
Mesoporous Networks of N-Vinylpyrrolidone with (di)Methacrylates as Precursors of Ecological Molecular Imprinted Polymers

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Preparation of Macromolecular Porogens—VP-TEGDM and VP-PEGMMA-TEGDM Branched Copolymers
2.3. Preparation of VP-TEGDM Polymer Composites with br-VP-TEGDM Polymer Additive
2.4. Preparation of VP-PEGMMA-TEGDM Polymer Composites with br-VP-PEGMMA-TEGDM Additive
2.5. Extraction of Sol Fractions from the Polymer Composites
2.6. IR- and 1H NMR Spectroscopy
2.7. Elemental Analysis
2.8. Size-Exclusion Chromatography
2.9. Transmission Electron Microscopy
2.10. Dynamic Light Scattering
2.11. Differential Scanning Calorimetry
2.12. Physical Mechanical Measurements
2.13. Scanning Electron Microscopy
2.14. Measurement of the Specific Surface Area of Porous Polymer Matrices
2.15. Electronic Absorption Spectroscopy
2.16. Electrochemical Studies
2.17. Quantum Chemical Modeling of the Structure of RB Complexes with the VP Monomer and a VP-VP-VP Site of the Copolymer
2.18. Preparation of VP-TEGDM Porous Copolymers for Studying RB Adsorption from Aqueous Solutions
2.19. Preparation of VP-PEGMMA-TEGDM Porous Copolymers for Studying RB Adsorption from Aqueous Solutions
2.20. Experimental Procedure for RB Adsorption from Aqueous Solutions by Porous Polymer Matrices
2.21. Experimental Technique for RB Desorption from VP-TEGDM Polymer Matrices in Aqueous Solutions
2.22. Experimental Technique for RB Desorption from VP-PEGMMA-TEGDM Polymer Matrices in Aqueous Solutions
2.23. Determination of the Specific Surface Area from the Data of RB Adsorption by Polymer Matrices
3. Results and Discussion
3.1. Synthesis and Characteristics of Branched Copolymers of VP as Potential Macromolecular Porogens during the Formation of Polymer Matrices
