity detections developed for the determination of inorganic anions and organic acids in waters (B) [79]. (Reproduced under permission from ACS, licence is not required).
4.2. On-Line (In-Capillary) Pretreatment Operations
Simpler procedures, in terms of their technical development, seem to be operations performed in the separation capillary by appropriate changes to the electric field and the sequence and kind of introduced solutions. The optimization of such procedures is, admittedly, very tedious, because of the complexity of the physico-chemical phenomena taking place during such operations.
An increase to the volume of the sample introduced into capillary results in worsening of the separation efficiency. Certain methods of the introduction of larger amounts of analytes from the sample to be analyzed without deterioration of the resolution, with simultaneous preconcentration of analyte(s), have been developed. For this purpose, a stacking effect can be employed by combining capillary zone electrophoresis with isotachophoresis. In both cases, the electromigration phenomenon in the capillary under applied high voltage is utilized. The preconcentration of the analyte as effect of stacking is based on its concentration in narrow segments of the interphase, separating zones of weak and strong electric fields so the stacking effect can be created by introducing a sample solution of much lower conductivity than that one used in BGE. For example, the field amplified sample stacking technique for on-line sample preconcentration was applied in the analysis of five sulfur-containing anions, and the LOD values (from 0.02 to 0.12 μg mL−1) were improved by about 4-fold [21]. The CE runs were performed by introducing a water plug prior to the electrokinetic injection of the sample. The same technique was also employed in trace bromate enrichment using OT-CEC as a separation method, with a 10-fold improvement of the LOD [25].
A pressure-assisted electrokinetic injection (PAEKI) has been used for the on-line pre-concentration of arsenic (As(III) and As(V)), selenium (Se(IV) and Se(VI)) and bromate (BrO3−) [71]. This technique utilizes the principles of both counter-current electro-concentration and stacking. During the electrokinetic injection of the sample, the velocity of the electroosmotic flow is balanced to an external hydrodynamic pressure to create a stationary boundary at the inlet of the capillary where the target analytes accumulate, according to the principles of stacking. For on-line enrichment coupled with CE-ESI-MS/MS, sensitivity LODs of 1–3 ng·mL−1 were achieved, which were below the maximum contaminant levels in drinking water for all five anions studied. This technique seems to be very promising, as the enhancement depends on the sampling time only, and it is not limited by the capillary dimensions.
Several on-line sample preconcentration techniques, such as large volume sample stacking with an electroosmotic flow pump (LVSEP), field amplified sample injection (FASI), transient isotachophoresis (tITP), and electrokinetic supercharging (EKS) combining FASI and tITP, and counter flow CF-EKS, were compared to improve the sensitivity of detection of arsenic speciation in CE-UV analysis [33]. For separation, a cross-linking, fluorocarbon polymer-coated fused silica capillary (μSiL-FC) was used to reduce the EOF with a buffer of a high ionic strength. Figure 4 presents electropherograms of arsenic species obtained without preconcentration, with full injection and removal of the sample matrix (LVSEP). Enrichment in the range of 130–180-fold was possible using this procedure. The best results of the enrichment were achieved using the CF-EKS procedure. A larger quantity of the sample was injected during the FASI process due to sample zone movement, which was exactly counter-balanced by a counter flow, using pressure. Enrichment factors of 45,000, 18,000, 6300 and 7100 were obtained for the standards of As(V), MMA, As(III) and DMA respectively. For natural water samples, due to the presence of complex matrix components, the enrichment factors decreased, but nevertheless the limits of detection obtained with UV absorbance detection were in the range of 2–9 ppb of As.
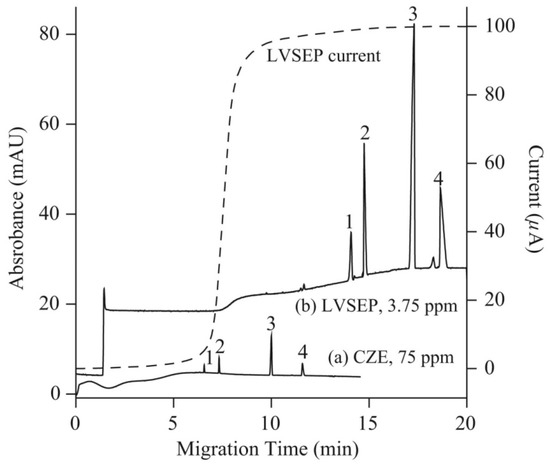
Figure 4.
Electropherograms recorded for arsenic speciation using CE-UV without (a) and with (b) large volume sample stacking with an electroosmotic flow pump (LVSEP) (b) [33]. In (a): recording for 75 ppm As of each As species (1; As(V), 2; MMA, 3; As(III), 4; DMA) in deionized water injected at 0.5 psi for 5 s, and (b): recording LVSEP of 3.75 ppm of each As species in 0.1 mM sodium phosphate buffer with full injection. CE conditions: μSiL-FC coated capillary—60 (50) cm, 50 μm I.D., BGE—100 mM sodium phosphate at pH 9.6, V: 20 kV, UV detection at 200 nm. Dotted line: electric current. (Reproduced under permission from Elsevier. License Number 5173120740442).
An on-line column-coupled capillary isotachophoresis–capillary zone electrophoresis (ITP-CZE) automated analyzer was reported upon for bromate determination with the use of an isotachophoretic separation step, and sufficient resolutions of bromate from the macro-constituents, i.e., sulfate, chloride and nitrate, as well as from the micro-constituents, i.e., nitrite, fluoride, phosphate were obtained [24]. A leading type of tITP–CZE method was applied for fluoride determination in seawater [26]. Chloride acted as the leading ion, and 2,6-pyridinedicarboxylic acid (PDA) in BGE as the terminating ion. In optimized separation conditions and using indirect UV detection, an LOD of 24 μg·L−1 was achieved.
One can also find in the literature examples of a combination of off-line preconcen-tration processes with an on-line stacking strategy employed for further improvement of the detection limits, for trace analysis by CE. For instance, an SPE with nanometer-sized Al2O3 as a sorbent, together with field-amplified sample stacking, was used to preconcentrate inorganic selenium species [31]. The enrichment factors related to two preconcentration steps were 41367-fold and 61935-fold for Se(IV) and Se(VI), respectively. By the combination of off-line SPE and on-line stacking, the LODs of the developed methods were greatly improved, namely to 57 and 71 ng·L−1 for Se(IV) and Se(VI), respectively. However, the processing time of the whole method was rather long, due to the time-consuming off-line SPE step (over 80 min per sample).
In a CE-ICP-MS system, a stacking step was carried out together with dispersive solid–phase extraction (DSPE), which was successfully applied in the determination of ultra-trace levels of MeHg, EtHg and Hg(II) [65]. Thiol cotton particles pretreated with mercapto-acetic acid were used as the solid phase in the off-line method for preconcentration. The stacking step enhanced the sensitivity of the CE-ICP-MS 25-fold, 29-fold and 27-fold for MeHg, EtHg and Hg(II), respectively. Using both methods of preconcentration, ultra-high sensitivity was achieved with LODs in the range of 9–11 ng·L−1. In another work dealing with mercury speciation, a different sample preconcentration technique was also used, namely phase transfer-based liquid–liquid–liquid microextraction (PT-LLLME), together with large volume sample stacking (LVSS) [29]. LVSS with polar switching was performed for the matrix removal. Under optimized conditions, enrichment factor values ranging from 160 to 478 were obtained for the extraction step of the target mercury species. By combining PT/MS-LLLME with LVSS-CE/UV, the enrichment factor values were magnified from 2250 to 12,138, and the limits of detection were obtained at the ppb level (1.40–5.21 μg·L−1).
5. Simultaneous Determination of Anions and Cations
One attractive feature of capillary electrophoresis in water analysis applications is the possibility of simultaneous determinations of anionic and cationic species. Such a possibility is provided to some extent by multicomponent techniques of atomic emission spectroscopy, but with a single signal for the total content of all species containing particular element. Such determinations can also be carried out by the appropriate configurations of ion chromatography setups, such as in a non-suppressed mode of IC connecting anion-exchange and cation-exchange columns [80], by a two dimensional ion chromatography using one-valve switching and two columns [81], or in dual-capillary ion chromatography system [82].
