
Calcium Phosphate-Coated Lipid Nanoparticles as a Potential Tool in Bone Diseases Therapy

 ,
,  ,
,  ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of NPs and Screening of Charging Agents
2.3. Purification of NPs
2.4. Characterization of NPs
2.4.1. Particle Size and Zeta Potential Measurements
2.4.2. SEM and FESEM Characterization
2.4.3. TEM Characterization
2.5. CaP Coating
- Method A
- Method B
2.6. Coating Evaluation
2.6.1. Particle Size and Zeta Potential Measurements
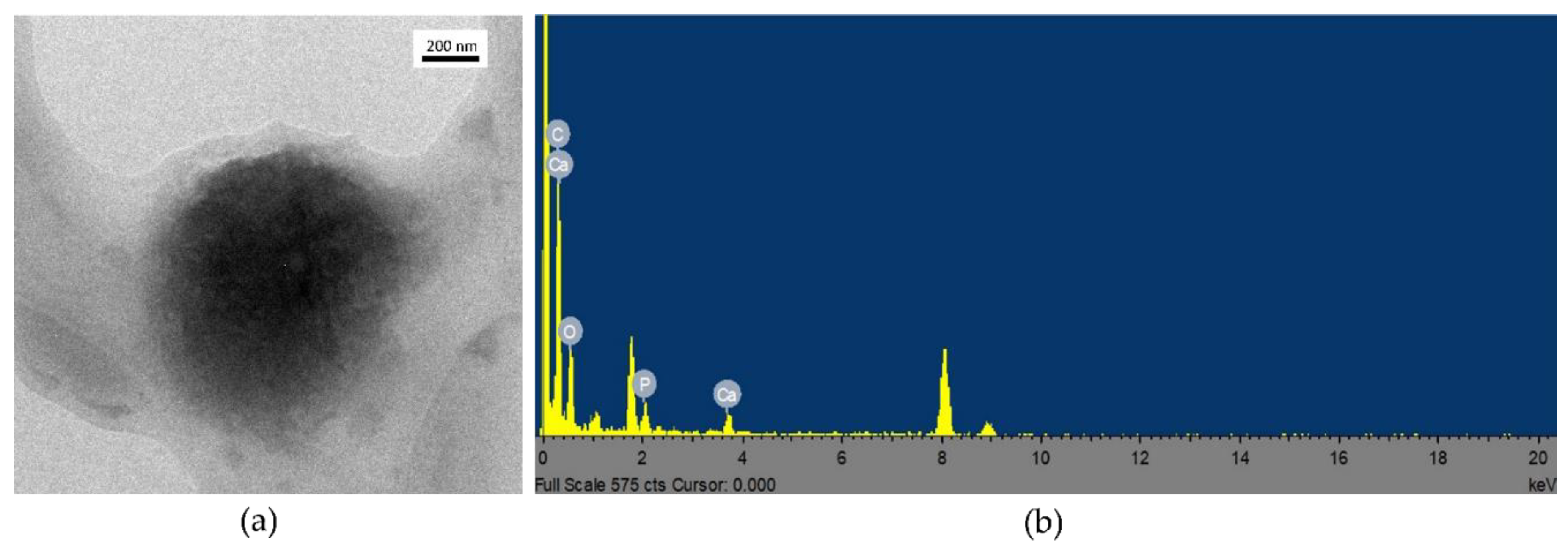
2.6.2. SEM-EDS and TEM-EDS Analyses
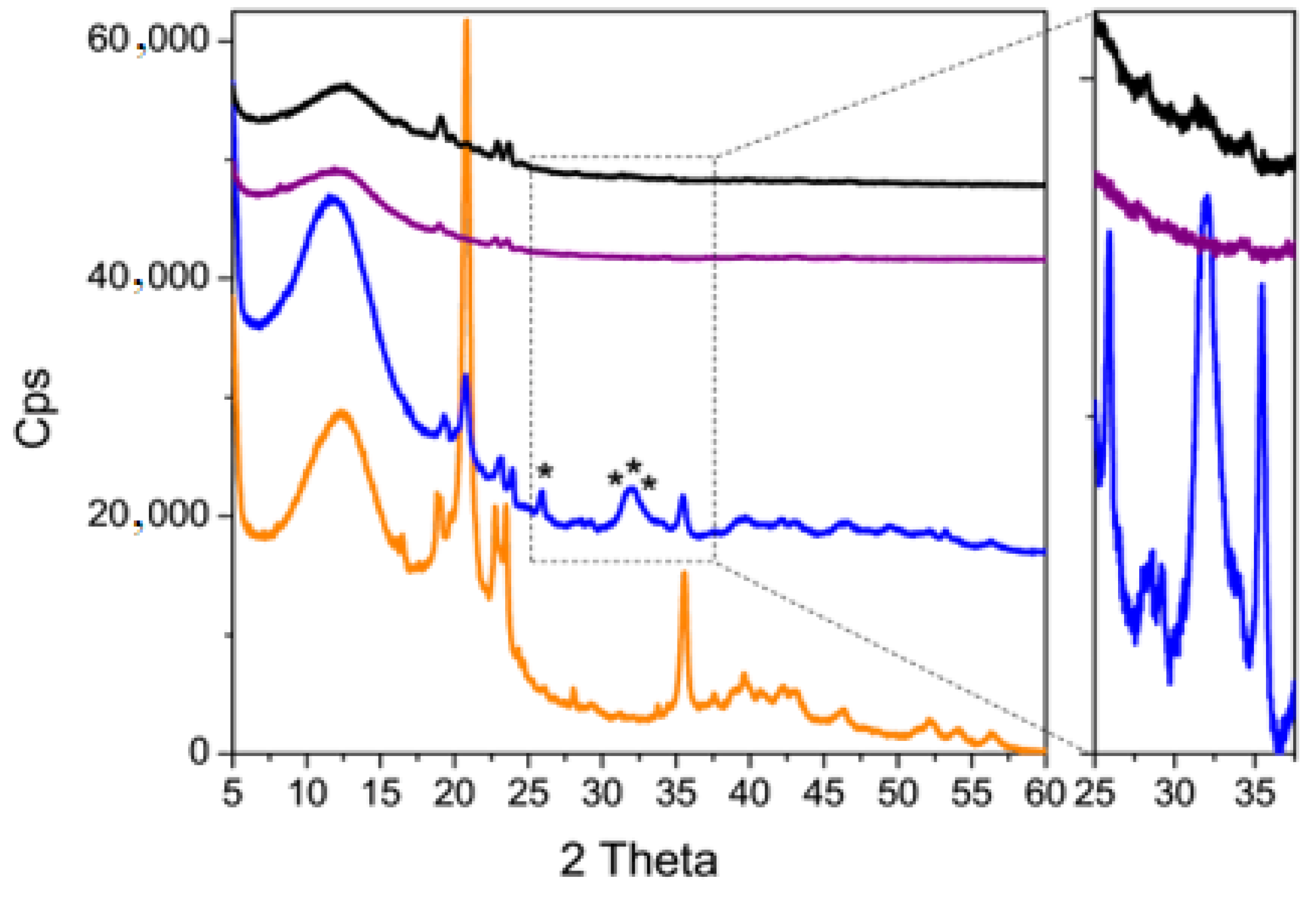
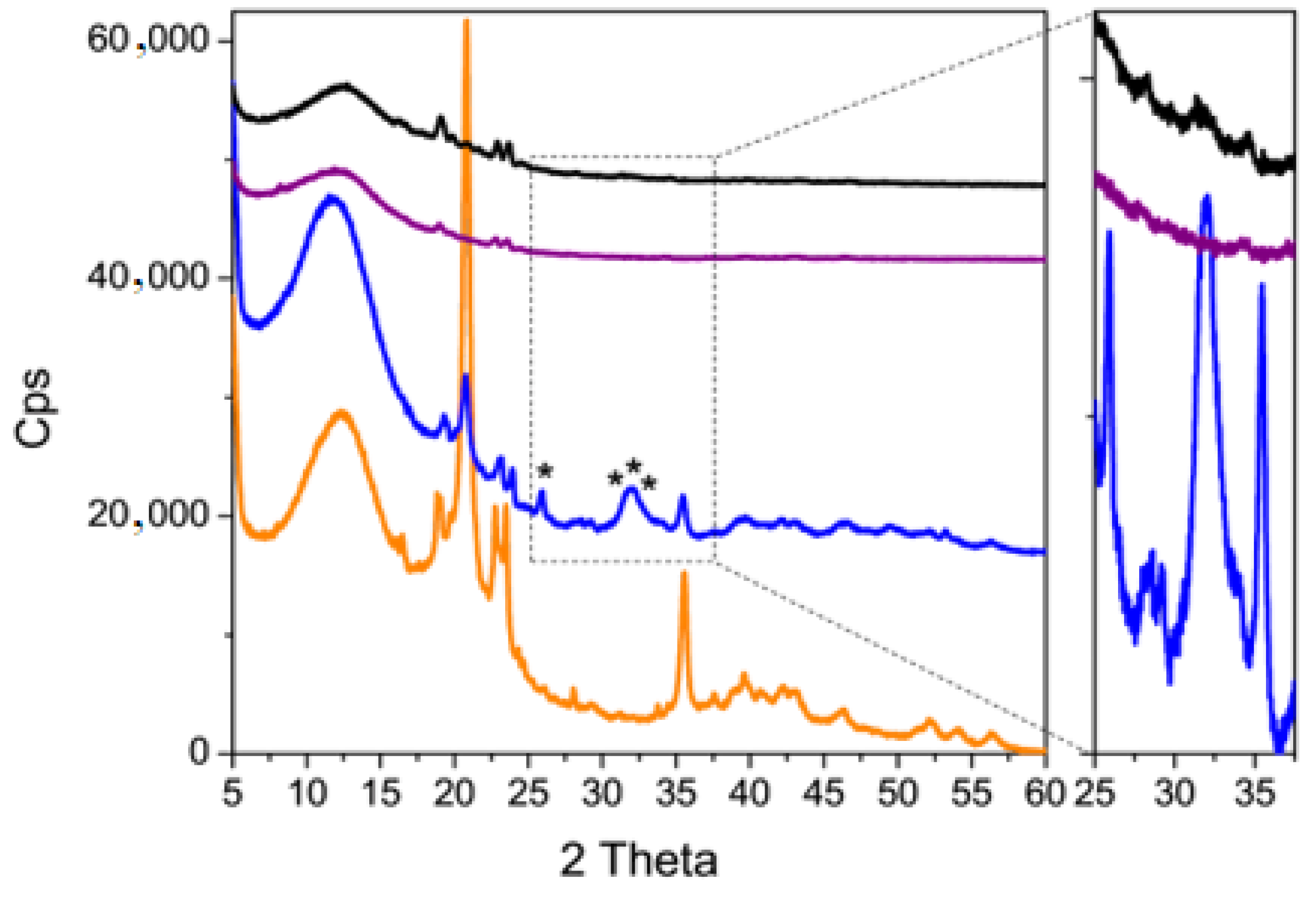
2.6.3. X-ray Diffraction
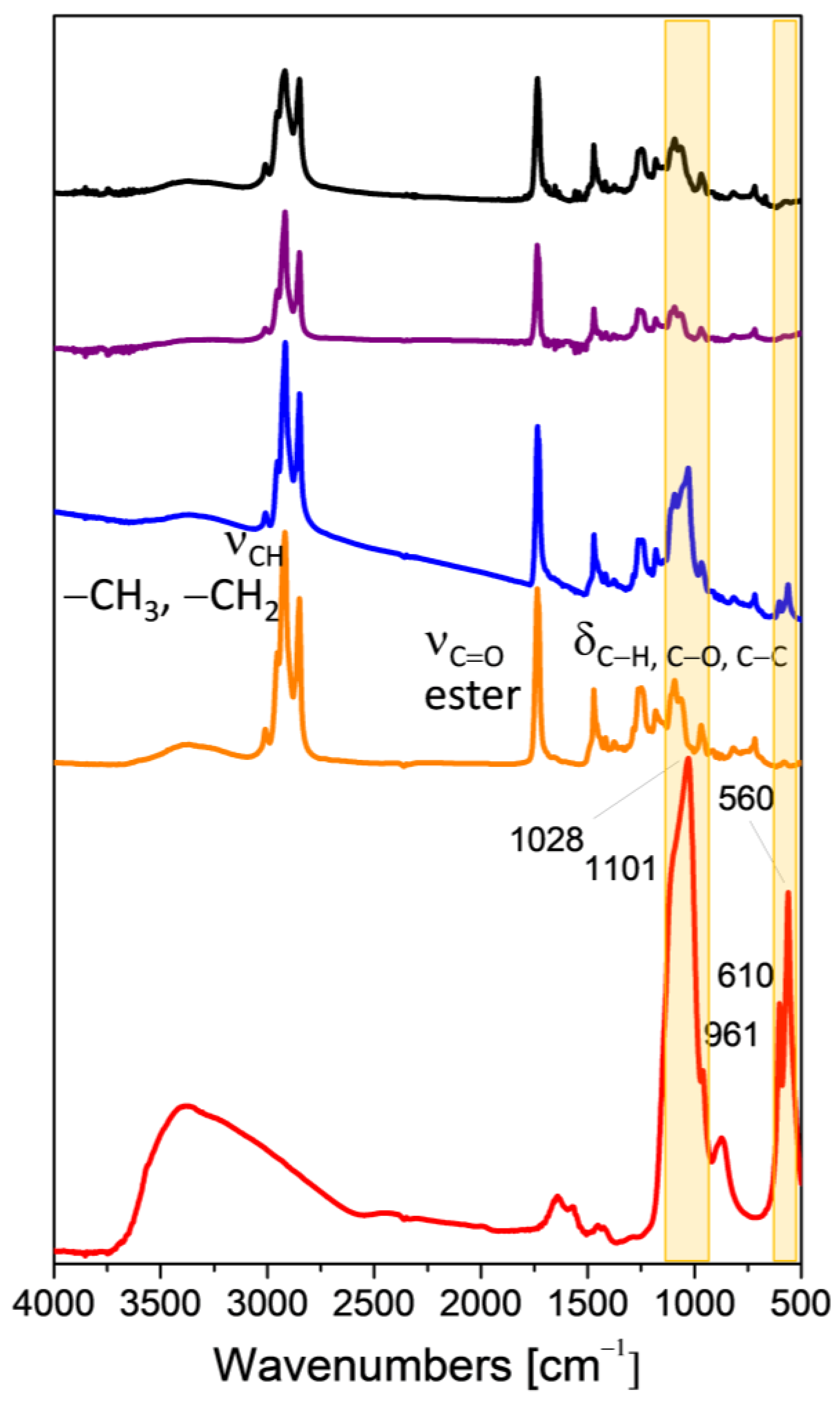
2.6.4. FTIR Spectroscopy
2.7. Stability Studies
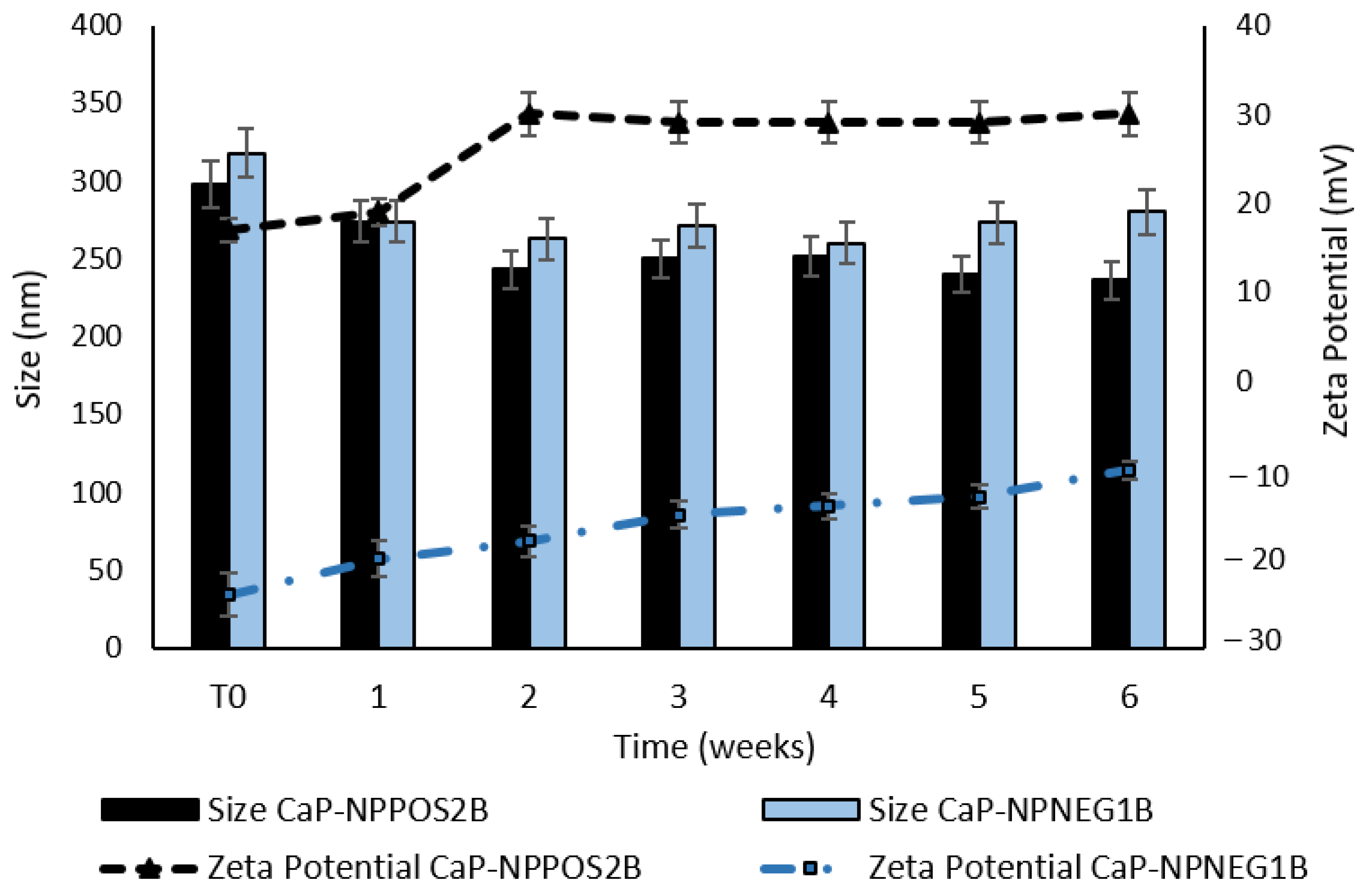
2.7.1. Size and Zeta Potential
2.7.2. Freeze-Drying/Re-Suspension of NPs
2.8. Incorporation of Sudan Red III and Determination of Entrapment Efficiency
2.9. In Vitro Cell Studies
2.9.1. Cell Culture
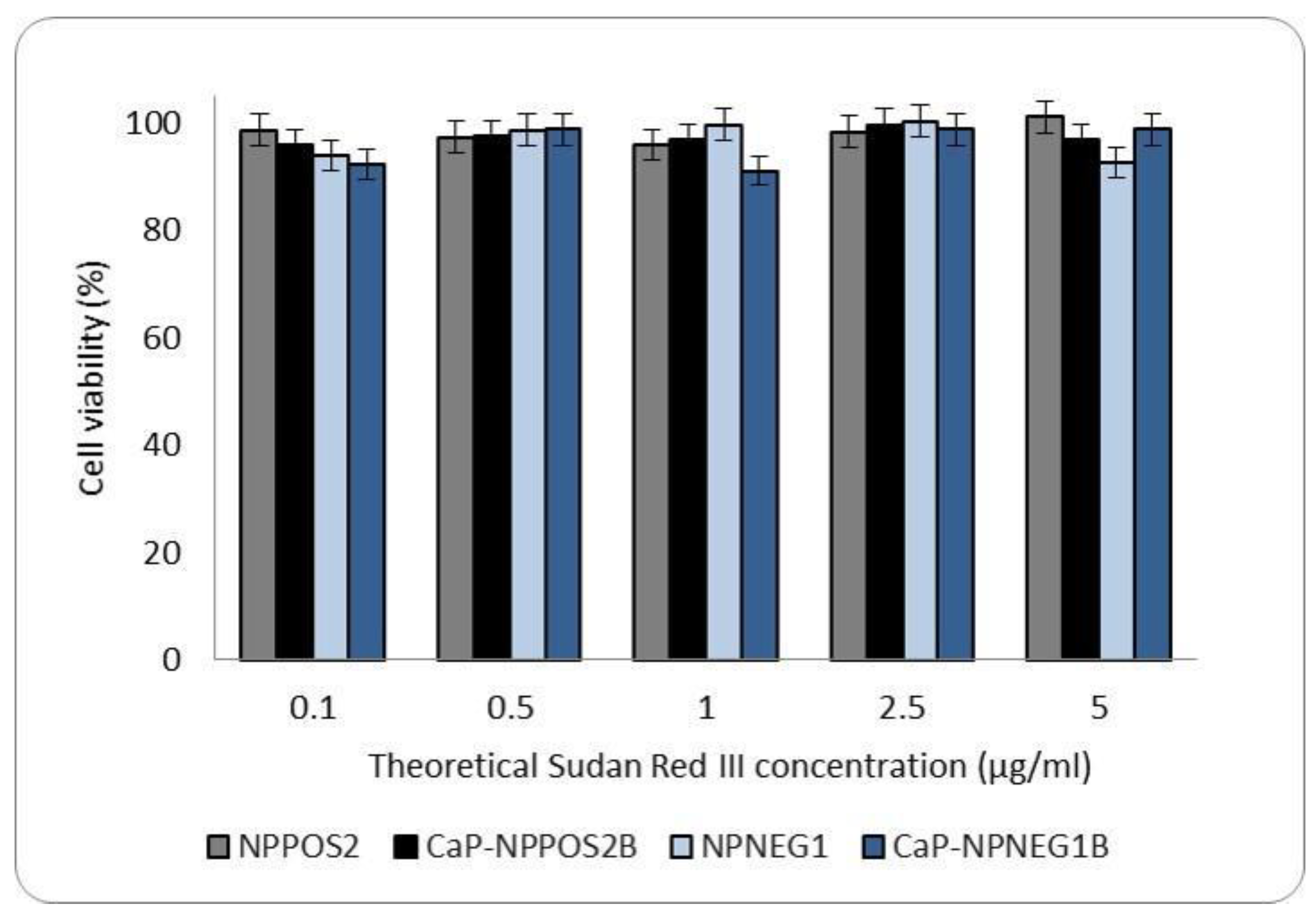
2.9.2. Cytotoxicity
2.9.3. Sudan Red III Cellular Uptake
2.9.4. Statistical Analysis
3. Results and Discussion
3.1. Preparation of NPs and Screening of Charging Agents
3.2. Characterization of NPs
3.2.1. Particle Size and Zeta Potential Measurements