3.2. FTIR and 1H NMR Spectroscopy Data Indicate the Monomeric Composition of the Copolymers
3.3. Formation of Polymer Composites and Obtaining Porous Polymer Matrices
3.4. Sol-Gel Analysis of the Polymer Composites and Their Structures
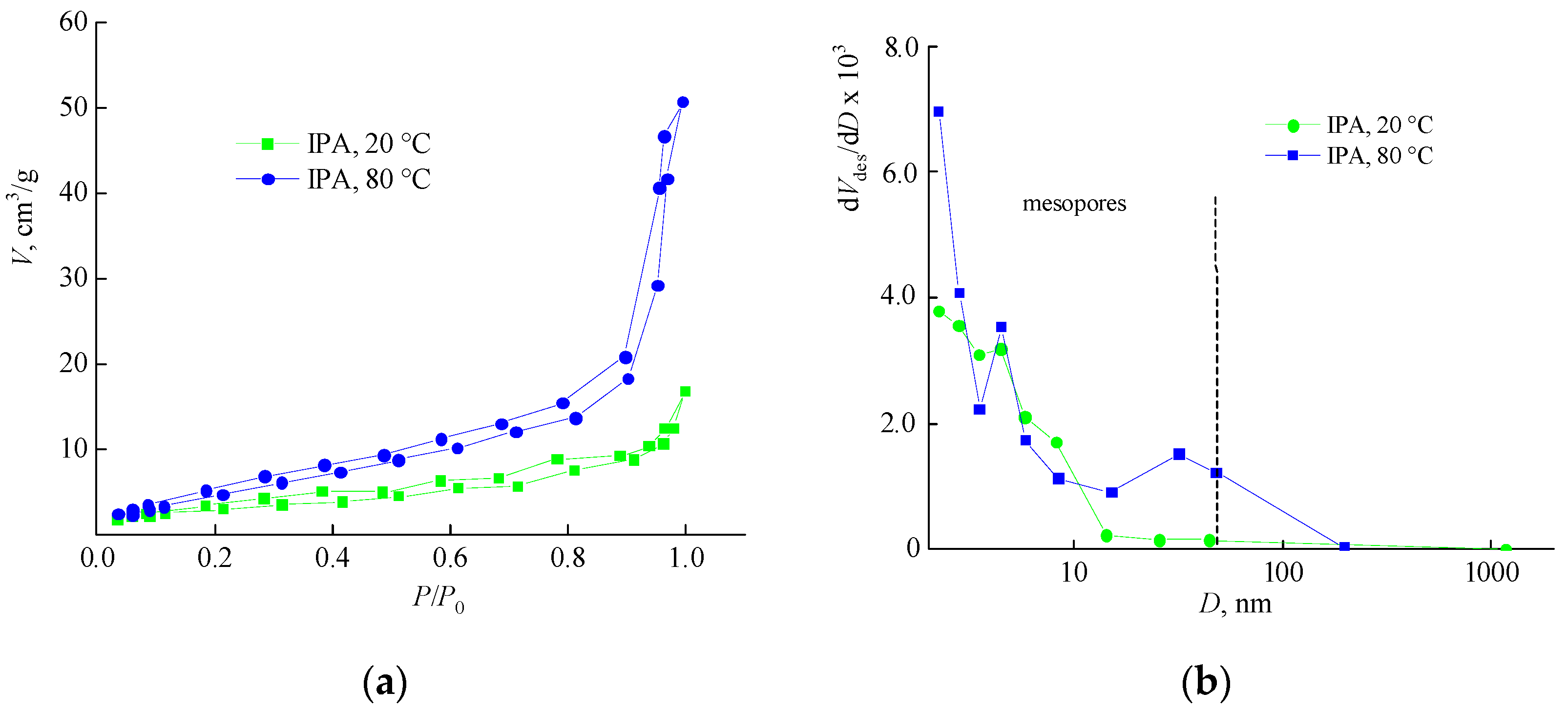
3.5. Analysis of the Porous Structure of Polymer Matrices by Low-Temperature Nitrogen Adsorption and Dye Adsorption from Aqueous Solution
3.6. Adsorption of Rose Bengal from Aqueous Solutions by Polymer Matrices
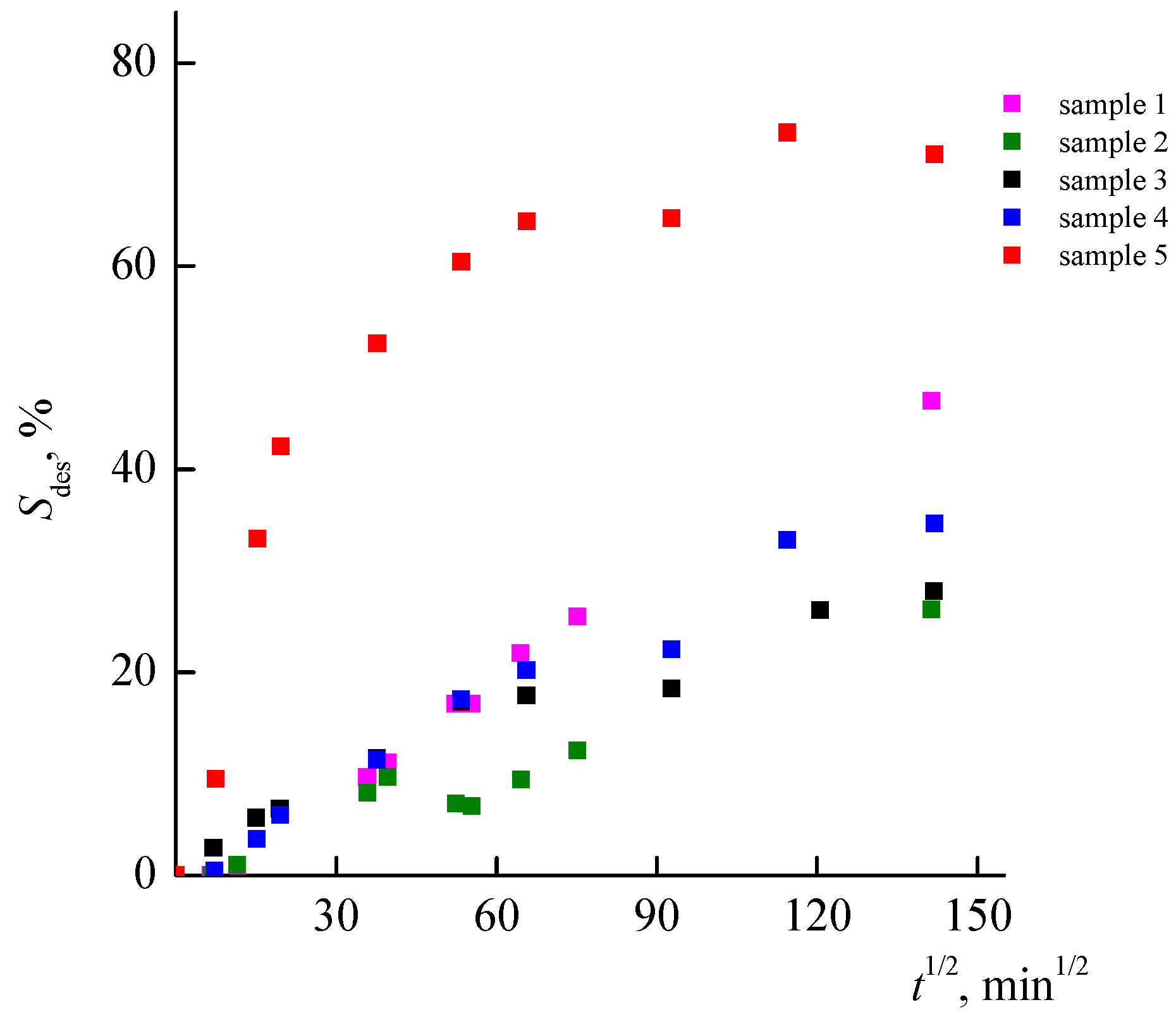
3.7. Desorption of RB from Nanoporous Polymer Matrices
3.8. Electrochemical Behavior of Free and Bound RB in Aqueous Solution
3.9. Quantum Chemical Modeling the Structure of H-Complex of RB with VP Monomer and VP-VP-VP Site of the Copolymer
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wulff, G.; Sarhan, A. Use of polymers with enzyme-analogous structures for the resolution of racemates. Angew. Chem. 1972, 11, 341–344. [Google Scholar]
- Arshady, R.; Mosbach, M. Synthesis of substrate-selective polymers by host-guest polymerization. Macromol. Chem. Chem. Phys.-Macromol. Chem. 1981, 182, 687–692. [Google Scholar] [CrossRef]
- Hasanah, A.N.; Soni, D.; Pratiwi, R.; Rahayu, D.; Megantara, S.; Mutakin. Synthesis of diazepam-imprinted polymers with two functional monomers in chloroform using a bulk polymerization method. J. Chem. 2020, 2020, 7282415. [Google Scholar] [CrossRef]
- Keçili, R.; Hussain, C.M. Recent progress of imprinted nanomaterials in analytical chemistry. Int. J. Anal. Chem. 2018, 2018, 8503853. [Google Scholar] [CrossRef]
- Dinc, M.; Esen, C.; Mizaikoff, B. Recent advances on core–shell magnetic molecularly imprinted polymers for biomacromolecules. TrAC-Trends Anal. Chem. 2019, 114, 202–217. [Google Scholar] [CrossRef]