Simultaneous CE determinations can be carried out in dual capillary systems, but a unique feature of the CE technique using appropriate measuring methodology is the possibility of the simultaneous determination of anions and cations in one separation capillary in a single run. This is possible with the application of a large electroosmotic flow and a normal mode of polarization, when cations migrate to the detector together with EOF, when the EOF is larger than the mobilities of anions, and when anions migrate to the detector. It has to be admitted, however, that due to the large mobility of inorganic ions, satisfactory separation under such conditions is difficult to optimize, and different strategies to cope with this difficulty have been reported, see e.g., review [83]. Some attempts reported so far were based on a special injection method. For instance, the dual opposite-end hydrodynamic injection of the sample was proposed for simultaneous inorganic ions in drinking water [62]. Two sample plugs were placed in the two opposite ends of the capillary prior to the separation. Due to the migration of cations and anions in opposite directions, the optimization of the C4D detector position along the capillary was necessary in order to achieve a satisfactory separation of ions. Using PVA-coated fused silica capillary of 60 cm length, 11 cations and anions were separated in less than 3 min with LODs in the range 0.07–2 mg·L−1.
One very helpful methos of manipulations with sample processing, injections and mechanization of whole analytical procedure is the hyphenation of CE setups to flow injection systems, see e.g., review [84]. For instance, in the application of sequential injection analysis (SIA) systems combined with CE setups, the flexible manipulation of the sample plug is possible, leading to the ability to precisely control the pressure for hydrodynamic injection, automated flushing of the separation capillary, and two-way pumping. Use of the SIA makes it possible to change the measurement conditions during the run to optimize the separation. SIA-CE was reported with different detection techniques, but in inorganic ion determination C4D is predominant. For instance, for the determination of inorganic ions in creek water, the SIA-CE system was employed with a T-junction as the interface and a C4D detector with two cells [48]. All operations of the procedure were automatically controlled, the same BGE was used for all ions, and the cations and anions were determined sequentially by changing the polarity of the separation voltage. The same authors also developed other configurations of SIA-CE systems with one or two C4D detectors for the simultaneous separation of major inorganic cations and anions [58]. For the analysis of tap water, dual single-end injection CE in one capillary with two C4D detectors was employed. Both samples were injected at the same end of the capillary, but one sample plug was delivered to the other end of the capillary by the use of pressure. Baseline separations were achieved for 9 cations and 5 anions in a single run of 4 min, with LODs in the 1.5–2.0 μM range for the anions and in the 0.3–1.5 μM range for the cations. In Figure 5, a schematic diagram of this SIA-CE system and electropherograms of tap water samples are presented.
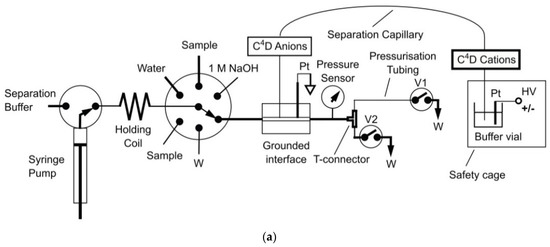
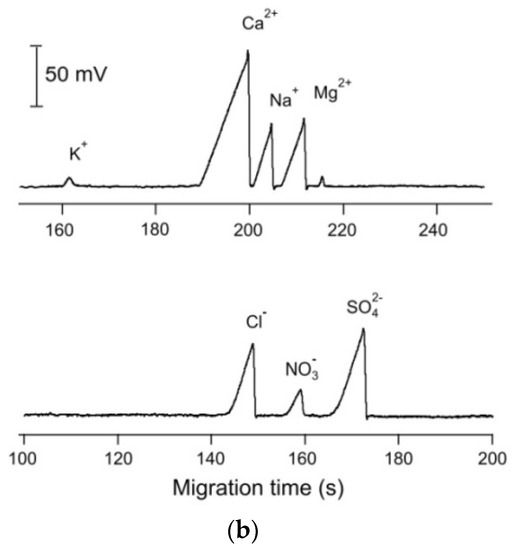
Figure 5.
Schematic diagram of an SIA-CE system with dual contactless C4D detectors for simultaneous determination of anions and cations (a), and an example of its application in the analysis of tap water (b) [58]. In A: C4D: contactless conductivity detector; HV: high-voltage power supply; W: waste; and V1, V2: stop valves. In B: tap water of pH 4 was filtered and diluted 5 times before injections. Both sample plugs were injected at the grounded end of the capillary, with the plug of the anion sample delivered to the HV-end of the capillary with a pressure of 4.5 bar within 90 s. CE conditions: capillary—50 cm, 10 μm I.D. (Leff for cations—43 cm and Leff for anions—35 cm), BGE—12 mM His, 2 mM 18-crown-6, pH 4, E = 400 V/cm. (Reproduced under permission from Elsevier. License Number 5173120143146).
Through detailed optimization of the SIA-CE setup with C4D detection (capillary length, pressure levels and separation time), it was possible to achieve the separation of inorganic cations and anions in wastewater in one run (Figure 6) [61]. The EOF was reversed by adding CTAB to the BGE, and the anion separation was performed using negative polarization. In order to move the cations towards the detector and prevent them from exiting the capillary at the injection end, a hydrodynamic flow towards the detector end was superimposed.
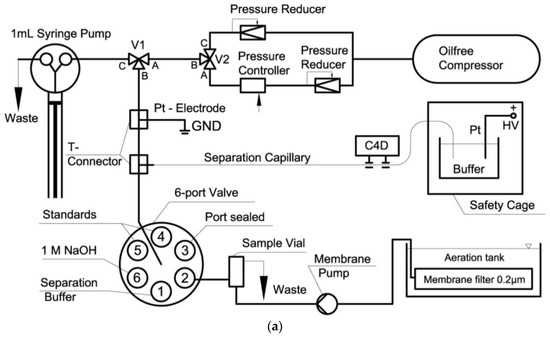
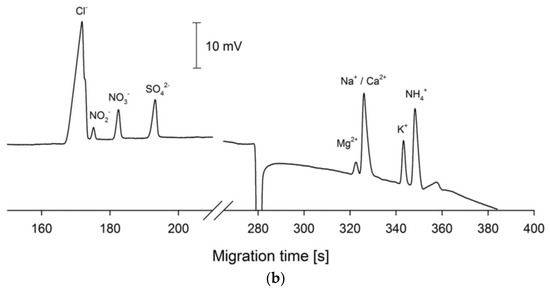
Figure 6.
Schematic diagram of a SI-CE system with contactless C4D conductivity detection developed for the determination of nitrogen-containing ions (a), and sample electropherograms recorded from wastewater samples (b) [61]. CE conditions in (b): capillary—68 cm, 20 μm I.D., BGE—100 mM His, 100 mM MES, 0.13 mM CTAB, 1.5 mM 18-crown-6, pH 6, V: +24 kV. (Reproduced under permission from Elsevier. License Number 5173120520906).
In the capillary filling method, the separation capillary is filled with a sample containing anions and cations dissolved in BGE, and after a high voltage application, the anions and cations move to the appropriate electrode, based on their mobilities under conditions with suppressed EOF [85]. The analytes form boundaries and migrate towards the C4D detector. Using the capillary with an effective length 20 cm for cations and 15 cm for anions, 8 ions (Cl−, NO3−, SO42− and Na+, K+, NH4+, Ca2+, Mg2+) were successfully separated and detected at ppm levels within 100 s in water samples. In another attempt, the separation of cations and anions was achieved by gradient elution moving boundary electrophoresis (GEMBE) [57]. GEMBE is a technique that involves electrophoretic separation in a short-length capillary using continuous injection against a variable counter-flow, which is a combination of the EOF and a pressure-controlled, variable hydrodynamic flow. Each analyte is introduced sequentially in order of decreasing electrophoretic mobility because the pressure-driven flow is reduced over time and the resolution is controlled by the pressure gradient. Seven anionic and cationic analytes were separated using non-linear gradients in less than 7 min, with LOD values in the low μM to sub-μM range.
