
Role of Specialized Pro-Resolving Mediators in Modifying Host Defense and Decreasing Bacterial Virulence



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
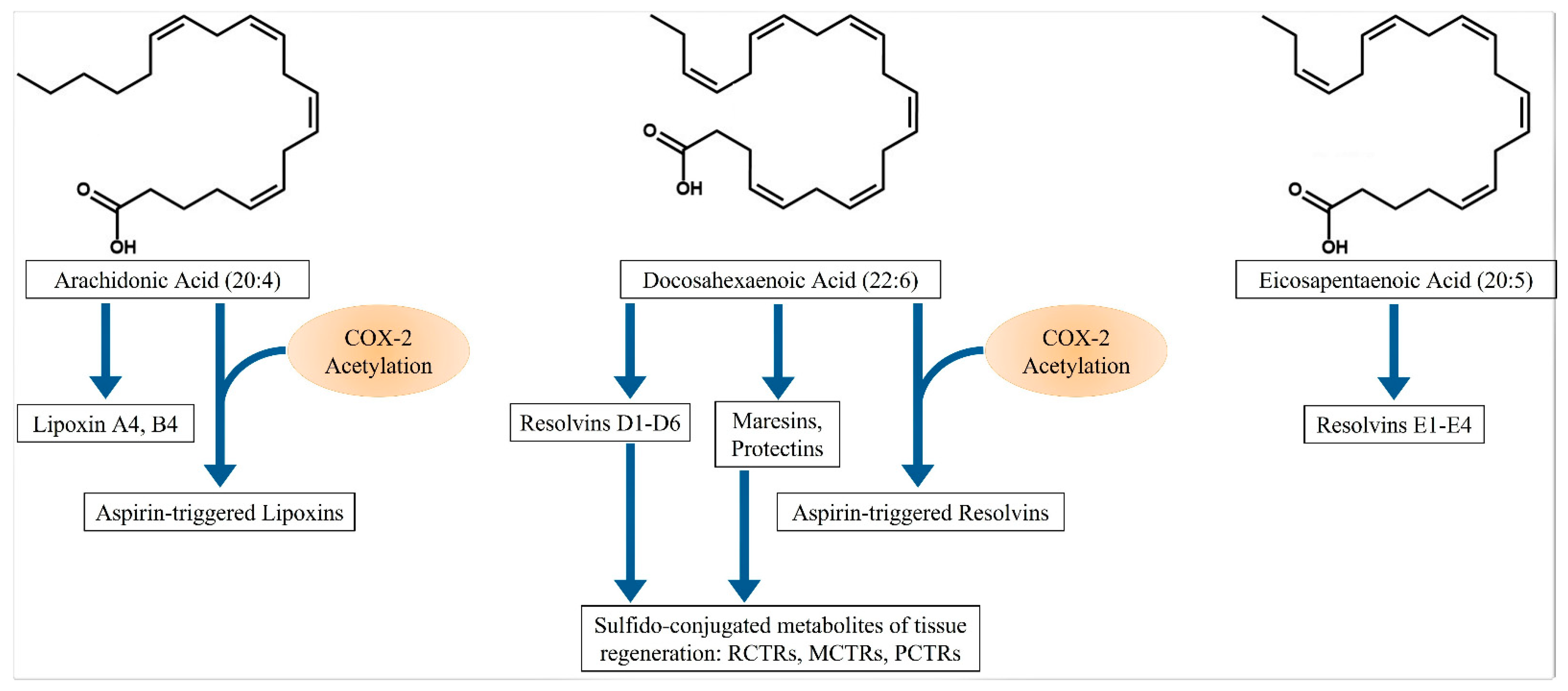
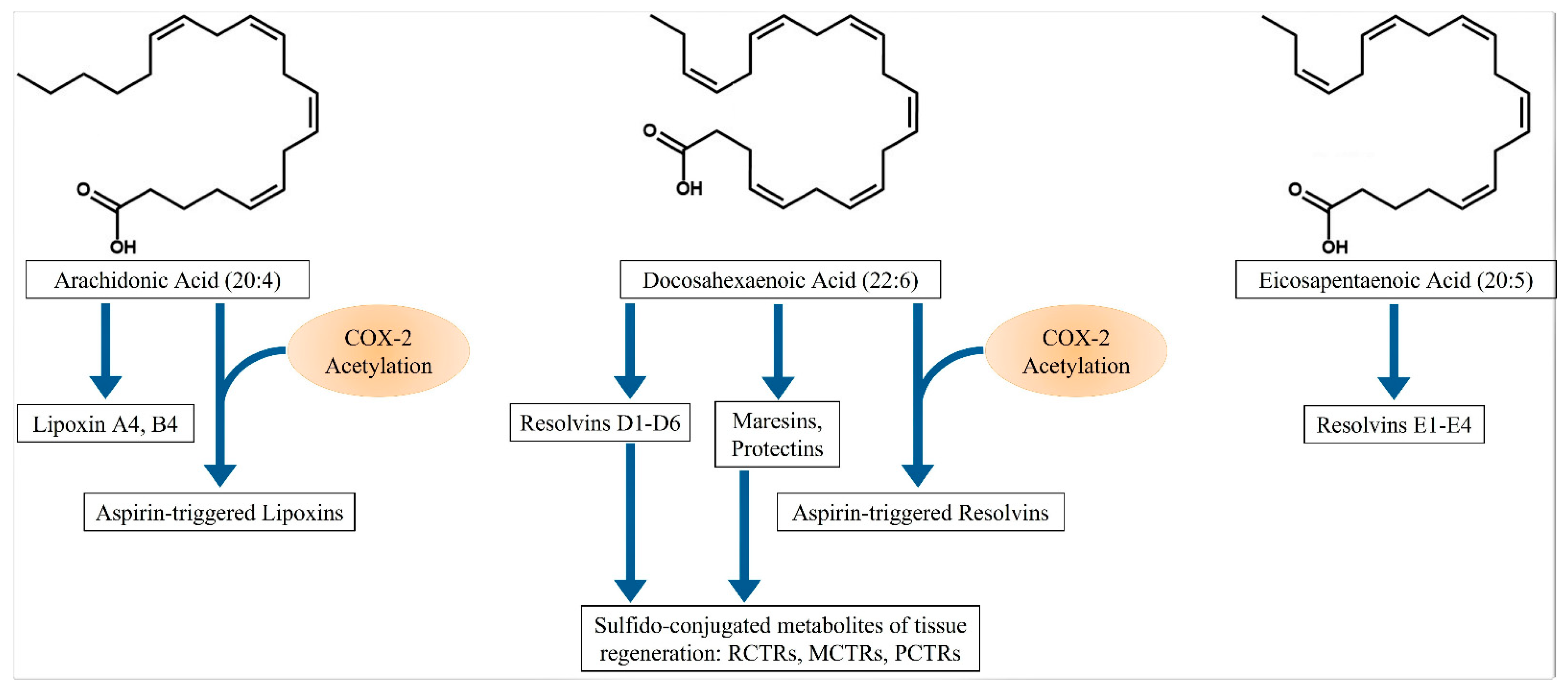
2. Specialized Proresolving Mediators (SPMs)
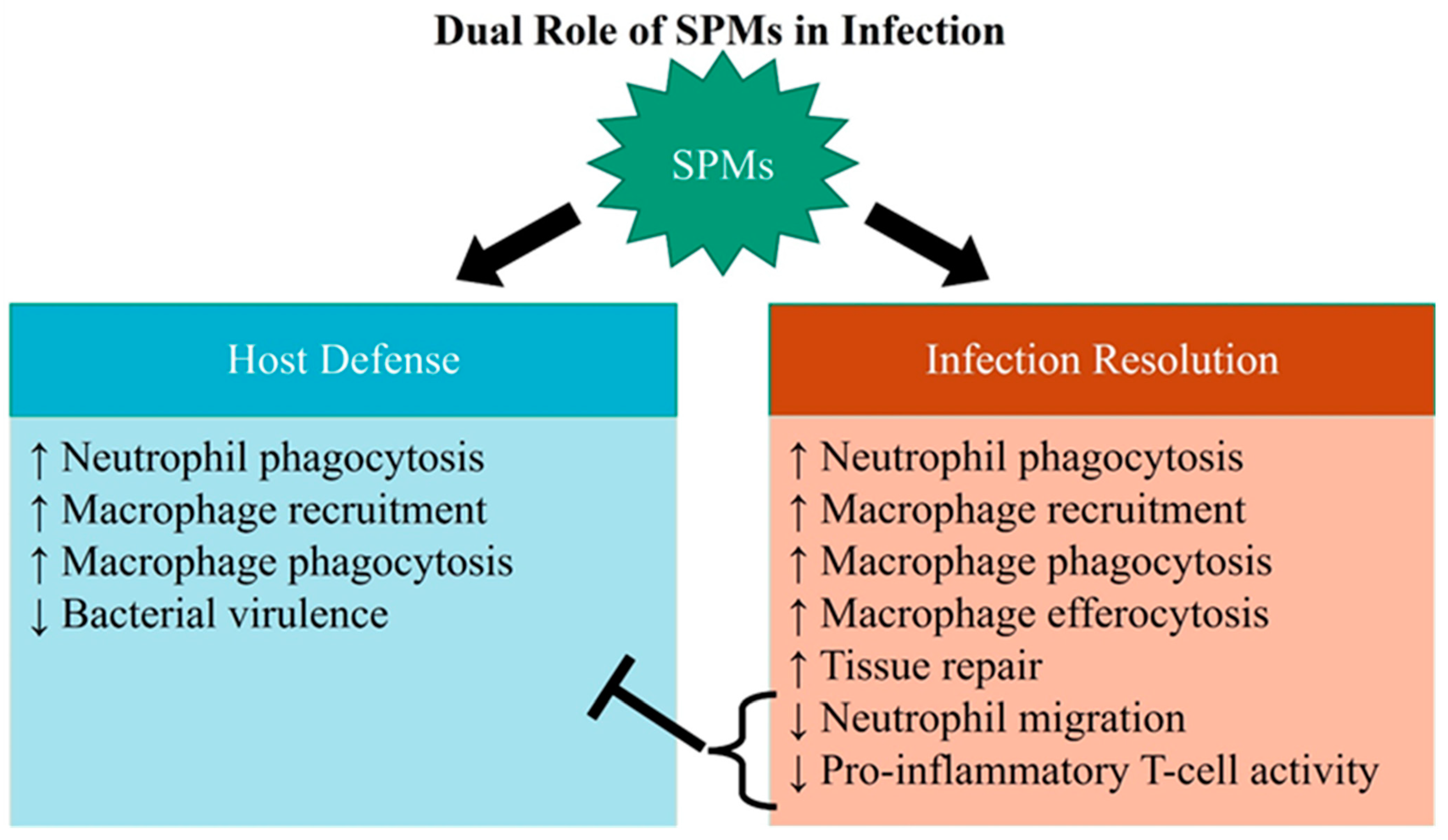
3. SPM Actions in Infectious Inflammation Resolution
4. SPM Levels in Bacterial Infections
5. SPM-Mediated Effects on Neutrophil Activity
6. SPM Effects on Macrophage Activity
7. SPM Action on T and B-Cells
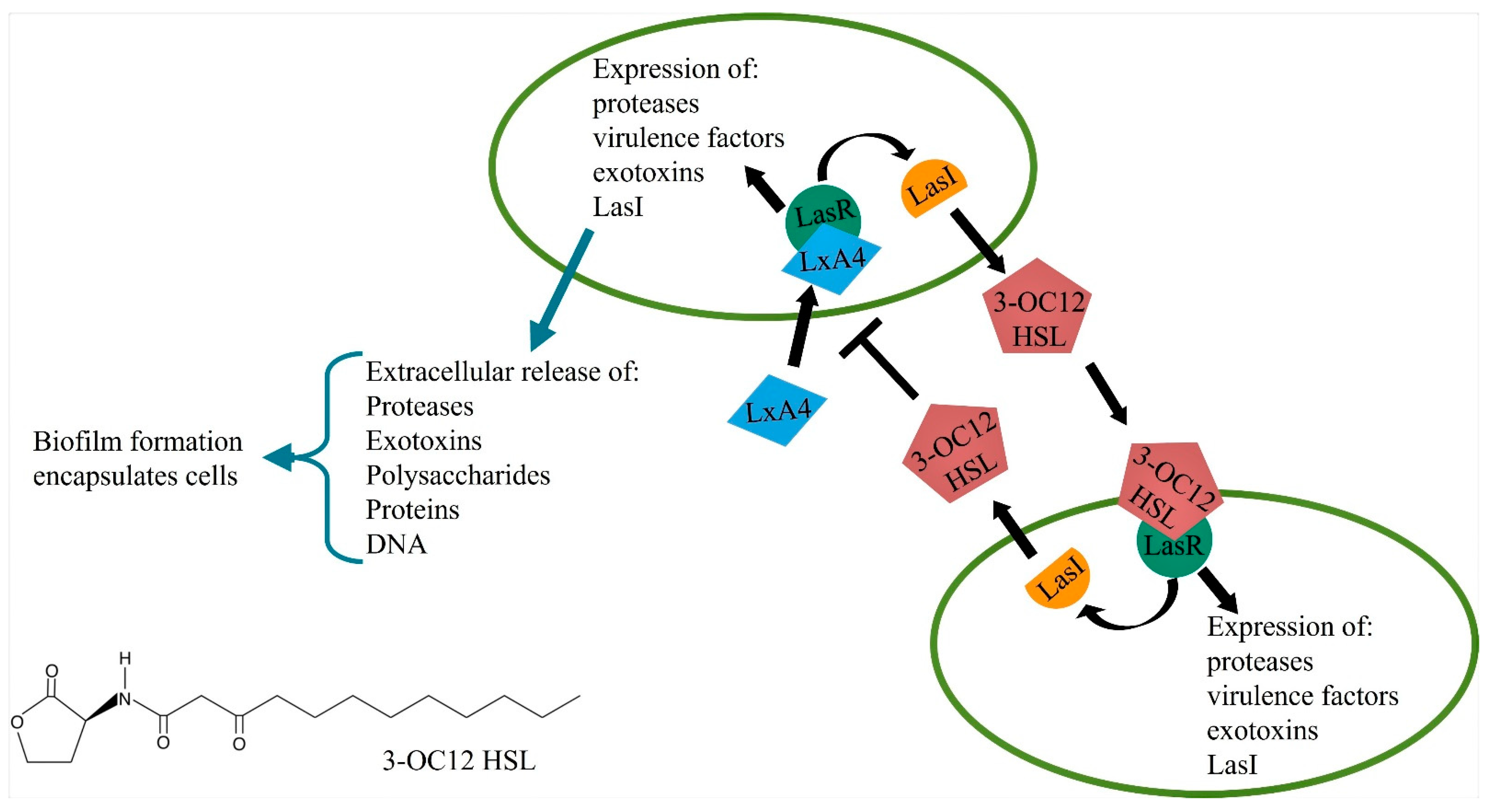
8. Quorum Sensing and Bacterial Virulence
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Van der Poll, T.; Opal, S.M. Host-pathogen interactions in sepsis. Lancet Infect. Dis. 2008, 8, 32–43. [Google Scholar] [CrossRef]
- Tsujimoto, H.; Ono, S.; Efron, P.A.; Scumpia, P.O.; Moldawer, L.L.; Mochizuki, H. Role of Toll-like receptors in the development of sepsis. Shock 2008, 29, 315–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, S.; Medzhitov, R. Toll-like receptors and their role in experimental models of microbial infection. Genes Immun. 2003, 4, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; DeLeo, F.R. Neutrophil apoptosis and the resolution of infection. Immunol. Res. 2009, 43, 25–61. [Google Scholar] [CrossRef]
- Mu, X.; Li, Y.; Fan, G.C. Tissue-Resident Macrophages in the Control of Infection and Resolution of Inflammation. Shock 2021, 55, 14–23. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Invest. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Dalli, J.; Chiang, N.; Serhan, C.N. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat. Med. 2015, 21, 1071–1075. [Google Scholar] [CrossRef]
- Leuti, A.; Maccarrone, M.; Chiurchiu, V. Proresolving Lipid Mediators: Endogenous Modulators of Oxidative Stress. Oxidative Med. Cell. Longev. 2019, 2019, 8107265. [Google Scholar] [CrossRef]
- Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Arita, M.; Hong, S.; Gotlinger, K. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their endogenous aspirin-triggered epimers. Lipids 2004, 39, 1125–1132. [Google Scholar] [CrossRef]
- Serhan, C.N.; Krishnamoorthy, S.; Recchiuti, A.; Chiang, N. Novel anti-inflammatory--pro-resolving mediators and their receptors. Curr. Top. Med. Chem. 2011, 11, 629–647. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Xie, Y.K.; Zhang, Z.J.; Wang, Z.; Xu, Z.Z.; Ji, R.R. GPR37 regulates macrophage phagocytosis and resolution of inflammatory pain. J. Clin. Invest. 2018, 128, 3568–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, N.; Libreros, S.; Norris, P.C.; de la Rosa, X.; Serhan, C.N. Maresin 1 activates LGR6 receptor promoting phagocyte immunoresolvent functions. J. Clin. Invest. 2019, 129, 5294–5311. [Google Scholar] [CrossRef] [Green Version]
- de la Rosa, X.; Norris, P.C.; Chiang, N.; Rodriguez, A.R.; Spur, B.W.; Serhan, C.N. Identification and Complete Stereochemical Assignments of the New Resolvin Conjugates in Tissue Regeneration in Human Tissues that Stimulate Proresolving Phagocyte Functions and Tissue Regeneration. Am. J. Pathol. 2018, 188, 950–966. [Google Scholar] [CrossRef] [Green Version]