- Piletsky, S.; Canfarotta, F.; Poma, A.; Bossi, A.M.; Piletsky, S. Molecularly imprinted polymers for cell recognition. Trends Biotechnol. 2020, 38, 368–387. [Google Scholar] [CrossRef] [PubMed]
- Muratsugu, S.; Shirai, S.; Tada, M. Recent progress in molecularly imprinted approach for catalysis. Tetrahedron Lett. 2020, 61, 151603. [Google Scholar] [CrossRef]
- Mokhtari, P.; Ghaedi, M. Water compatible molecularly imprinted polymer for controlled release of riboflavin as drug delivery system. Eur. Polym. J. 2019, 118, 614–618. [Google Scholar] [CrossRef]
- Lulinski, P. Molecularly imprinted polymers based drug delivery devices: A way to application in modern pharmacotherapy. A review. Mater. Sci. Eng. C 2017, 76, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.K.; Yan, W.Y.; Guo, C.X.; Zhang, J.H.; Yu, L.G.; Zhang, G.H.; Wang, X.M.; Fang, G.Z.; Sun, D.D. Magnetic molecularly imprinted electrochemical sensors: A review. Anal. Chim. Acta 2020, 1106, 1–21. [Google Scholar] [CrossRef]
- Huang, D.L.; Tang, Z.H.; Peng, Z.W.; Lai, C.; Zeng, G.M.; Zhang, C.; Xu, P.A.; Cheng, M.; Wan, J.; Wang, R.Z. Fabrication of water-compatible molecularly imprinted polymer based on β-cyclodextrin modified magnetic chitosan and its application for selective removal of bisphenol A from aqueous solution. J. Taiwan Inst. Chem. Eng. 2017, 77, 113–121. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.W.; Hu, Y.L.; Li, G.K.; Chen, Y.Q. Preparation of magnetic indole-3-acetic acid imprinted polymer beads with 4-vinylpyridine and beta-cyclodextrin as binary monomer via microwave heating-initiated polymerization and their application to trace analysis of auxins in plant tissues. J. Chromatogr. A 2010, 1217, 7337–7344. [Google Scholar] [CrossRef]