The analytical literature provides numerous examples of dual-channel CE systems developed for the simultaneous determination of anions and cations with two capillaries [44,54,55,56]. A system with simultaneous hydrodynamic injection into both capillaries was reported, and the application of such a system was reported for the monitoring of ammonia, nitrite and nitrate during the denitrification of sewage in an activated sludge reactor [55]. Another dual-channel CE system was developed with electrokinetic injection for the separation of anions and cations in mineral fertilizer solution samples [44]. As the amount of injected sample in electrokinetic injection is highly dependent on the conductivity of each sample, careful optimization of measuring conditions is needed. Due to short capillaries (effective length 8 cm), base-line separation of all ions (NH4+, K+, Ca2+, Mg2+, Sr2+, Ba2+, Cl−, NO3−, SO42−, ClO3− and F−) was achieved in less than 1 min.
In a purpose-made, automated SIA-CE setup with two capillaries and two C4D detectors, a sample was simultaneously injected electrokinetically onto two separate capillaries for independent separation [54]. In order to improve the separation, coated capillaries (35 cm effective length for the cations and 28 cm for the anions) were used. Parallel separation of 11 anions and 12 cations was achieved, in a total analysis time of 3.5 min. Detailed information about applications of CE for the simultaneous determination of inorganic anions and cations in water samples are shown in Table 2.
Table 2.
Capillary electrophoretic methods developed for simultaneous determination of inorganic anions and cations in waters.
The development of new designs and applications of microfluidic devices in recent decades is one of the most intensive directions of scientific research in analytical chemistry [86]. They are being developed both for fast single and multicomponent determinations based on CE separations [87], including programmable and reconfigurable chips [88] and paper-based microsystems [89]. Most commonly, they are designed for medical diagnostic needs, see e.g., [90], but they are also designed for environmental monitoring [91,92], including water analysis [93,94,95]. This pronounced development trend has resulted in thousands of papers now being published annually (more than 8000 papers per year in 2019–2020).
Microchip capillary electrophoresis (MCE) offers some advantages over conventional, larger scale capillary electrophoresis techniques, including the integration of different separation functions onto the chip, the consumption of smaller amounts of samples and reagents and shorter analysis time, as separations within a minute or few seconds have been reported [95]. It has to be emphasized, however, that the method of sample introduction, complexity of the sample matrix, and need for determinations of low-concentration of analytes are still crucial challenges in the use of a microfluidic devices for environmental analysis, hence their acceptance in routine laboratories is still limited.
So far, only a few applications have been presented for determinations of inorganic ions in water samples. Obviously, the most challenging task is to develop sufficiently sensitive detection devices which are compatible with the size of the miniaturized separation units. Although different detection methods can be used in microfluidic devices, the detectors successfully used for the determination of inorganic ions in waters were most commonly conductivity detectors in the contact [96,97,98,99] or contactless modes [100,101,102], mostly because of their simplicity of fabrication and the possibility of direct incorporation onto a chip. Capacitively-coupled contactless conductivity detection (C4D) is the most widely used and commercially available method.
The MCE systems for inorganic anions offer sensitivity at the μM level. For instance, in a developed dual ‘top–bottom’ C4D cell configuration used with a chip made from thin plastic sheets of 125 μm (Figure 7), LOD values of 0.3 and 0.15 μM were obtained for the determination of cations and anions, respectively, in drinking water samples [101]. A novel concept of contactless conductivity detection based on an indium tin oxide-coated poly(ethylene terephthalate) film was also reported for the detection of metal ions, to enhance the efficiency of prototyping and fabricating a desired microchip [98]. The application of amperometric detection in a microchip CE was reported with the use of either graphite pencil leads [103], or screen-printed carbon-based electrodes [104] as the working electrodes. In both cases, the determination of nitrite in well water with an LOD 2.8 μM [103], and in drinking water [104] was reported. Very recently, LIF detection was employed in microchip electrophoresis for the simultaneous trace determination of silver and mercury ions [104]. The developed analytical procedure also involved an off-line preconcentration step with the use of a stirring bar modified with encoded hairpin DNA probes for the specific extraction of analytes.
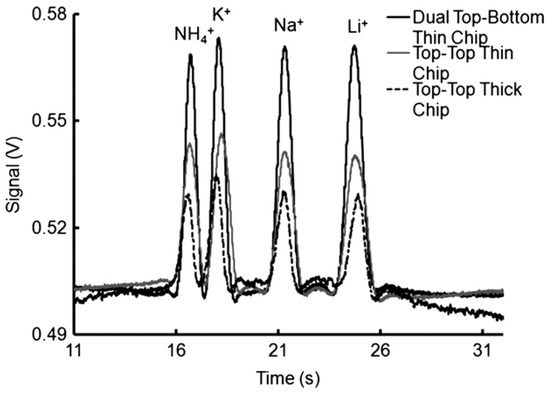
Figure 7.
Comparison of the applications of different detector cell designs in CEs of inorganic cations with contactless C4D conductivity detection [101]. Electropherograms recorded for 0.5 mM cations (K+, Ca+, Mg+, Li+) using electrodes in dual top–bottom (black line), top–top (gray line) positions using thin chips, and in the conventional top–top configuration with thick chips (dotted line). CE conditions: channel—85 (65) mm × 50 × 50 μm, BGE—20 mM MES/His, 2 mM 18-crown-6, pH 6.4, injection at 0.5 kV for 5 s, V: +3 kV. C4D detector: a sinusoidal excitation signal of 22 Vpp (peak-to-peak) at 300 kHz; electrode distance, 1 mm; electrode width, 1 mm. (Reproduced under permission from John Wiley and Sons. License Number 5173110627787).
Most CE microchips have been laboratory-made from polymer materials, and poly(di-methylsiloxane) (PDMS) [96,98,100] and poly(methyl methacrylate) (PMMA) [97,99,101] are the two most commonly used. For the same purpose, commercially available borosilicate glass chips can be used [102].
MCEs have been applied for the determination of anions in drinking water [96,97,104] and river water [100,102], as well as for the determination of heavy metals cations in river water [98], or alkali and alkaline earth metals in drinking water [101]. The ITP-CZE column-coupled microchip was used for NH4+ determination in waste water [99]. The sample was preconcentrated and cleaned up in the isotachophoretic capillary, and in the next capillary the cations were separated. Determination of trace ammonium in the presence of other major cations up to 400 to 800 times higher concentrations was possible. Detailed information about the applications of CE microfluidics in water analysis are listed in Table 3.
Table 3.
Application of microfluidic chips for capillary electrophoretic analysis of waters.
The development of robust, portable analytical measuring systems for different applications, including environmental monitoring is currently a very remarkable trend thanks to recent progress in micromechanics, material science and electronics. Portability allows analyses to be carried out outside of the laboratory, preventing or minimizing the risk of contaminating the sample and leading to faster response times at a lower cost. Portable instruments should, therefore, be lightweight and have small dimensions; they should be easy to transport, and should have independent power sources [106]. Recent trends in the design of portable capillary and microchip electrophoretic systems for field analysis have been presented in several reviews [16,95,107].
Based on the designs reported so far for such systems, conductivity detection is well suited to portable CE systems for the determination of ionic species because of their straightforward miniaturization and very limited electrical power consumption. A portable CE instrument with a C4D detector was successfully applied in the investigation of sediment porewaters [8,52]. The separation and quantitative determination of major inorganic anions and cations, including Mn(II) and Fe(II) ions, was achieved in less than 15 min. The sediment samples were collected from lakes and analyzed immediately in the field. By avoiding the transport of the sample, its composition was preserved, which made it possible to study geochemical processes. The electropherograms of the sediment porewater samples are shown in Figure 8 [52].