3.2.2. SEM Characterization
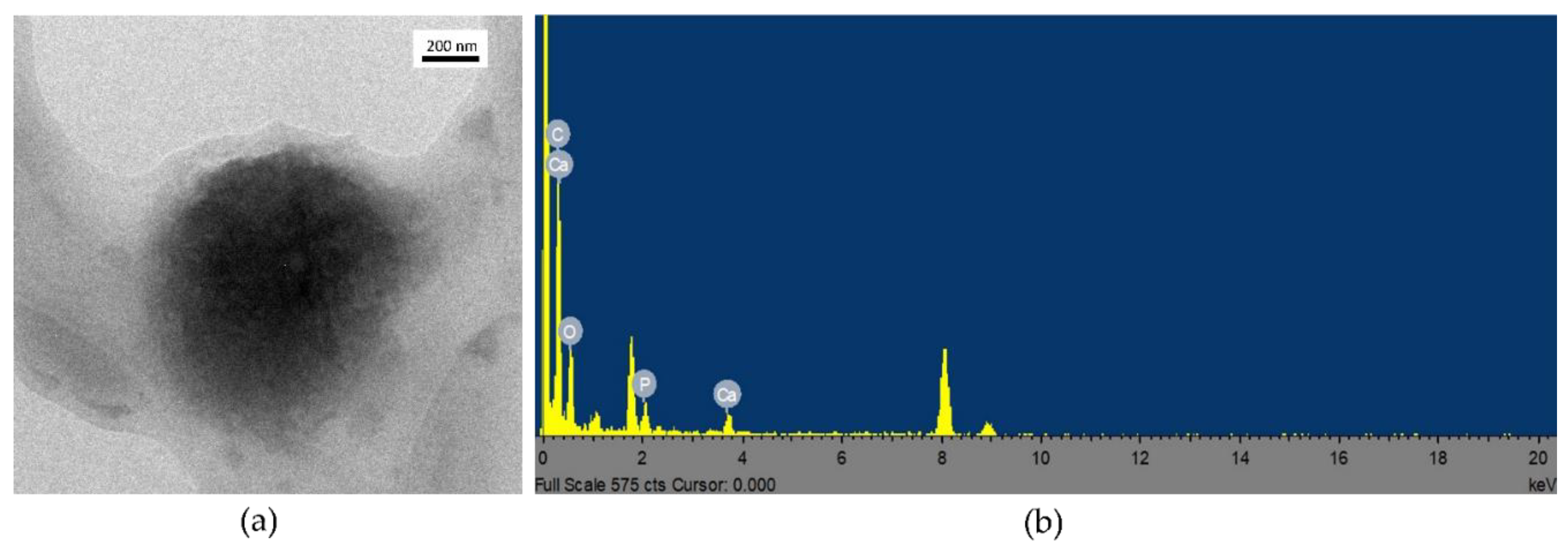
3.3. Coating Evaluation
3.4. Stability Studies
3.5. Incorporation of Sudan Red III and Determination of Entrapment Efficiency
3.6. In Vitro Cell Studies
3.6.1. Cytotoxicity
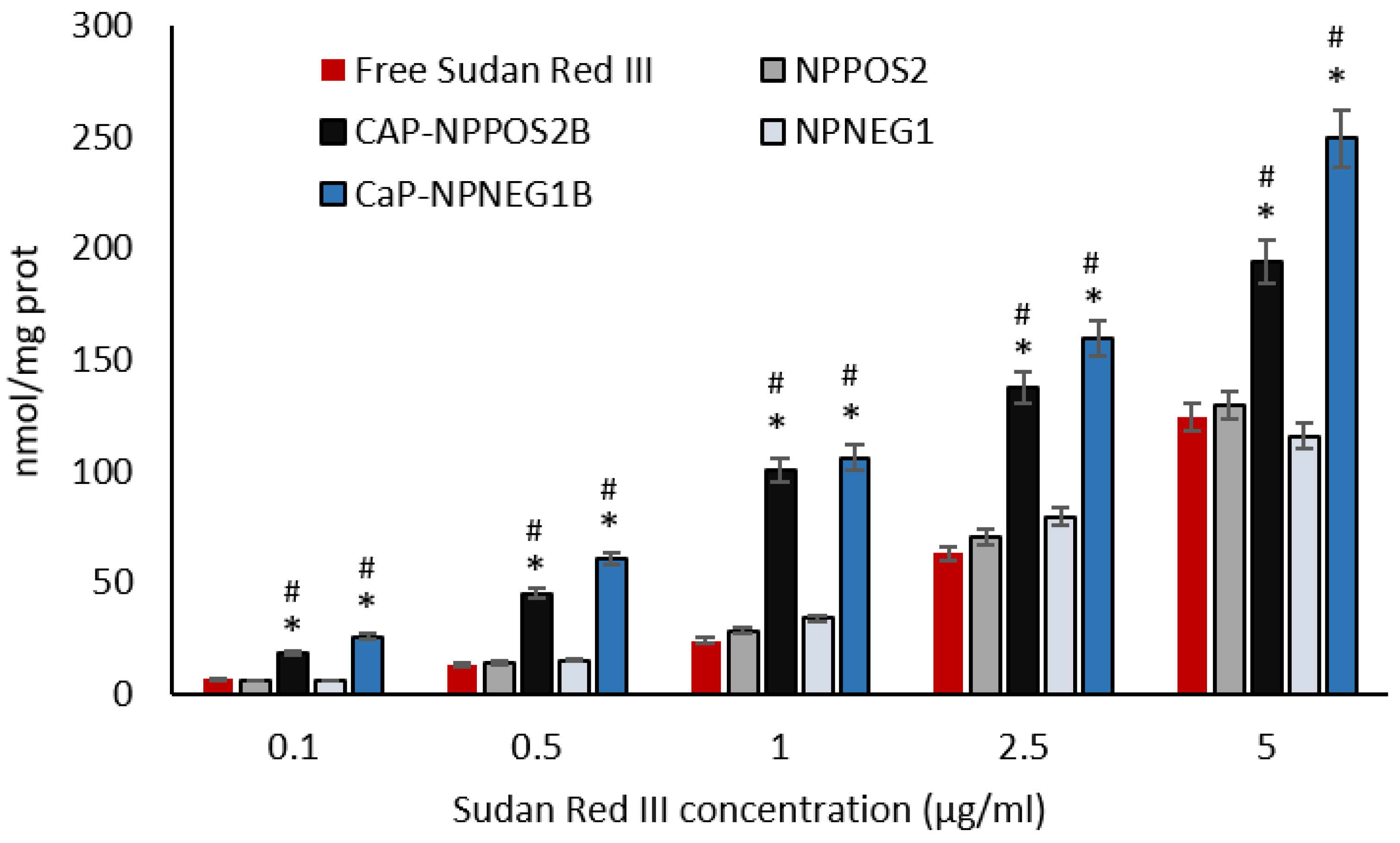
3.6.2. Sudan Red III Cellular Uptake
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eliaz, N.; Metoki, N. Calcium phosphate bioceramics: A review of their history, structure, properties, coating technologies and biomedical applications. Materials 2017, 10, 334. [Google Scholar] [CrossRef] [Green Version]
- Ginebra, M.P.; Canal, C.; Espanol, M.; Pastorino, D.; Montufar, E.B. Calcium phosphate cements as drug delivery materials. Adv. Drug Deliv. Rev. 2012, 64, 1090–1110. [Google Scholar] [CrossRef] [PubMed]
- Verron, E.; Khairoun, I.; Guicheux, J.; Bouler, J.-M. Calcium phosphate biomaterials as bone drug delivery systems: A review. Drug Discov. Today 2010, 15, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhao, L.; Liu, J.; Weir, M.D.; Zhou, X.; Xu, H.H.K. Bone tissue engineering via nanostructured calcium phosphate biomaterials and stem cells. Bone Res. 2014, 2, 14017–14031. [Google Scholar] [CrossRef] [PubMed]