- Urraca, J.L.; Hall, A.J.; Moreno-Bondi, M.C.; Sellergren, B. A Stoichiometric Molecularly imprinted polymer for the class-selective recognition of antibiotics in aqueous media. Angew. Chem. Int. Ed. 2006, 45, 5158–5161. [Google Scholar] [CrossRef]
- Oral, E.; Peppas, N.A. Hydrophilic Molecularly Imprinted poly(hydroxyethyl-methacrylate) polymers. J. Biomed. Mater. Res. A 2006, 78, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Manesiotis, P.; Borrelli, C.; Aureliano, C.S.A.; Svensson, C.; Sellergren, B. Water-Compatible Imprinted Polymers for Selective Depletion of Riboflavine from Beverages. J. Mater. Chem. 2009, 19, 6185–6193. [Google Scholar] [CrossRef]
- Kubo, T.; Hosoya, K.; Nomachi, M.; Tanaka, N.; Kaya, K. Preparation of a Novel Molecularly Imprinted Polymer Using a Water-Soluble Crosslinking Agent. Anal. Bioanal. Chem. 2005, 382, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Sarpong, K.A.; Xu, W.; Huang, W.; Yang, W. The development of molecularly imprinted polymers in the clean-up of water pollutants: A review. Amer. J. Analyt. Chem. 2019, 10, 202–226. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Xu, F.; Sun, B.; Fu, R.; He, H.; Matyjaszewski, K. Design and preparation of porous polymers. Chem. Rev. 2012, 112, 3959–4015. [Google Scholar] [CrossRef]
- Nemanash, M.; Noh, J.-H.; Meijboom, R. Dendrimers as alternative templates and pore-directing agents for the synthesis of micro- and mesoporous materials. J. Mater. Sci. 2018, 53, 12663–12678. [Google Scholar] [CrossRef]
- Kurmaz, S.V.; Kochneva, I.S.; Perepelitsina, E.O.; Bubnova, M.L.; Bakova, G.M.; Knerel’man, E.I.; Davydova, G.I. Ethyl acrylate copolymers as promising porogens for the synthesis of polydimethacrylates with controlled porous structures. Polym. Sci. 2013, 55, 712–720. [Google Scholar] [CrossRef]