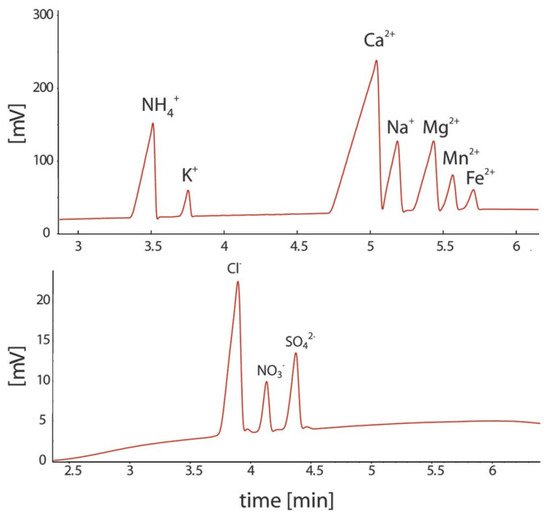
Figure 8.
Electropherograms recorded for the CE determination of anions and cations in sediment pore water using a portable CE system with contactless C4D conductivity detection and hydrodynamic injection [52]. Sample: 700 μM NH4+, 100 μM K+, 1500 μM Ca2+, 500 μM Na+, 320 μM Mg2+, 120 μM Mn2+, and 70 μM Fe2+ for the cations and 610 μM Cl−, 120 μM NO3−, and μM SO42− for the anions. CE conditions: capillary—55 cm, 50 μm I.D., BGE—11 mM His, 50 mM AcOH, 1.5 mM 18-crown-6, 0.1 citric acid, V: ±15 kV. (Permission from RSC is not required).
The same research group also proposed a dual-capillary portable instrument for the simultaneous determination of inorganic anions and cations in similar samples [63]. The instrument has a compact design, with dimensions of 52 × 34 × 18 cm, and a weight of less than 15 kg. Each channel has a separate buffer container to allow for the independent optimization of separation conditions. The system is controlled by a computer and works automatically. The electropherograms of a sample of mining pond water are shown in Figure 9. Because the migration time for the copper ion was relatively long compared to the other analytes, pressure assistance was applied to shorten the analysis time.
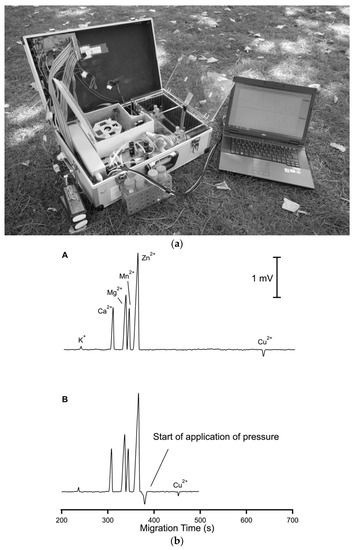
Figure 9.
Photograph of a thermostatted dual-channel portable capillary system with contactless C4D conductivity detection for the concurrent determination of anions and cations (a) and sample electropherogram recorded for 1:100 diluted mining pond water (b) [63]. In a: the syringe pump and valves are in the front, the thermostatted compartment is in the center and the high voltage compartments are to the right; all compartments have been opened. In b: ions separation (A) without and (B) with pressure assistance. CE conditions: capillary—80 (65) cm, 25 μm I.D., BGE—9 mM His, 4.6 mM lactate, 25 mM acetic acid, and 1 mM 18-crown-6 ether, V: −25 kV, injection—50 kPa. Co-flow: 70 kPa applied after 375 s. (Reproduced under permission from John Wiley and Sons. License number 5173110276810).
Another type of portable CE-C4D instrument was designed with the use of automated injection [50]. This portable instrument can operate continuously for 9 h in battery-powered mode. The system can be optimized for different samples, and very fast separations of Cl−, NO3−, SO42− (within 17 s) and high reproducibility were reported. Its use in the field was demonstrated for the determination of phosphates at a sewage treatment plant and a tap water sample spiked with 1 μM NO2− (Figure 10). For a 4s sample injection time, chloride, nitrate, and sulfate were satisfactorily separated, but a peak for nitrite was not detected due to its concentration being below the detection limit. The signal for nitrite can be also detected by increasing the injection time up to 10 s and increasing the pressure.
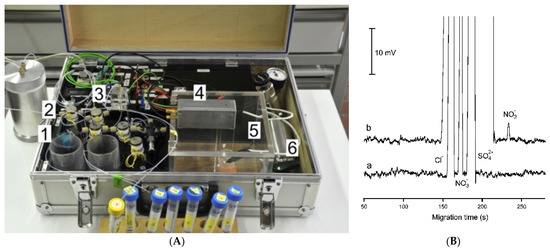
Figure 10.
Photograph (A), and sample recording of an electropherogram applied in water analysis (B) of a laboratory-made portable capillary electrophoresis system with contactless conductivity detection [50]. In (A): (1) membrane pump, (2) valves, (3) splitter, (4) detector, (5) safety cage for application of high voltage, (6) pressurized air. In (B): recoding of the CE analysis of a tap water sample spiked with 1 μM NO2−. (a) Normal injection volume: 1 bar, 150 μL, splitting valve set to 0.15, 4 s. (b) Large volume injection: pressure, 1 bar; sample loop, 150 μL; splitting valve set to 0.10; injection time, 10 s. CE conditions: capillary—60 (36) cm, 50 μm I.D., BGE—12 mM His adjusted to pH 4 with AcOH, 2 mM of 18-crown-6, V: +15 kV. (Reproduced under permission from ACS).
A fully automated, portable capillary electrophoresis analyzer can be also compatible with different detectors—ESI-MS, C4D and LIF—for potential in-situ deployment in spaceflight missions and exploration [108]. The implementation of these detectors can enable the analysis of a wide range of organic compounds potentially indicative of life, as well as inorganic compounds that can serve as indicators of habitability. Combining microfluidic and pneumatic circuits with rotary valves ensures full automation and good repeatability.
6. Conclusions and Perspectives
Capillary electrophoresis is an analytical technique which exhibits numerous advantages from the point of view of potential applications in the analysis of waters of different origin. The use of appropriate detectors allows its application in the determination of numerous organic compounds, and of inorganic ions, which is the subject of this review of original works published in recent decades. The possibility of the application of preconcentration in off-line mode, and especially on-line (in-capillary) provides opportunities of the application for the determination of trace level analytes. CE instrumentation can be more easily miniaturized to a microfluidic format than high-performance chromatographic HPLC or ion-chromatography instrumentation.
In the determination of inorganic ions the application of conductivity detection, particularly contactless C4D detection, which responds to all ions is especially suitable. The simultaneous determination of anions and cations can be carried out in the same capillary, and usually in shorter time than ion chromatography. Moreover, CE instrumentation is generally less expensive than ion chromatography, and the scaling down of the instruments is easier in the case of CE than for ion chromatography.
Admittedly, this technique is not commonly employed in routinely for water quality control, despite the development of numerous CE methods for water analysis and the aforementioned advantages. This seems to be mostly due to much wider availability of commercial instrumentation for HPLC than for CE, which can be easily adapted for ion chromatography without purchasing a dedicated ion chromatograph. The more difficult optimization of experimental conditions for CE determinations may be a second reason which inhibits the routine application of CE, compared to chromatography, but this is a subjective opinion. It can be assumed, however, that the publication of reviews on that subject may provide a valuable impact by means of widening the interest in the use of CE for water analysis. Several trends in CE development for water analysis remain to be emphasized. One of them is the design of multi-dimensional instrumental setups. For instance, the IC x CE-MS system was developed for simultaneous determinations of anions and cations, and has been employed for the speciation of arsenic [109]. There is also A review on systems combining chromatography with electrophoresis [110]. In a search for new detection techniques, a surface-enhanced Raman scattering-based microfluidic CE system has been reported, with applications including DNA-functionalized substrate use in the determination of Hg(II) in sewage water with an impressive LOD of 1 pM [111]. One example of efficient analyte preconcentration is electrokinetic supercharging, which obtained an enrichment factor of 500,000 in the determination of rare-earth metal ions [112]. This is certainly among the valuable ways to improve the limits of detection, e.g., in the determination of inorganic analytes in sea waters [113]. In the increasing applications of CE microfluidics (with some attempts to describe it as “nano-capillary electrophoresis” [114]), and the improvements of in-capillary sample processing methods, one can notice some perspectives of new routine applications of CE in water analysis.