- Surmenev, R.A.; Surmeneva, M.A.; Ivanova, A.A. Significance of calcium phosphate coatings for the enhancement of new bone osteogenesis—A review. Acta Biomater. 2014, 10, 557–579. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Wu, C.; Chen, J.; Xiao, Y. Nanotechnology in the targeted drug delivery for bone diseases and bone regeneration. Int. J. Nanomed. 2013, 8, 2305–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chindamo, G.; Sapino, S.; Peira, E.; Chirio, D.; Gonzalez, M.C.; Gallarate, M. Bone diseases: Current approach and future perspectives in drug delivery systems for bone targeted therapeutics. Nanomaterials 2020, 10, 875. [Google Scholar] [CrossRef]
- Bellido, T.; Plotkin, L.I. Novel actions of bisphosphonates in bone: Preservation of osteoblast and osteocyte viability. Bone 2011, 49, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, X.; Deng, T.; Yao, P.; Song, H.; Zhou, S.; Yan, W. Development of drug loaded nanoparticles binding to hydroxyapatite based on a bisphosphonate modified nonionic surfactant. J. Nanomater. 2015, 16, 145. [Google Scholar] [CrossRef]
- Wang, G.; Mostafa, N.Z.; Incani, V.; Kucharski, C.; Uludaĝ, H. Bisphosphonate-decorated lipid nanoparticles designed as drug carriers for bone diseases. J. Biomed. Mater. Res. Part A 2012, 100, 684–693. [Google Scholar] [CrossRef]