- Kurmaz, S.V.; Grubenko, G.A.; Knerelman, E.I.; Davydova, G.I.; Torbov, V.I.; Dremova, N.N. Promising macromolecular nanoobjects for the template synthesis of network copolymers with mesoporous structure. Mendeleev Commun. 2014, 24, 125–127. [Google Scholar] [CrossRef]
- Fadeeva, N.V.; Kurmaz, S.V.; Knerelman, E.I.; Davydova, G.I.; Torbov, V.I.; Dremova, N.N. New polymer materials with controlled nanoporous structure based on N-vinylpyrrolidone. Russ. Chem. Bull. 2016, 65, 2089–2096. [Google Scholar] [CrossRef]
- Fadeeva, N.V.; Kurmaz, S.V.; Knerelman, E.I.; Davydova, G.I.; Torbov, V.I.; Dremova, N.N. Nanoporous polymer networks based on N-vinylpyrrolidone. Polym. Sci. Ser. B 2017, 59, 257–267. [Google Scholar] [CrossRef]
- Kurmaz, S.V.; Fadeeva, N.V.; Knerelman, E.I.; Davydova, G.I.; Torbov, V.I.; Dremova, N.N. Nanoporous polymer networks of N−vinylpyrrolidone with dimethacrylates of various polarity. Synthesis, structure, and properties. J. Polym. Res. 2019, 26, 153–165. [Google Scholar] [CrossRef]
- Kurmaz, S.V.; Pyryaev, A.N. Synthesis of N-vinyl-2-pyrrolidone-based branched copolymers via crosslinking free-radical copolymerization in the presence of a chain-transfer agent. Polym. Sci. 2010, 52, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, B.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurmaz, S.V.; Fadeeva, N.V.; Ignat’ev, V.M.; Kurmaz, V.A.; Kurochkin, S.A.; Emel’yanova, N.S. Structure and state of water in branched N-vinylpyrrolidone copolymers as carriers of a hydrophilic biologically active compound. Molecules 2020, 25, 6015. [Google Scholar] [CrossRef] [PubMed]
- Todd, A.; Keith, T.K. AIMAll, Version 10.05.04; Gristmill Software: Overland Park, KS, USA, 2010; Available online: aim.tkgristmill.com (accessed on 1 November 2021).
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com (accessed on 1 November 2021).