Author Contributions
Conceptualization, M.T. and E.P.; methodology, M.T.; investigation, E.P.; writing, M.T. and E.P.; initial draft analysis, M.T.; editing, E.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Viratanen, R. Zone electrophoresis in a narrow-bore tube employing potentiometric detection—Theoretical and experimental study. Acta Polyt. Scand. Chem. Incl. Metall. Ser. 1974, 123, 1–67. [Google Scholar]
- Jorgenson, J.W.; Lukacs, K.D. Zone electrophoresis in open-tubular glass capillaries. Anal. Chem. 1981, 53, 1298–1302. [Google Scholar] [CrossRef]
- Voeten, R.L.; Ventouri, I.K.; Haselberg, R.; Somsen, G.W. Capillary electrophoresis: Trends and recent advances. Anal. Chem. 2018, 90, 1464–1481. [Google Scholar] [CrossRef] [PubMed]
- Choi, O.-K.; Cho, J.-S. Analysis of inorganic anions in various drinking waters by capillary electrophoresis. Anal. Sci. Technol. 1995, 8, 835–841. [Google Scholar]
- Haddad, P.R. Comparison of ion chromatography and capillary electrophoresis for the determination of inorganic ions. J. Chromatogr. A 1997, 770, 281–290. [Google Scholar] [CrossRef]
- Pacakova, V.; Stulik, K. Capillary electrophoresis of inorganic anions and its comparison with ion chromatography. J. Chromatogr. A 1997, 789, 169–180. [Google Scholar] [CrossRef]
- Breadmore, M.C. Capillary and microchip electrophoresis: Challenging the common conceptions. J. Chromatogr. A 2012, 1221, 42–55. [Google Scholar] [CrossRef]
- Torres, N.T.; Hauser, P.C.; Furrer, G.; Brandl, H.; Muller, B. Sediment porewater extraction and analysis combining filter tube samplers and capillary electrophoresis. Environ. Sci. Process. Impacts 2013, 15, 715–720. [Google Scholar] [CrossRef]
- Timerbaev, A.R. Recent advances and trends in capillary electrophoresis of inorganic ions. Electrophoresis 2002, 23, 3884–3906. [Google Scholar] [CrossRef]
- Timerbaev, A.R. Capillary electrophoresis of inorganic ions: An update. Electrophoresis 2004, 25, 4008–4031. [Google Scholar] [CrossRef]
- Kubáň, P.; Timerbaev, A.R. Inorganic analysis using CE: Advanced methodologies to face old challenges. Electrophoresis 2014, 35, 225–233. [Google Scholar] [CrossRef]
- Mala, Z.; Gebauer, P. Recent progress in analytical capillary isotachophoresis. Electrophoresis 2019, 40, 55–64. [Google Scholar] [CrossRef]
- Fukushi, K.; Takeda, S.; Chayama, K.; Walida, S. Application of capillary electrophoresis to the analysis of inorganic ions in environmental samples. J. Chromatogr. A 1999, 834, 349–362. [Google Scholar] [CrossRef]
- Valsecchi, S.M.; Polsello, S. Analysis of inorganic species in environmental samples by capillary electrophoresis. J. Chromatogr. A 1999, 834, 363–385. [Google Scholar] [CrossRef]
- Ali, I.; Aboul-Enein, H.Y. Determination of metal ions in water, soil and sediment by capillary electrophoresis. Anal. Lett. 2002, 35, 2053–2076. [Google Scholar] [CrossRef]
- Lewis, A.P.; Cranny, A.; Harris, N.R.; Green, N.G.; Wharton, J.A.; Wood, R.J.K.; Stokes, K.R. Review on the development of truly portable and in-situ capillary electrophoresis systems. Meas. Sci. Technol. 2013, 24, 042001. [Google Scholar] [CrossRef]
- El Fellah, S.; Duporté, G.; Sirén, H. Steroid hormones, inorganic ions and botrydial in drinking water. Determination with capillary electrophoresis and liquid chromatography-orbitrap high resolution mass spectrometry. Microchem. J. 2017, 133, 126–136. [Google Scholar] [CrossRef]
- Kowalski, P.; Olędzka, I.; Plenis, A.; Bączek, T. Dynamic double coating, electrophoretic method with indirect detection for the simultaneous quantification of mono- and divalent cations in various water samples. Electrophoresis 2017, 38, 477–485. [Google Scholar] [CrossRef]
- Varden, L.; Bou-Abdallah, F. Detection and separation of inorganic cations in natural, potable, and wastewater samples using capillary zone electrophoresis with indirect UV detection. Am. J. Analyt. Chem. 2017, 8, 81–94. [Google Scholar] [CrossRef][Green Version]
- Lancioni, C.; Aspromonte, J.; Tascon, M.; Gagliardi, L.G. Development of a background electrolyte for the determination of inorganic cations in high ionic strength samples by capillary electrophoresis with indirect UV-absorption detection. J. Chromatogr. A 2021, 1645, 462091. [Google Scholar] [CrossRef]
- Pappoe, M.; Bottaro, C.S. Systematic optimization of a pyromellitic acid background electrolyte for capillary electrophoresis with indirect UV-vis detection and online preconcentration analysis of thiosalt anions in treated mine tailings. Anal. Meth. 2014, 6, 9305–9312. [Google Scholar] [CrossRef]
- Donkor, K.K.; Guo, Z.C.; Soliman, L.C.; Law, Y.T.; Risley, J.M.; Schmidt, K.J.; Crabtree, H.J.; Warrender, N.A. Determination of sulfate and chloride ions in highly saline oilfield water by capillary electrophoresis using bilayer-coated capillaries and indirect absorption detection. Intern. J. Environ. Anal. Chem. 2015, 95, 175–186. [Google Scholar] [CrossRef]
- Wang, Q.-P.; Chen, Z.-L.; Chen, G.-N.; Lin, J.-M. Simultaneous determination of phosphate and calcium in river water samples by capillary zone electrophoresis with UV detection. Intern. J. Environ. Anal. Chem. 2011, 91, 255–262. [Google Scholar] [CrossRef]
- Marák, J.; Staňová, A.; Vaváková, V.; Hrenáková, M.; Kaniansky, D. On-line capillary isotachophoresis–capillary zone electrophoresis analysis of bromate in drinking waters in an automated analyzer with coupled columns and photometric detection. J. Chromatogr. A 2012, 1267, 252–258. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, F.; Meng, L.; Tang, W.; Xia, Y.; Wu, Y.; Zhang, S. Preparation and application of trimethylamine amination polychloromethyl styrene nanolatex coated capillary column for the determination of bromate by field-amplified sample stacking open-tubular capillary electrochromatography. Electrophoresis 2013, 34, 1312–1318. [Google Scholar] [CrossRef]
- Fukushi, K.; Fujita, Y.; Nonogaki, J.; Tsujimoto, J.; Hattori, T.; Inui, H.; Beškoski, V.P.; Hotta, H.; Hayashi, M.; Nakano, T. Capillary zone electrophoresis determination of fluoride in seawater using transient isotachophoresis. Anal. Bioanal. Chem. 2018, 410, 1825–1831. [Google Scholar] [CrossRef]
- Yang, F.; Li, J.; Lu, W.; Wen, Y.; Cai, X.; You, J.; Ma, J.; Ding, Y.; Chen, L. Speciation analysis of mercury in water samples by dispersive liquid–liquid microextraction coupled to capillary electrophoresis. Electrophoresis 2014, 35, 474–481. [Google Scholar] [CrossRef]
- Li, P.; He, M.; Chen, B.; Hu, B. Automated dynamic hollow fiber liquid–liquid–liquid microextraction combined with capillary electrophoresis for speciation of mercury in biological and environmental samples. J. Chromatogr. A 2015, 1415, 48–56. [Google Scholar] [CrossRef]