- Dalli, J.; Chiang, N.; Serhan, C.N. Identification of 14-series sulfido-conjugated mediators that promote resolution of infection and organ protection. Proc. Natl. Acad. Sci. USA 2014, 111, E4753–E4761. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.; de la Rosa, X.; Libreros, S.; Pan, H.; Dreyfuss, J.M.; Serhan, C.N. Cysteinyl-specialized proresolving mediators link resolution of infectious inflammation and tissue regeneration via TRAF3 activation. Proc. Natl. Acad. Sci. USA 2021, 118, e2013374118. [Google Scholar] [CrossRef]
- Mitchell, S.; Thomas, G.; Harvey, K.; Cottell, D.; Reville, K.; Berlasconi, G.; Petasis, N.A.; Erwig, L.; Rees, A.J.; Savill, J.; et al. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: Stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J. Am. Soc. Nephrol. 2002, 13, 2497–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, J.M.; Chiang, N.; Arita, M.; Serhan, C.N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007, 447, 869–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libreros, S.; Shay, A.E.; Nshimiyimana, R.; Fichtner, D.; Martin, M.J.; Wourms, N.; Serhan, C.N. A New E-Series Resolvin: RvE4 Stereochemistry and Function in Efferocytosis of Inflammation-Resolution. Front. Immunol. 2020, 11, 631319. [Google Scholar] [CrossRef]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.L. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasuga, K.; Yang, R.; Porter, T.F.; Agrawal, N.; Petasis, N.A.; Irimia, D.; Toner, M.; Serhan, C.N. Rapid appearance of resolvin precursors in inflammatory exudates: Novel mechanisms in resolution. J. Immunol. 2008, 181, 8677–8687. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.; Serhan, C.N. Specialized pro-resolving mediator network: An update on production and actions. Essays Biochem. 2020, 64, 443–462. [Google Scholar] [CrossRef]
- Dalli, J.; Kraft, B.D.; Colas, R.A.; Shinohara, M.; Fredenburgh, L.E.; Hess, D.R.; Chiang, N.; Welty-Wolf, K.; Choi, A.M.; Piantadosi, C.A.; et al. The Regulation of Proresolving Lipid Mediator Profiles in Baboon Pneumonia by Inhaled Carbon Monoxide. Am. J. Respir. Cell Mol. Biol. 2015, 53, 314–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codagnone, M.; Cianci, E.; Lamolinara, A.; Mari, V.C.; Nespoli, A.; Isopi, E.; Mattoscio, D.; Arita, M.; Bragonzi, A.; Iezzi, M.; et al. Resolvin D1 enhances the resolution of lung inflammation caused by long-term Pseudomonas aeruginosa infection. Mucosal Immunol. 2018, 11, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werz, O.; Gerstmeier, J.; Libreros, S.; De la Rosa, X.; Werner, M.; Norris, P.C.; Chiang, N.; Serhan, C.N. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat. Commun. 2018, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Fredman, G.; Backhed, F.; Oh, S.F.; Vickery, T.; Schmidt, B.A.; Serhan, C.N. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 2012, 484, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Dalli, J.; Colas, R.A.; Quintana, C.; Barragan-Bradford, D.; Hurwitz, S.; Levy, B.D.; Choi, A.M.; Serhan, C.N.; Baron, R.M. Human Sepsis Eicosanoid and Proresolving Lipid Mediator Temporal Profiles: Correlations With Survival and Clinical Outcomes. Crit. Care Med. 2017, 45, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Barden, A.; Mas, E.; Croft, K.D.; Phillips, M.; Mori, T.A. Short-term n-3 fatty acid supplementation but not aspirin increases plasma proresolving mediators of inflammation. J. Lipid Res. 2014, 55, 2401–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mas, E.; Croft, K.D.; Zahra, P.; Barden, A.; Mori, T.A. Resolvins D1, D2, and other mediators of self-limited resolution of inflammation in human blood following n-3 fatty acid supplementation. Clin. Chem. 2012, 58, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.; Dichter, E.; Lacorte, G.; Kerner, D.; Spur, B.; Rodriguez, A.; Yin, K. Lipoxin a4 increases survival by decreasing systemic inflammation and bacterial load in sepsis. Shock 2011, 36, 410–416. [Google Scholar] [CrossRef]
- Chiang, N.; de la Rosa, X.; Libreros, S.; Serhan, C.N. Novel Resolvin D2 Receptor Axis in Infectious Inflammation. J. Immunol. 2017, 198, 842–851. [Google Scholar] [CrossRef] [Green Version]
- Santos, S.S.; Brunialti, M.K.; Rigato, O.; Machado, F.R.; Silva, E.; Salomao, R. Generation of nitric oxide and reactive oxygen species by neutrophils and monocytes from septic patients and association with outcomes. Shock 2012, 38, 18–23. [Google Scholar] [CrossRef]
- Camicia, G.; Pozner, R.; de Larranaga, G. Neutrophil extracellular traps in sepsis. Shock 2014, 42, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Kovach, M.A.; Standiford, T.J. The function of neutrophils in sepsis. Curr. Opin. Infect. Dis. 2012, 25, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, L.M.; Neff, T.A.; Neff, S.B.; Younger, J.G.; Olle, E.W.; Gao, H.; Pianko, M.J.; Bernacki, K.D.; Sarma, J.V.; Ward, P.A. Harmful and protective roles of neutrophils in sepsis. Shock 2005, 24, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Brain, S.D.; Pearson, J.D.; Edgeworth, J.D.; Lewis, S.M.; Treacher, D.F. Neutrophils in development of multiple organ failure in sepsis. Lancet 2006, 368, 157–169. [Google Scholar] [CrossRef]
- Martin, E.L.; Souza, D.G.; Fagundes, C.T.; Amaral, F.A.; Assenzio, B.; Puntorieri, V.; Del Sorbo, L.; Fanelli, V.; Bosco, M.; Delsedime, L.; et al. Phosphoinositide-3 kinase gamma activity contributes to sepsis and organ damage by altering neutrophil recruitment. Am. J. Respir. Crit. Care Med. 2010, 182, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Rios-Santos, F.; Alves-Filho, J.C.; Souto, F.O.; Spiller, F.; Freitas, A.; Lotufo, C.M.; Soares, M.B.; Dos Santos, R.R.; Teixeira, M.M.; Cunha, F.Q. Down-regulation of CXCR2 on neutrophils in severe sepsis is mediated by inducible nitric oxide synthase-derived nitric oxide. Am. J. Respir. Crit. Care Med. 2007, 175, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Delano, M.J.; Thayer, T.; Gabrilovich, S.; Kelly-Scumpia, K.M.; Winfield, R.D.; Scumpia, P.O.; Cuenca, A.G.; Warner, E.; Wallet, S.M.; Wallet, M.A.; et al. Sepsis induces early alterations in innate immunity that impact mortality to secondary infection. J. Immunol. 2011, 186, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Medvedev, A.E. Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J. Leukoc. Biol. 2011, 90, 1141–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darcy, C.J.; Minigo, G.; Piera, K.A.; Davis, J.S.; McNeil, Y.R.; Chen, Y.; Volkheimer, A.D.; Weinberg, J.B.; Anstey, N.M.; Woodberry, T. Neutrophils with myeloid derived suppressor function deplete arginine and constrain T cell function in septic shock patients. Crit. Care 2014, 18, R163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taneja, R.; Parodo, J.; Jia, S.H.; Kapus, A.; Rotstein, O.D.; Marshall, J.C. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit. Care Med. 2004, 32, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Fialkow, L.; Fochesatto Filho, L.; Bozzetti, M.C.; Milani, A.R.; Rodrigues Filho, E.M.; Ladniuk, R.M.; Pierozan, P.; de Moura, R.M.; Prolla, J.C.; Vachon, E.; et al. Neutrophil apoptosis: A marker of disease severity in sepsis and sepsis-induced acute respiratory distress syndrome. Crit. Care 2006, 10, R155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekheri, M.; El Kebir, D.; Edner, N.; Filep, J.G. 15-Epi-LXA4 and 17-epi-RvD1 restore TLR9-mediated impaired neutrophil phagocytosis and accelerate resolution of lung inflammation. Proc. Natl. Acad. Sci. USA 2020, 117, 7971–7980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Kebir, D.; Gjorstrup, P.; Filep, J.G. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 14983–14988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannenberg, G.L.; Chiang, N.; Ariel, A.; Arita, M.; Tjonahen, E.; Gotlinger, K.H.; Hong, S.; Serhan, C.N. Molecular circuits of resolution: Formation and actions of resolvins and protectins. J. Immunol. 2005, 174, 4345–4355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Maddox, J.F.; Petasis, N.A.; Akritopoulou-Zanze, I.; Papayianni, A.; Brady, H.R.; Colgan, S.P.; Madara, J.L. Design of lipoxin A4 stable analogs that block transmigration and adhesion of human neutrophils. Biochemistry 1995, 34, 14609–14615. [Google Scholar] [CrossRef] [PubMed]
- Papayianni, A.; Serhan, C.N.; Brady, H.R. Lipoxin A4 and B4 inhibit leukotriene-stimulated interactions of human neutrophils and endothelial cells. J. Immunol. 1996, 156, 2264–2272. [Google Scholar] [PubMed]
- Sun, Y.P.; Oh, S.F.; Uddin, J.; Yang, R.; Gotlinger, K.; Campbell, E.; Colgan, S.P.; Petasis, N.A.; Serhan, C.N. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J. Biol. Chem. 2007, 282, 9323–9334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, H.; Fukunaga, K.; Arita, M.; Arai, H.; Nakanishi, H.; Taguchi, R.; Miyasho, T.; Takamiya, R.; Asano, K.; Ishizaka, A.; et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J. Immunol. 2010, 184, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulnour, R.E.; Sham, H.P.; Douda, D.N.; Colas, R.A.; Dalli, J.; Bai, Y.; Ai, X.; Serhan, C.N.; Levy, B.D. Aspirin-triggered resolvin D1 is produced during self-resolving gram-negative bacterial pneumonia and regulates host immune responses for the resolution of lung inflammation. Mucosal Immunol. 2016, 9, 1278–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, E.R.; Croze, R.H.; Fang, X.; Matthay, M.A.; Gotts, J.E. Inhibition of the lipoxin A4 and resolvin D1 receptor impairs host response to acute lung injury caused by pneumococcal pneumonia in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L1085–L1092. [Google Scholar] [CrossRef] [PubMed]