- Kurmaz, S.V.; Fadeeva, N.V.; Knerelman, E.I.; Davydova, G.I. Preparation of porous polymer networks of N-vinylpyrrolidone with triethylene glycol dimethacrylate and determination of their specific surface area using Rose Bengal dye. Russ. J. Appl. Chem. 2018, 91, 115–122. [Google Scholar] [CrossRef]
- Vlasova, I.M.; Polyansky, D.V.; Vlasov, A.A.; Saletsky, A.M. Investigation of rotational diffusion of the Rose Bengal fluorescent nanomarker in human serum albumin solutions. Mosc. Univ. Phys. Bull. 2013, 68, 231–234. [Google Scholar] [CrossRef]
- Roshchupkin, V.P.; Kurmaz, S.V. State-of-the-art in the studies of three-dimensional radical copolymerization. Uspekhi Khimii 2004, 73, 247–274. [Google Scholar] [CrossRef]
- Vlakh, E.G.; Korzhikov, V.A.; Hubina, A.V.; Tennikova, T.B. Moleclar imprinting: A tool of modern chemistry for the preparation of highly selective monolithic sorbents. Russ. Chem. Rev. 2015, 84, 952–980. [Google Scholar] [CrossRef]
- Gregg, S.J.; Sing, K.S.W. Adsorption, Surface Area and Porosity, 2nd ed.; Academic Press: New York, NY, USA, 1982; p. 306. [Google Scholar]
- Fini, P.; Catucci, M.; Castagnolo, M.; Cosma, P.; Pluchinotta, V.; Agostiano, A. Spectroscopic investigation of Rose Bengal/cyclodextrin interactions in aqueous solution: The case of the hydroxypropyl-cyclodextrins. J. Phenom. Macrocycl. Chem. 2007, 57, 663–668. [Google Scholar] [CrossRef]
- Batistela, V.R.; Pellosi, D.S.; de Souza, F.D.; da Costa, W.F.; de Oliveira Santin, S.M.; de Souza, V.R.; Caetano, W.; Moisés de Oliveir, H.P.; Scarminio, I.S.; Hioka, N. pKa determinations of xanthene derivates in aqueous solutions by multivariate analysis applied to UV–Vis spectrophotometric data. Spectrochim. Acta 2011, 79, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Urano, Y.; Setsukinai, K.; Takakusa, H.; Kojima, H.; Kikuchi, K.; Ohkubo, K.; Fukuzumi, S.; Nagano, T. Rational principles for modulating fluorescence properties of fluorescein. J. Am. Chem. Soc. 2004, 126, 14079–14085. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, I.M.; Vlasova, A.A.; Kuleshova, A.A.; Gordeyeva, Y.A.; Saletskiy, A.M. Constants of the Formation of Complexes with Nanomarkers of the Fluorescein Family and Bovine Serum Albumin in Aqueous Solutions. Russ. J. Phys. Chem. A 2020, 94, 152–157. [Google Scholar] [CrossRef]
- Ishchenko, A.A.; Shapovalov, S.A. Heterogeneous Association of the Ions of Dyes in Solutions. J. Appl. Spectrosc. 2004, 71, 605–629. [Google Scholar] [CrossRef]
- Demartis, S.; Obinu, A.; Gavini, E.; Giunchedi, P.; Rassu, G. Nanotechnology-based rose Bengal: A broad-spectrum biomedical tool. Dye. Pigment. 2021, 188, AN 109236. [Google Scholar] [CrossRef]
- Alexandrino, F.J.R.; Bezerra, E.M.; Da Costa, R.F.; Cavalcante, L.R.L.; Sales, F.A.M.; Francisco, T.S.; Rodrigues, L.K.A.; de Brito, D.H.A.; Ricardo, N.M.P.S.; Costa, S.N.; et al. Rose Bengal incorporated to alpha-cyclodextrin microparticles for photodynamic therapy against the cariogenic microorganism Streptococcus mutans. Photodiagnosis Photodyn. Ther. 2019, 25, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Deshagani, S.; Ghosal, P.; Deepa, M. Redox active and electrically conducting cobalt telluride Nanorods/Poly(1-aminoanthraquinone) composite and photoactive Rose Bengal dye based photo-supercapacitor. Appl. Mater. Today 2020, 19, 100592–100604. [Google Scholar] [CrossRef]