- Li, P.; Zhang, X.; Hu, B. Phase transfer membrane supported liquid–liquid–liquid microextraction combined with large volume sample injection capillary electrophoresis–ultraviolet detection for the speciation of inorganic and organic mercury. J. Chromatogr. A 2011, 1218, 9414–9421. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Peng, M.; Hou, X.; Zheng, C.; Long, Z. Improved hollow fiber supported liquid–liquid–liquid membrane microextraction for speciation of inorganic and organic mercury by capillary electrophoresis. Anal. Methods 2013, 5, 1185–1191. [Google Scholar] [CrossRef]
- Duan, J.; Hu, B.; He, M. Nanometer-sized alumina packed microcolumn solid-phase extraction combined with field-amplified sample stacking-capillary electrophoresis for the speciation analysis of inorganic selenium in environmental water samples. Electrophoresis 2012, 33, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Choi, K.; Kim, J.; Sung, I.-H.; Chung, D.S. Sensitive arsenic analysis by carrier-mediated counter-transport single drop microextraction coupled with capillary electrophoresis. Microchem. J. 2013, 106, 220–225. [Google Scholar] [CrossRef]
- Lee, H.G.; Kwon, J.Y.; Chung, D.S. Sensitive arsenic speciation by capillary electrophoresis using UV absorbance detection with on-line sample preconcentration techniques. Talanta 2018, 181, 366–372. [Google Scholar] [CrossRef]
- Saito, S.; Nakano, Y.; Hikichi, A.; Suzuki, R.; Yoshimoto, K.; Maeda, M.; Aoyamab, M.; Shibukawa, M. Ultrasensitive CE for heavy metal ions using the variations in the chemical structures formed from new octadentate fluorescent probes and cationic polymers. Analyst 2011, 136, 2697–2707. [Google Scholar] [CrossRef]
- Pei, L.; Schmidt, K.J.; Crabtree, H.J.; Lucy, C.A. Determination of inorganic anions in oilfield water using capillary electrophoresis with indirect fluorescence detection. Anal. Methods 2015, 7, 8689–8696. [Google Scholar] [CrossRef]
- Trojanowicz, M. Recent developments in electrochemical flow detections—A Review, Part I. Flow analysis and capillary electrophoresis. Anal. Chim. Acta 2009, 653, 36–58. [Google Scholar] [CrossRef]
- Zemann, A.J.; Schnell, E.; Volgger, D.; Bonn, G.K. Contactless conductivity detection for capillary electrophoresis. Anal. Chem. 1998, 70, 563–567. [Google Scholar] [CrossRef]
- Da Silva, J.A.F.; Do Lago, C.L. An oscillometric detector for capillary electrophoresis. Anal. Chem. 1998, 70, 4339–4343. [Google Scholar] [CrossRef]
- Kubáň, P.; Hauser, P.C. Contactless conductivity detection for analytical techniques—Developments from 2014 to 2016. Electrophoresis 2017, 38, 95–114. [Google Scholar] [CrossRef] [PubMed]
- Kubáň, P.; Hauser, P.C. 20th anniversary of axial capacitively coupled contactless conductivity detection in capillary electrophoresis. Trends Anal. Chem. 2018, 102, 311–321. [Google Scholar] [CrossRef]
- Silva, M.; Mendiguchía, C.; Moreno, C.; Kubáň, P. Electromembrane extraction and capillary electrophoresis with capacitively coupled contactless conductivity detection: Multi-extraction capabilities to analyses trace metals from saline samples. Electrophoresis 2018, 39, 2152–2159. [Google Scholar] [CrossRef]
- Ferreira Santos, M.S.; Cordeiro, T.G.; Noell, A.C.; Garcia, C.D.; Mora, M.F. Analysis of inorganic cations and amino acids in high salinity samples by capillary electrophoresis and conductivity detection: Implications for in-situ exploration of ocean worlds. Electrophoresis 2018, 39, 2890–2897. [Google Scholar] [CrossRef]
- Kubáň, P.; Boček, P. Preconcentration in micro-electromembrane extraction across free liquid membranes. Anal. Chim. Acta 2014, 848, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Opekar, F.; Tůma, P. Dual-channel capillary electrophoresis for simultaneous determination of cations and anions. J. Chromatogr. A 2016, 1446, 158–163. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, L.; Zhang, D.; Ge, X.; Ye, J.; Chu, Q. Sensitive determination of bromate in water samples by capillary electrophoresis coupled with electromembrane extraction. Food Anal. Methods 2016, 9, 393–400. [Google Scholar] [CrossRef]
- Kuban, P.; Strieglerova, L.; Gebauer, P.; Bocek, P. Electromembrane extraction of heavy metal cations followed by capillary electrophoresis with capacitively coupled contactless conductivity detection. Electrophoresis 2011, 32, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Duong, H.A.; Le, M.D.; Nguyen, K.D.M.; Hauser, P.C.; Pham, H.V.; Mai, T.D. In-house-made capillary electrophoresis instruments coupled with contactless conductivity detection as a simple and inexpensive solution for water analysis: A case study in Vietnam. Environ. Sci. Process. Impacts 2015, 17, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Mai, T.D.; Schmid, S.; Muller, B.; Hauser, P.C. Capillary electrophoresis with contactless conductivity detection coupled to a sequential injection analysis manifold for extended automated monitoring applications. Anal. Chim. Acta 2010, 665, 1–6. [Google Scholar] [CrossRef]
- Mai, T.D.; Le, M.D.; Sáiz, J.; Duong, H.A.; Koenka, I.J.; Pham, H.V.; Hauser, P.C. Triple-channel portable capillary electrophoresis instrument with individual background electrolytes for the concurrent separations of anionic and cationic species. Anal. Chim. Acta 2016, 911, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Mai, T.D.; Pham, T.T.T.; Pham, H.V.; Sáiz, J.; Ruiz, C.G.; Hauser, P.C. Portable capillary electrophoresis instrument with automated injector and contactless conductivity detection. Anal. Chem. 2013, 85, 2333–2339. [Google Scholar] [CrossRef]
- Sáiz, J.; Koenka, I.J.; Garćıa-Ruiz, C.; Müller, B.; Chwalek, T.; Hauser, P.C. Micro-injector for capillary electrophoresis. Electrophoresis 2015, 36, 1941–1944. [Google Scholar] [CrossRef]
- Torres, N.T.; Och, L.M.; Hauser, P.C.; Furrer, G.; Brandl, H.; Vologina, E.; Sturm, M.; Burgmanna, H.; Muller, B. Early diagenetic processes generate iron and manganese oxide layers in the sediments of Lake Baikal, Siberia. Environ. Sci. Process. Impacts 2014, 16, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Kiplagat, I.K.; Doan, T.K.O.; Kubáň, P.; Boček, P. Trace determination of perchlorate using electromembrane extraction and capillary electrophoresis with capacitively coupled contactless conductivity detection. Electrophoresis 2011, 32, 3008–3015. [Google Scholar] [CrossRef]
- Gaudry, A.J.; Guijt, R.M.; Mack, M.; Hutchinson, J.P.; Johns, C.; Hilder, E.F.; Dicinoski, G.W.; Nesterenko, P.N.; Haddad, P.R.; Breadmore, M.C. On-line simultaneous and rapid separation of anions and cations from a single sample using dual-capillary sequential injection-capillary electrophoresis. Anal. Chim. Acta 2013, 781, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.T.T.; Mai, T.D.; Nguyen, T.D.; Sáiz, J.; Pham, H.V.; Hauser, P.C. Automated dual capillary electrophoresis system with hydrodynamic injection for the concurrent determination of cations and anions. Anal. Chim. Acta 2014, 841, 77–83. [Google Scholar] [CrossRef]
- Koenka, I.J.; Mai, T.D.; Hauser, P.C.; Sáiz, J. Simultaneous separation of cations and anions in capillary electrophoresis—Recent applications. Anal. Methods 2016, 8, 1452–1456. [Google Scholar] [CrossRef]