- Salamanna, F.; Giavaresi, G.; Parrilli, A.; Torricelli, P.; Boanini, E.; Bigi, A.; Fini, M. Antiresorptive properties of strontium substituted and alendronate functionalized hydroxyapatite nanocrystals in an ovariectomized rat spinal arthrodesis model. Mater. Sci. Eng. C 2019, 95, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.T.; Gray, B.L.; Wingert, P.A.; Ostafin, A.E. Assembly of aqueous-cored calcium phosphate nanoparticles for drug delivery. Chem. Mater. 2004, 16, 4942–4947. [Google Scholar] [CrossRef]
- Yeo, C.H.; Zein, S.H.S.; Ahmad, A.L.; McPhail, D.S. Comparison of DOPA and DPPA liposome templates for the synthesis of calcium phosphate nanoshells. Ceram. Int. 2012, 38, 561–570. [Google Scholar] [CrossRef]
- Rivero Berti, I.; Dell’ Arciprete, M.L.; Dittler, M.L.; Miñan, A.; Fernández Lorenzo de Mele, M.; Gonzalez, M. Delivery of fluorophores by calcium phosphate-coated nanoliposomes and interaction with Staphylococcus aureus biofilms. Colloids Surf. B Biointerfaces 2016, 142, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, L.; Gallarate, M. Lipid nanoparticles: State of the art, new preparation methods and challenges in drug delivery. Expert Opin. Drug Deliv. 2012, 9, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Harde, H.; Das, M.; Jain, S. Solid lipid nanoparticles: An oral bioavailability enhancer vehicle. Expert Opin. Drug Deliv. 2011, 8, 1407–1424. [Google Scholar] [CrossRef]