- Midya, A.; Ghosh, R.; Santra, S.; Ray, S.K.; Guha, P.K. Reduced graphene oxide-rose bengal hybrid film for improved ammonia detection with low humidity interference at room temperature. Mater. Res. Express 2016, 3, AN 025101. [Google Scholar] [CrossRef]
- Linden, S.M.; Neckers, D.C. Bleaching studies of rose-bengal onium salts. J. Am. Chem. Soc. 1988, 110, 1257–1260. [Google Scholar] [CrossRef]
- Andrieux, F.P.L.; Boxall, C. Electrochemical studies of Rose Bengal using the electrochemical quartz crystal microbalance. ECS Trans. 2007, 6, 67–77. [Google Scholar] [CrossRef]
- Fini, P.; Longobardi, F.; Catucci, L.; Cosma, P.; Agostiano, A. Spectroscopic and electrochemical study of Rose Bengal in aqueous solutions of cyclodextrins. Bioelectrochemistry 2004, 63, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Fini, P.; Loseto, R.; Catucci, L.; Cosma, P.; Agostiano, A. Study on the aggregation and electrochemical properties of Rose Bengal in aqueous solution of cyclodextrins. Bielectrochemistry 2007, 70, 44–49. [Google Scholar] [CrossRef]
- Konev, D.V.; Lizgina, K.V.; Istakova, O.I.; Baulin, V.E.; Kalashnikova, I.P.; Devillers, C.H.; Vorotyntsev, M.A. Electropolymerization of magnesium 5, 15-di (N-methoxyphenyl) porphine. Russ. J. Electrochem. 2016, 52, 1150–1158. [Google Scholar] [CrossRef]
- Kurmaz, S.V.; Gak, V.Y.; Kurmaz, V.A.; Konev, D.V. Preparation and Properties of Hybrid Nanostructures of Zinc Tetraphenylporphyrinate and an Amphiphilic Copolymer of N-Vinylpyrrolidone in a Neutral Aqueous Buffer Solution. Russ. J. Phys. Chem. A 2018, 92, 329–333. [Google Scholar] [CrossRef]
- Kurmaz, S.V.; Konev, D.V.; Sen’, V.D.; Kurmaz, V.A.; Kulikov, A.V. Preparation and characterization of stable water soluble hybrid nanostructures of hydrophobic compounds by encapsulation into nanoparticles of amphiphilic N-vinylpyrrolidone copolymers of new generation. In Proceedings of the IOP Conference Series: Materials Science and Engineering, Moscow, Russia, 30 October–1 November 2019. [Google Scholar] [CrossRef]
- Kurmaz, S.V.; Fadeeva, N.V.; Fedorov, B.S.; Kozub, G.I.; Emel’yanova, N.S.; Kurmaz, V.A.; Manzhos, R.A.; Balakina, A.A.; Terentyev, A.A. New antitumor hybrid materials based on PtIV organic complex and polymer nanoparticles consisting of N-vinylpyrrolidone and (di)methacrylates. Mendeleev Commun. 2020, 30, 22–24. [Google Scholar] [CrossRef]
- Kurmaz, S.V.; Fadeeva, N.V.; Fedorov, B.S.; Kozub, G.I.; Kurmaz, V.A.; Ignat’ev, V.M.; Emel’yanova, N.S. Amphiphilic copolymers of N-vinylpyrrolidone with (di)methacrylates as promising carriers for the platinum(IV) complex with antitumor activity. Russ. Chem. Bull. 2021, 70, 1832–1844. [Google Scholar] [CrossRef]
- Maréchal, Y. The Hydrogen Bond and the Water Molecule; Elsevier Science: Amsterdam, The Netherlands; Boston, MA, USA, 2007. [Google Scholar]