- El Kebir, D.; Jozsef, L.; Pan, W.; Wang, L.; Petasis, N.A.; Serhan, C.N.; Filep, J.G. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 2009, 180, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Walker, J.; Spur, B.; Rodriguez, A.; Yin, K. Effects of Lipoxin A4 on antimicrobial actions of neutrophils in sepsis. Prostaglandins Leukot. Essent. Fat. Acids 2015, 94, 55–64. [Google Scholar] [CrossRef]
- Wu, B.; Capilato, J.; Pham, M.P.; Walker, J.; Spur, B.; Rodriguez, A.; Perez, L.J.; Yin, K. Lipoxin A4 augments host defense in sepsis and reduces Pseudomonas aeruginosa virulence through quorum sensing inhibition. FASEB J. 2016, 30, 2400–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, E.; Schlatterer, K.; Beck, C.; Peschel, A.; Kretschmer, D. Formyl-Peptide Receptor Activation Enhances Phagocytosis of Community-Acquired Methicillin-Resistant Staphylococcus aureus. J. Infect. Dis. 2020, 221, 668–678. [Google Scholar] [CrossRef]
- Pouliot, M.; Clish, C.B.; Petasis, N.A.; Van Dyke, T.E.; Serhan, C.N. Lipoxin A(4) analogues inhibit leukocyte recruitment to Porphyromonas gingivalis: A role for cyclooxygenase-2 and lipoxins in periodontal disease. Biochemistry 2000, 39, 4761–4768. [Google Scholar] [CrossRef]
- Hasturk, H.; Schulte, F.; Martins, M.; Sherzai, H.; Floros, C.; Cugini, M.; Chiu, C.J.; Hardt, M.; Van Dyke, T. Safety and Preliminary Efficacy of a Novel Host-Modulatory Therapy for Reducing Gingival Inflammation. Front. Immunol. 2021, 12, 704163. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Liu, S.; Su, X.; Pan, P.; Zhang, L.; Hu, Y.; Tan, H.; Wu, D.; Liu, B.; Li, H.; Li, H.; et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci. Rep. 2016, 6, 37252. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Koike, Y.; Shimura, T.; Okigami, M.; Ide, S.; Toiyama, Y.; Okugawa, Y.; Inoue, Y.; Araki, T.; Uchida, K.; et al. In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PLoS ONE 2014, 9, e111888. [Google Scholar] [CrossRef] [PubMed]
- Lefrancais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 2018, 3, e98178. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [Green Version]
- Cherpokova, D.; Jouvene, C.C.; Libreros, S.; DeRoo, E.P.; Chu, L.; de la Rosa, X.; Norris, P.C.; Wagner, D.D.; Serhan, C.N. Resolvin D4 attenuates the severity of pathological thrombosis in mice. Blood 2019, 134, 1458–1468. [Google Scholar] [CrossRef]
- Isopi, E.; Mattoscio, D.; Codagnone, M.; Mari, V.C.; Lamolinara, A.; Patruno, S.; D’Aurora, M.; Cianci, E.; Nespoli, A.; Franchi, S.; et al. Resolvin D1 Reduces Lung Infection and Inflammation Activating Resolution in Cystic Fibrosis. Front. Immunol. 2020, 11, 581. [Google Scholar] [CrossRef]
- Sordi, R.; Menezes-de-Lima, O., Jr.; Horewicz, V.; Scheschowitsch, K.; Santos, L.F.; Assreuy, J. Dual role of lipoxin A4 in pneumosepsis pathogenesis. Int. Immunopharmacol. 2013, 17, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Maddox, J.F.; Serhan, C.N. Lipoxin A4 and B4 are potent stimuli for human monocyte migration and adhesion: Selective inactivation by dehydrogenation and reduction. J. Exp. Med. 1996, 183, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Godson, C.; Mitchell, S.; Harvey, K.; Petasis, N.A.; Hogg, N.; Brady, H.R. Cutting edge: Lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 2000, 164, 1663–1667. [Google Scholar] [CrossRef]