- Müller, R.H.; Radtke, M.; Wissing, S.A. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Proc. Adv. Drug Deliv. Rev. 2002, 4, S131–S155. [Google Scholar] [CrossRef]
- Wissing, S.A.; Kayser, O.; Müller, R.H. Solid lipid nanoparticles for parenteral drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 1257–1272. [Google Scholar] [CrossRef] [PubMed]
- Chirio, D.; Peira, E.; Sapino, S.; Dianzani, C.; Barge, A.; Muntoni, E.; Morel, S.; Gallarate, M. Stearoyl-chitosan coated nanoparticles obtained by microemulsion cold dilution technique. Int. J. Mol. Sci. 2018, 19, 3833. [Google Scholar] [CrossRef] [Green Version]
- Chirio, D.; Peira, E.; Dianzani, C.; Muntoni, E.; Gigliotti, C.L.; Ferrara, B.; Sapino, S.; Chindamo, G.; Gallarate, M. Development of solid lipid nanoparticles by cold dilution of microemulsions: Curcumin loading, preliminary in vitro studies, and biodistribution. Nanomaterials 2019, 9, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.-B.; Takagi, C.; Wu, C.-J.; Mizukami, H.; Ostafin, A. Synthesis of calcium phosphate controllable coating thickness on oil-in-water nanoemulsion with performance of oxygen release as oxygen carrier. J. Biomater. Nanobiotechnol. 