- Flanigan, P.M.; Ross, D.; Shackman, J.G. Determination of inorganic ions in mineral water by gradient elution moving boundary electrophoresis. Electrophoresis 2010, 31, 3466–3474. [Google Scholar] [CrossRef]
- Mai, T.D.; Hauser, P.C. Simultaneous separations of cations and anions by capillary electrophoresis with contactless conductivity detection employing a sequential injection analysis manifold for flexible manipulation of sample plugs. J. Chromatogr. A 2012, 1267, 266–272. [Google Scholar] [CrossRef]
- Liu, S.; Pan, Z.; Liang, Y.; Li, F.; Breadmore, M.C.; Zhang, M. An electrophoretic ion analyzer for on-site autonomous water monitoring. J. Chromatogr. A 2021, 1637, 461791. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, N.; Cabot, J.M.; Lam, S.C.; Rodriguez, E.S.; Paull, B. Selective capillary electrophoresis separation of mono and divalent cations within a high-surface area-to-volume ratio multi-lumen capillary. Anal. Chim. Acta 2019, 1051, 41–48. [Google Scholar] [CrossRef]
- Fuiko, R.; Saracevic, E.; Koenka, I.J.; Hauser, P.C.; Krampe, J. Capillary electrophoresis for continuous nitrogen quantification in wastewater treatment processes. Talanta 2019, 195, 366–371. [Google Scholar] [CrossRef]
- Neaga, I.-O.; Iacob, B.C.; Bodoki, E. The analysis of small ions with physiological implications using capillary electrophoresis with contactless conductivity detection. J. Liq. Chromatogr. Relat. Technol. 2014, 37, 2072–2090. [Google Scholar] [CrossRef]
- Koenka, I.J.; Kung, N.; Kuban, P.; Chwalek, T.; Furrer, G.; Wehrli, B.; Muller, B.; Hauser, P.C. Thermostated dual-channel portable capillary electrophoresis instrument. Electrophoresis 2016, 37, 2368–2375. [Google Scholar] [CrossRef]
- Tian, Z.; Qin, W. Capillary electrophoretic separation of anions in dimethylformamide–acetic acid medium. Anal. Methods 2014, 6, 5353–5359. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, X.; Mo, F.; Huang, L.; Wu, Z.; Wu, Y.; Xu, L.; Fu, F. Ultra-sensitive speciation analysis of mercury by CE-ICP-MS together with field-amplified sample stacking injection and dispersive solid-phase extraction. Electrophoresis 2016, 37, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Li, B.-H. Rapid speciation analysis of mercury by short column capillary electrophoresis on-line coupled with inductively coupled plasma mass spectrometry. Anal. Methods 2011, 3, 116–121. [Google Scholar] [CrossRef]
- Liu, L.; He, B.; Yun, Z.; Sun, J.; Jiang, G. Speciation analysis of arsenic compounds by capillary electrophoresis on-line coupled with inductively coupled plasma mass spectrometry using a novel interface. J. Chromatogr. A 2013, 1304, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yun, Z.; He, B.; Jiang, G. Efficient interface for online coupling of capillary electrophoresis with inductively coupled plasma—Mass spectrometry and its application in simultaneous speciation analysis of arsenic and selenium. Anal. Chem. 2014, 86, 8167–8175. [Google Scholar] [CrossRef]
- Zhou, D.; Lin, Y.; Long, H.; Xu, Y.; Wang, B.; Xian, L.; Xia, C.H.; Hou, X.; Zheng, C.h. Simultaneous total and speciation analysis of rhenium by capillary electrophoresis-inductively coupled plasma mass spectrometry. Spectrochim. Acta Part B Atmo. Spectrosc. 2021, 180, 106211. [Google Scholar] [CrossRef]
- Dominguez-Alvarez, J. Capillary electrophoresis coupled to electrospray mass spectrometry for the determination of organic and inorganic arsenic compounds in water samples. Talanta 2020, 212, 120803. [Google Scholar] [CrossRef]
- Zhang, H.; Gavina, J.; Feng, Y.-L. Understanding mechanisms of pressure-assisted electrokinetic injection: Application to analysis of bromate, arsenic and selenium species in drinking water by capillary electrophoresis-mass spectrometry. J. Chromatogr. A 2011, 1218, 3095–3104. [Google Scholar] [CrossRef]
- Gaspar, A.; Pesti, A.; Szabo, M.; Kecskemeti, A. Determination of chlorine species by capillary electrophoresis—Mass spectrometry. Electrophoresis 2019, 40, 2637–2643. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Deng, B.; Shen, C.; Long, C.; Deng, Q.; Tao, C. Selenium speciation using capillary electrophoresis coupled with modified electrothermal atomic absorption spectrometry after selective extraction with 5-sulfosalicylic acid functionalized magnetic nanoparticles. J. Chromatogr. A 2015, 1395, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.; Qin, X.; Xiao, Y.; Wang, Y.; Yin, H.; Xu, X.; Shen, C. Interface of on line coupling capillary electrophoresis with hydride generation electrothermal atomic absorption spectrometry and its application to arsenic speciation in sediment. Talanta 2013, 109, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Chen, Z.; Gjelstad, A.; Pedersen-Bjergaard, S.; Shen, X. Electromembrane extraction. Trends Anal. Chem. 2017, 95, 47–56. [Google Scholar] [CrossRef]
- Oedit, A.; Ramautar, R.; Hankemeier, T.; Lindenburg, P.W. Electroextraction and electromembrane extraction: Advances in hyphenation to analytical techniques. Electrophoresis 2016, 37, 1170–1186. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Kim, S.J.; Jin, Y.G.; Jang, Y.O.; Kim, J.S.; Chung, D.S. Single drop microextraction using commercial capillary electrophoresis instruments. Anal. Chem. 2009, 81, 225–230. [Google Scholar] [CrossRef]
- Alothman, Z.A.; Dawod, M.; Kim, J.; Chung, D.S. Single-drop microextraction as a powerful pretreatment tool for capillary electrophoresis: A review. Anal. Chim. Acta 2012, 739, 14–24. [Google Scholar] [CrossRef]
- Ranjbar, L.; Gaudry, A.J.; Breadmore, M.C.; Shellie, R.A. Online comprehensive two-dimensional ion chromatography × capillary electrophoresis. Anal. Chem. 2015, 87, 8673–8678. [Google Scholar] [CrossRef]
- Meng, H.-B.; Wang, T.-R.; Guo, B.-Y.; Hashi, Y.; Guo, C.-X.; Lin, J.-M. Simultaneous determination of inorganic anions and cations in explosive residues by ion chromatography. Talanta 2008, 76, 241–245. [Google Scholar] [CrossRef]
- Fa, Y.; Yu, Y.; Li, F.; Du, F.; Liang, X.; Liu, H. Simultaneous detection of anions and cations in mineral water by two dimensional ion chromatography. J. Chromatogr. A 2018, 1554, 123–127. [Google Scholar] [CrossRef]
- Rodriguez, E.S.; Plummer, C.; Nation, M.; Moy, A.; Curran, M.; Haddad, P.R.; Paull, B. Sub- 1 mL sample requirement for simultaneous determination of 17 organic and inorganic anions and cations in Antarctic ice core samples by dual capillary ion chromatography. Anal. Chim. Acta 2019, 1063, 167–177. [Google Scholar] [CrossRef]
- Sáiz, J.; Koenka, I.J.; Mai, T.D.; Hauser, P.C.; García-Ruiz, C. Simultaneous separation of cations and anions in capillary electrophoresis. Trends Anal. Chem. 2014, 62, 162–172. [Google Scholar] [CrossRef]
- Clavijo, S.; Avivar, J.; Suárez, R.; Cerdá, V. Analytical strategies for coupling separation and flow-injection techniques. Trends Anal. Chem. 2015, 67, 26–33. [Google Scholar] [CrossRef]