- Reville, K.; Crean, J.K.; Vivers, S.; Dransfield, I.; Godson, C. Lipoxin A4 redistributes myosin IIA and Cdc42 in macrophages: Implications for phagocytosis of apoptotic leukocytes. J. Immunol. 2006, 176, 1878–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recchiuti, A.; Codagnone, M.; Pierdomenico, A.M.; Rossi, C.; Mari, V.C.; Cianci, E.; Simiele, F.; Gatta, V.; Romano, M. Immunoresolving actions of oral resolvin D1 include selective regulation of the transcription machinery in resolution-phase mouse macrophages. FASEB J. 2014, 28, 3090–3102. [Google Scholar] [CrossRef] [Green Version]
- Rittirsch, D.; Huber-Lang, M.S.; Flierl, M.A.; Ward, P.A. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2009, 4, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muenzer, J.T.; Davis, C.G.; Dunne, B.S.; Unsinger, J.; Dunne, W.M.; Hotchkiss, R.S. Pneumonia after cecal ligation and puncture: A clinically relevant “two-hit” model of sepsis. Shock 2006, 26, 565–570. [Google Scholar] [CrossRef]
- Remick, D.G.; Newcomb, D.E.; Bolgos, G.L.; Call, D.R. Comparison of the mortality and inflammatory response of two models of sepsis: Lipopolysaccharide vs. cecal ligation and puncture. Shock 2000, 13, 110–116. [Google Scholar] [CrossRef]
- Ayala, A.; Perrin, M.M.; Kisala, J.M.; Ertel, W.; Chaudry, I.H. Polymicrobial sepsis selectively activates peritoneal but not alveolar macrophages to release inflammatory mediators (interleukins-1 and -6 and tumor necrosis factor). Circ. Shock 1992, 36, 191–199. [Google Scholar]
- Pahuja, M.; Tran, C.; Wang, H.; Yin, K. Alveolar macrophage suppression in sepsis is associated with high mobility group box 1 transmigration. Shock 2008, 29, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Spite, M.; Norling, L.V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N.A.; Flower, R.J.; Perretti, M.; Serhan, C.N. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 2009, 461, 1287–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, N.; Dalli, J.; Colas, R.A.; Serhan, C.N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 2015, 212, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wang, Y.; Ma, Z.; Ma, M.; Wang, D.; Xie, G.; Yin, Y.; Zhang, P.; Tao, K. Maresin 1 Mitigates Inflammatory Response and Protects Mice from Sepsis. Mediat. Inflamm. 2016, 2016, 3798465. [Google Scholar] [CrossRef] [PubMed]
- Croasdell, A.; Sime, P.J.; Phipps, R.P. Resolvin D2 decreases TLR4 expression to mediate resolution in human monocytes. FASEB J. 2016, 30, 3181–3193. [Google Scholar] [CrossRef] [Green Version]
- Fredman, G.; Oh, S.F.; Ayilavarapu, S.; Hasturk, H.; Serhan, C.N.; Van Dyke, T.E. Impaired phagocytosis in localized aggressive periodontitis: Rescue by Resolvin E1. PLoS ONE 2011, 6, e24422. [Google Scholar] [CrossRef] [PubMed]