2016, 7, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Müller, R.H.; Radtke, H.; Wissing, S.A. Nanostructured lipid matrices for improved microencapsulation of drugs. Int. J. Pharm. 2002, 42, 121–128. [Google Scholar] [CrossRef]
- Hou, D.Z.; Xie, C.S.; Huang, K.J.; Zhu, C.H. The production and characteristics of solid lipid nanoparticles (SLNs). Biomaterials 2003, 24, 1781–1785. [Google Scholar] [CrossRef]
- Ślósarczyk, A.; Paluszkiewicz, C.; Gawlicki, M.; Paszkiewicz, Z. The FTIR spectroscopy and QXRD studies of calcium phosphate based materials produced from the powder precursors with different Ca/P ratios. Ceram. Int. 1997, 23, 297–304. [Google Scholar] [CrossRef]
- Laurens, L.M.L.; Wolfrum, E.J. Feasibility of spectroscopic characterization of algal lipids: Chemometric correlation of NIR and FTIR Spectra with exogenous lipids in algal biomass. Bioenergy Res. 2011, 4, 22–35. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients (mg) | µEPOS1/ NPPOS1 | µEPOS2/ NPPOS2 | µENEG1/ NPNEG1 | µENEG2/ NPNEG2 | µENEG3/ NPNEG3 | µENEG4/ NPNEG4 | µENEG5/ NPNEG5 | ||
|---|---|---|---|---|---|---|---|---|---|
| NPs | µEs | Trilaurin | 60 | 60 | 60 | 60 | 60 | 60 | 60 |
| s-EA | 200 | 200 | 200 | 200 | 200 | 200 | 200 | ||
| Epikuron®200 | 150 | 150 | 170 | 150 | 150 | 150 | 150 | ||