- Yamamoto, S.; Fujiwara, H.; Maruyama, K.; Tanaka, Y.; Kinoshita, M.; Suzuki, S. Simultaneous determination of inorganic anions and cations in water and biological samples by capillary electrophoresis with a capacitive coupled contactless conductivity detector using capillary filling method. Anal. Sci. 2019, 35, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Hens, A.; Fernandez-Romero, J.M. Microfluidic systems in analytical chemistry. In Encyclopedia of Analytical Chemistry; Wiley Online Library: Hoboken, NJ, USA, 2017. [Google Scholar] [CrossRef]
- Ragab, M.A.A.; El-Kimary, E.I. Recent advances and applications of microfluidic capillary electrophoresis: A comprehensive review (2017–Mid 2019). Crit. Rev. Anal. Chem. 2020. [Google Scholar] [CrossRef]
- Renaudot, R.; Agache, V.; Fouillet, Y.; Laffite, G.; Bisceglia, E.; Jalabert, L.; Kumemura, M.; Collard, D.; Fujita, H. A programmable and reconfigurable microfluidic chip. Lab Chip 2013, 13, 4517–4524. [Google Scholar] [CrossRef] [PubMed]
- Sriram, G.; Bhat, M.P.; Patil, P.; Uthappa, U.T.; Jung, H.-Y.; Altalhi, T.; Kumeria, T.; Aminabhavi, T.M.; Pai, R.K.; Kurkuri, M.D. Paper-based microfluidic analytical devices for colorimetric detection of toxic ions: A review. Trends Anal. Chem. 2017, 93, 212–227. [Google Scholar] [CrossRef]
- Konry, T.; Bale, S.S.; Bhushan, A.; Shen, K.; Seker, E.; Polyak, B.; Yarmush, M. Particles and microfluidics merged: Perspectives of highly sensitive diagnostic detection. Microchim. Acta 2012, 176, 251–269. [Google Scholar] [CrossRef]
- Marle, L.; Greenway, G.M. Microfluidic devices for environmental monitoring. Trends Anal. Chem. 2005, 24, 795–802. [Google Scholar] [CrossRef]
- Kudr, J.; Zitka, O.; Klimanek, M.; Vrba, R.; Adam, V. Microfluidic electrochemical devices for pollution analysis—A review. Sens. Actuat. B Chem. 2017, 246, 578–590. [Google Scholar] [CrossRef]
- Bridle, H.; Miller, B.; Desmulliez, M.P.Y. Application of microfluidics in waterborne pathogen monitoring: A review. Water Res. 2014, 55, 256–271. [Google Scholar] [CrossRef]
- Nightingale, A.M.; Beaton, A.D.; Mowlen, M.C. Trends in microfluidic systems for in situ chemical analysis of natural waters. Sens. Actuat. B: Chem. 2015, 221, 1398–1405. [Google Scholar] [CrossRef]
- Zhang, M.; Phung, S.C.; Smejkal, P.; Guijt, R.M.; Breadmore, M.C. Recent trends in capillary and micro-chip electrophoretic instrumentation for field-analysis. Trends Environ. Anal. Chem. 2018, 18, 1–10. [Google Scholar] [CrossRef]
- Gertsch, J.C.; Noblitt, S.D.; Cropek, D.M.; Henry, C.S. Rapid analysis of perchlorate in drinking water at parts per billion levels using microchip electrophoresis. Anal. Chem. 2010, 82, 3426–3429. [Google Scholar] [CrossRef] [PubMed]
- Masár, M.; Bomastyk, B.; Bodor, R.; Horčičiak, M.; Danč, L.; Troška, P.; Kuss, H.-M. Determination of chloride, sulfate and nitrate in drinking water by microchip electrophoresis. Microchim Acta 2012, 177, 309–316. [Google Scholar] [CrossRef]
- Yan, X.; Liu, W.; Yuan, Y.; Chen, C. Indium tin oxide coated PET film contactless conductivity detector for microchip capillary electrophoresis. Anal. Methods 2015, 7, 5295–5302. [Google Scholar] [CrossRef]
- Luc, M.; Kruk, P.; Masar, M. Determination of ammonium in wastewaters by capillary electrophoresis on a column-coupling chip with conductivity detection. J. Sep. Sci. 2011, 34, 1561–1567. [Google Scholar] [CrossRef]
- Koczka, P.I.; Bodoki, E.; Gaspár, A. Application of capacitively coupled contactless conductivity as an external detector for zone electrophoresis in poly(dimethylsiloxane) chips. Electrophoresis 2016, 37, 398–405. [Google Scholar] [CrossRef]
- Mahabadi, K.A.; Rodriguez, I.; Lim, C.Y.; Maurya, D.K.; Hauser, P.; De Rooij, N.F. Capacitively coupled contactless conductivity detection with dual top–bottom cell configuration for microchip electrophoresis. Electrophoresis 2010, 31, 1063–1070. [Google Scholar] [CrossRef]
- Freitas, C.B.; Moreira, R.C.; De Oliveira Tavares, M.G.; Coltro, W.K.T. Monitoring of nitrite, nitrate, chloride and sulfate in environmental samples using electrophoresis microchips coupled with contactless conductivity detection. Talanta 2016, 147, 335–341. [Google Scholar] [CrossRef]
- Da Silva, E.N.T.; Petroni, J.M.; Lucca, B.G.; Ferreira, V.S. Pencil graphite leads as simple amperometric sensors for microchip electrophoresis. Electrophoresis 2017, 38, 2733–2740. [Google Scholar] [CrossRef]
- Petroni, J.M.; Lucca, B.G.; Ferreira, V.S. Simple approach for the fabrication of screen-printed carbon-based electrode for amperometric detection on microchip electrophoresis. Anal. Chem. Acta 2017, 954, 88–96. [Google Scholar] [CrossRef]
- Chen, X.; Hong, F.; Zhang, W.; Wu, D.; Li, T.; Hu, F.; Gan, N.; Lin, J.; Wang, Q. Microchip electrophoresis based multiplexerd assay for silver and mercury ions simultaneous detection in complex samples using a stirring bar modified with encoded hairpin probes for specific extraction. J. Chromatogr. A 2019, 1589, 173–181. [Google Scholar] [CrossRef]
- Gałuszka, A.; Migaszewski, Z.M.; Namieśnik, J. Moving your laboratories to the field—Advantages and limitations of the use of field portable instruments in environmental sample analysis. Environ. Res. 2015, 140, 593–603. [Google Scholar] [CrossRef]
- Van Schepdael, A. Recent advances in portable analytical electromigration devices. Separations 2016, 3, 2. [Google Scholar] [CrossRef]
- Zamuruyev, K.; Ferreira Santos, M.S.; Mora, M.F.; Kurfman, E.A.; Noell, A.C.; Willis, P.A. Automated capillary electrophoresis system compatible with multiple detectors for potential in situ spaceflight missions. Anal. Chem. 2021, 93, 9647–9655. [Google Scholar] [CrossRef]
- Beutner, A.; Piendl, S.K.; Wet, S.; Matysik, F.-M. Methodical studies of the simultaneous determinations of anions and cations by ICxCE-MS using arsenic species as model analytes. Anal. Bioanal. Chem. 2018, 410, 6321–6330. [Google Scholar] [CrossRef] [PubMed]
- Ranjbar, L.; Foley, J.P.; Breadmore, M.C. Multidimensional liquid-phase separations combining both chromatography and electrophoresis—A review. Anal. Chim. Acta 2017, 959, 7–31. [Google Scholar] [CrossRef]
- Chen, N.; Meng, X.; Ding, P.; Su, Y.; Wang, H.; He, Y. Biomimetic preparation of core-shell structured surface-enhanced Raman scattering substrate with antifouling ability, good stability, and reliable quantitative capability. Electrophoresis 2019, 40, 2172–2179. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Nakamura, K.; Timerbaev, A.R.; Hirokawa, T. Another approach toward over 100,000-fold sensitivity increase in capillary electrophoresis: Electrokinetic supercharging with optimized sample injection. Anal. Chem. 2011, 83, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Fukushi, K.; Hirokawa, T.; Timerbaev, A.R. Recent developments of capillary electrophoresis in seawater analysis. J. Chromatogr. A 2019, 1606, 360240. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Alharbi, O.M.L.; Sanagi, M.M. Nano-capillary electrophoresis for environmental analysis. Environ. Chem. Lett. 2016, 14, 79–98. [Google Scholar] [CrossRef] [PubMed]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).