- Souza, D.G.; Fagundes, C.T.; Amaral, F.A.; Cisalpino, D.; Sousa, L.P.; Vieira, A.T.; Pinho, V.; Nicoli, J.R.; Vieira, L.Q.; Fierro, I.M.; et al. The required role of endogenously produced lipoxin A4 and annexin-1 for the production of IL-10 and inflammatory hyporesponsiveness in mice. J. Immunol. 2007, 179, 8533–8543. [Google Scholar] [CrossRef] [Green Version]
- van Vught, L.A.; Wiewel, M.A.; Hoogendijk, A.J.; Frencken, J.F.; Scicluna, B.P.; Klein Klouwenberg, P.M.C.; Zwinderman, A.H.; Lutter, R.; Horn, J.; Schultz, M.J.; et al. The Host Response in Patients with Sepsis Developing Intensive Care Unit-acquired Secondary Infections. Am. J. Respir. Crit. Care Med. 2017, 196, 458–470. [Google Scholar] [CrossRef]
- Abe, R.; Hirasawa, H.; Oda, S.; Sadahiro, T.; Nakamura, M.; Watanabe, E.; Nakada, T.A.; Hatano, M.; Tokuhisa, T. Up-regulation of interleukin-10 mRNA expression in peripheral leukocytes predicts poor outcome and diminished human leukocyte antigen-DR expression on monocytes in septic patients. J. Surg. Res. 2008, 147, 1–8. [Google Scholar] [CrossRef]
- Sfeir, T.; Saha, D.C.; Astiz, M.; Rackow, E.C. Role of interleukin-10 in monocyte hyporesponsiveness associated with septic shock. Crit. Care Med. 2001, 29, 129–133. [Google Scholar] [CrossRef]
- Vassiliou, E.K.; Kesler, O.M.; Tadros, J.H.; Ganea, D. Bone marrow-derived dendritic cells generated in the presence of resolvin E1 induce apoptosis of activated CD4+ T cells. J. Immunol. 2008, 181, 4534–4544. [Google Scholar] [CrossRef] [Green Version]
- Ariel, A.; Li, P.L.; Wang, W.; Tang, W.X.; Fredman, G.; Hong, S.; Gotlinger, K.H.; Serhan, C.N. The docosatriene protectin D1 is produced by TH2 skewing and promotes human T cell apoptosis via lipid raft clustering. J. Biol. Chem. 2005, 280, 43079–43086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiurchiu, V.; Leuti, A.; Dalli, J.; Jacobsson, A.; Battistini, L.; Maccarrone, M.; Serhan, C.N. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci. Transl. Med. 2016, 8, 353ra111. [Google Scholar] [CrossRef] [Green Version]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Oner, F.; Alvarez, C.; Yaghmoor, W.; Stephens, D.; Hasturk, H.; Firatli, E.; Kantarci, A. Resolvin E1 Regulates Th17 Function and T Cell Activation. Front. Immunol. 2021, 12, 637983. [Google Scholar] [CrossRef] [PubMed]
- Derada Troletti, C.; Enzmann, G.; Chiurchiu, V.; Kamermans, A.; Tietz, S.M.; Norris, P.C.; Jahromi, N.H.; Leuti, A.; van der Pol, S.M.A.; Schouten, M.; et al. Pro-resolving lipid mediator lipoxin A4 attenuates neuro-inflammation by modulating T cell responses and modifies the spinal cord lipidome. Cell Rep. 2021, 35, 109201. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Chen, H.; Li, Y.; Zhong, H.; Sun, W.; Wang, J.; Zhang, T.; Ma, J.; Yan, S.; Zhang, J.; et al. Maresin 1 improves the Treg/Th17 imbalance in rheumatoid arthritis through miR-21. Ann. Rheum. Dis. 2018, 77, 1644–1652. [Google Scholar] [CrossRef]
- Chiurchiu, V.; Leuti, A.; Saracini, S.; Fontana, D.; Finamore, P.; Giua, R.; Padovini, L.; Incalzi, R.A.; Maccarrone, M. Resolution of inflammation is altered in chronic heart failure and entails a dysfunctional responsiveness of T lymphocytes. FASEB J. 2019, 33, 909–916. [Google Scholar] [CrossRef]
- Bafica, A.; Scanga, C.A.; Serhan, C.; Machado, F.; White, S.; Sher, A.; Aliberti, J. Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production. J. Clin. Invest. 2005, 115, 1601–1606. [Google Scholar] [CrossRef]