| Cremophor®RH60 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | ||
| Propylene glycol | 200 | 200 | 100 | - | 200 | 200 | 200 | ||
| Benzyl alcohol | - | 40 | - | - | 20 | 20 | 20 | ||
| TC | 30 | - | - | 10 | - | 10 | 20 | ||
| Myristic acid | - | - | - | - | 10 | - | - | ||
| Stearic acid | - | - | - | - | - | 10 | - | ||
| Behenic acid | - | - | - | - | - | - | 10 | ||
| Stearylamine | 10 | 10 | - | - | - | - | - | ||
| s-Water | 700 | 700 | 700 | 700 | 700 | 700 | 700 | ||
| Dilution water (mL) | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Conditions | NPPOS2A-1 | NPPOS2A-2 | NPPOS2A-3 | NPPOS2A-4 | NPPOS2A-5 |
|---|---|---|---|---|---|
| 69 mM CaCl2 (mL) | 1 | 1 | 1 | 1 | 1 |
| 41 mM H3PO4 (mL) | 1 | 1 | 1 | 1 | 1 |
| NPPOS2 (mL) | 1 | 1 | 1 | 0.5 | 0.5 |
| Reaction time (min) | 120 | 180 | 240 | 180 | 240 |
| Samples | Mean Diameter (nm) ± S.E. (P.I) | Zeta Potential (mV) ± S.E. |
|---|---|---|
| NPPOS1 | 370.9 ± 5.3 (0.271) | +5.01 ± 1.91 |
| NPPOS2 | 205.6 ± 3.9 (0.199) | +34.03 ± 3.90 |
| NPNEG1 | 250.5 ± 0.5 (0.235) | −23.54 ± 1.79 |
| NPNEG2 | 252.1 ± 4.5 (0.182) | −15.63 ± 2.42 |
| NPNEG3 | 538.4 ± 33.0 (0.259) | −11.94 ± 2.47 |
| NPNEG4 | 511.0 ± 12.3 (0.190) | −12.07 ± 2.00 |
| NPNEG5 | 524.7 ± 4.2 (0.258) | −12.98 ± 0.95 |