- Koch, U.; Popelier, P.L.A. Conformational dependence of atomic multipole moments. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | Composition of Copolymer, mol.% | Mw × 10−3 | p | Tg, °C | Rh *, nm |
|---|---|---|---|---|---|
| br-VP-TEGDM * | 80.5:12.4:7.1 | 20.0 | 1.8 | 63.0 | 4.0 |
| br-VP-PEGMMA- TEGDM | 86.5:9.9:3.6 | 24.0 ** | - | 69.9 | 3.0 |
| Sample, NN | Monomer Mixture | Composition of Mixture, wt.% | Polymer Additive, 20 wt.% | Polymer Composite | Polymer Matrix |
|---|---|---|---|---|---|
| 1 | VP-TEGDM | 40:0:60 | br-VP-TEGDM | VP-TEGDM | VP-TEGDM |
| 2 | |||||
| 3 | VP-PEGMMA-TEGDM | 40:10:50 | br-VP-PEGMMA-TEGDM | VP-PEGMMA-TEGDM | VP-PEGMMA-TEGDM |
| 4 | |||||
| 5 | 40:20:40 |
| Polymer Matrices | Composition of Monomer-Polymer Mixtures, wt% | Extraction Time, h | Ssp, m2/g | Vp, cm3/g |
|---|---|---|---|---|
| VP-TEGDM * | (40:60):20 | 7.0 | 11.0 | 0.026 |
| 14.0 | 22.0 | 0.078 | ||
| VP-PEGMMA- TEGDM ** | (40:10:50):20 | 7.0 | 5.0 | 0.057 |
| 3.5 | 4.5 | 0.016 | ||
| (40:20:40):20 | 7.0 | 2.0 | 0.011 |
| Complexes | ρ(r), a.u. | 2 ρ(r), a.u. | Eb, kcal/mol | ΔHf, kcal/mol | ΔHf, kcal/mol (Excluding Energy of the Zero Vibrations) |
|---|---|---|---|---|---|
| bvpa1 | +0.009799 | +0.048321 | −2.12 | 0.072 | −1253 |
| bvpa2 | +0.008217 | +0.039760 | −1.63 | 0.201 | −1.105 |
| bvpb1 | +0.086677 | +0.175948 | −31.13 | −9.07 | −9.625 |
| bvpb2 | +0.011927 | +0.060106 | −2.82 | −0.604 | −1.835 |
| bvpb3 | +0.011781 | +0.052662 | −2.57 | −0.683 | −1.987 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurmaz, S.V.; Fadeeva, N.V.; Gorshkova, A.I.; Kurochkin, S.A.; Knerelman, E.I.; Davydova, G.I.; Torbov, V.I.; Dremova, N.N.; Konev, D.V.; Kurmaz, V.A.; et al. Mesoporous Networks of N-Vinylpyrrolidone with (di)Methacrylates as Precursors of Ecological Molecular Imprinted Polymers. Materials 2021, 14, 6757. https://doi.org/10.3390/ma14226757
Kurmaz SV, Fadeeva NV, Gorshkova AI, Kurochkin SA, Knerelman EI, Davydova GI, Torbov VI, Dremova NN, Konev DV, Kurmaz VA, et al. Mesoporous Networks of N-Vinylpyrrolidone with (di)Methacrylates as Precursors of Ecological Molecular Imprinted Polymers. Materials. 2021; 14(22):6757. https://doi.org/10.3390/ma14226757
Chicago/Turabian StyleKurmaz, Svetlana V., Natalia V. Fadeeva, Anna I. Gorshkova, Sergey A. Kurochkin, Eugenia I. Knerelman, Galina I. Davydova, Vladimir I. Torbov, Nadezhda N. Dremova, Dmitry V. Konev, Vladimir A. Kurmaz, and et al. 2021. "Mesoporous Networks of N-Vinylpyrrolidone with (di)Methacrylates as Precursors of Ecological Molecular Imprinted Polymers" Materials 14, no. 22: 6757. https://doi.org/10.3390/ma14226757
APA StyleKurmaz, S. V., Fadeeva, N. V., Gorshkova, A. I., Kurochkin, S. A., Knerelman, E. I., Davydova, G. I., Torbov, V. I., Dremova, N. N., Konev, D. V., Kurmaz, V. A., Ignatiev, V. M., & Emelyanova, N. S. (2021). Mesoporous Networks of N-Vinylpyrrolidone with (di)Methacrylates as Precursors of Ecological Molecular Imprinted Polymers. Materials, 14(22), 6757. https://doi.org/10.3390/ma14226757