- Ramon, S.; Gao, F.; Serhan, C.N.; Phipps, R.P. Specialized proresolving mediators enhance human B cell differentiation to antibody-secreting cells. J. Immunol. 2012, 189, 1036–1042. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Ramon, S.; Thatcher, T.H.; Woeller, C.F.; Sime, P.J.; Phipps, R.P. Specialized proresolving mediators (SPMs) inhibit human B-cell IgE production. Eur. J. Immunol. 2016, 46, 81–91. [Google Scholar] [CrossRef]
- Kim, N.; Thatcher, T.H.; Sime, P.J.; Phipps, R.P. Corticosteroids inhibit anti-IgE activities of specialized proresolving mediators on B cells from asthma patients. JCI Insight 2017, 2, e88588. [Google Scholar] [CrossRef] [Green Version]
- Ramon, S.; Bancos, S.; Serhan, C.N.; Phipps, R.P. Lipoxin A(4) modulates adaptive immunity by decreasing memory B-cell responses via an ALX/FPR2-dependent mechanism. Eur. J. Immunol. 2014, 44, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, P.N.; Koch, G.; Thompson, J.A.; Xavier, K.B.; Cool, R.H.; Quax, W.J. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2012, 76, 46–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Zhang, L. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell 2015, 6, 26–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Loughlin, C.T.; Miller, L.C.; Siryaporn, A.; Drescher, K.; Semmelhack, M.F.; Bassler, B.L. A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc. Natl. Acad. Sci. USA 2013, 110, 17981–17986. [Google Scholar] [CrossRef] [Green Version]
- Munguia, J.; Nizet, V. Pharmacological Targeting of the Host-Pathogen Interaction: Alternatives to Classical Antibiotics to Combat Drug-Resistant Superbugs. Trends Pharmacol. Sci. 2017, 38, 473–488. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.C.; Chan, K.G.; Chang, C.Y. Modulation of Host Biology by Pseudomonas aeruginosa Quorum Sensing Signal Molecules: Messengers or Traitors. Front. Microbiol. 2015, 6, 1226. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.M.; Walker, J.M.; Sundarasivarao, P.Y.K.; Spur, B.W.; Rodriguez, A.; Yin, K. Lipoxin A4 promotes reduction and antibiotic efficacy against Pseudomonas aeruginosa biofilm. Prostaglandins Other Lipid Mediat. 2021, 152, 106505. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thornton, J.M.; Yin, K. Role of Specialized Pro-Resolving Mediators in Modifying Host Defense and Decreasing Bacterial Virulence. Molecules 2021, 26, 6970. https://doi.org/10.3390/molecules26226970
Thornton JM, Yin K. Role of Specialized Pro-Resolving Mediators in Modifying Host Defense and Decreasing Bacterial Virulence. Molecules. 2021; 26(22):6970. https://doi.org/10.3390/molecules26226970
Chicago/Turabian StyleThornton, Julianne M., and Kingsley Yin. 2021. "Role of Specialized Pro-Resolving Mediators in Modifying Host Defense and Decreasing Bacterial Virulence" Molecules 26, no. 22: 6970. https://doi.org/10.3390/molecules26226970
APA StyleThornton, J. M., & Yin, K. (2021). Role of Specialized Pro-Resolving Mediators in Modifying Host Defense and Decreasing Bacterial Virulence. Molecules, 26(22), 6970. https://doi.org/10.3390/molecules26226970