| Samples | Mean Diameter (nm) ± S.E. (P.I) | Zeta Potential (mV) ± S.E. |
|---|---|---|
| NPPOS2 | 205.6 ± 3.9 (0.199) | +34.03 ± 3.90 |
| CaP-NPPOS2A-1 | 481.7 ± 9.8 (0.308) | +25.90 ± 1.24 |
| CaP-NPPOS2A-2 | 488.5 ± 15.2 (0.338) | +26.44 ± 1.35 |
| CaP-NPPOS2A-3 | 502.8 ± 14.2 (0.299) | +21.28 ± 3.55 |
| CaP-NPPOS2A-4 | 445.4 ± 25.0 (0.387) | +25.30 ± 2.02 |
| CaP-NPPOS2A-5 | 455.9 ± 7.5 (0.372) | +24.03 ± 2.88 |
| Samples | Mean Diameter (nm) ± S.E. (P.I) | Zeta Potential (mV) ± S.E. |
|---|---|---|
| NPPOS2 | 205.6 ± 3.9 (0.199) | +34.03 ± 3.90 |
| CaP-NPPOS2B | 298.7 ± 10.3 (0.284) | +17.61 ± 1.72 |
| NPNEG1 | 250.5 ± 0.5 (0.235) | −23.54 ± 1.79 |
| CaP-NPNEG1B | 318.2 ± 11.6 (0.278) | −23.43 ± 1.82 |
| Samples | Mean Diameter (nm) ± S.E. | Zeta Potential (mV) ± S.E. | |
|---|---|---|---|
| CaP-NPPOS2B | fresh | 298.7 ± 10.3 | +17.61 ± 1.72 |
| CaP-NPPOS2B | freeze-dried | 1040 ± 20.3 | +20.15 ± 2.7 |
| CaP-NPPOS2B + trehalose | freeze-dried | 320.3 ± 9.5 | +21.37 ± 2.5 |
| CaP-NPPOS2B + glucose | freeze-dried | 305.5 ± 8.9 | +19.56 ± 3.19 |
| CaP-NPNEG1B | fresh | 318.2 ± 11.6 | −23.43 ± 1.82 |
| CaP-NPNEG1B | freeze-dried | 1011.8 ± 97.9 | −19.47 ± 2.38 |
| CaP-NPNEG1B + trehalose | freeze-dried | 343.5 ± 10.0 | −20.42 ± 2.16 |
| CaP-NPNEG1B + glucose | freeze-dried | 322.0 ± 16.8 | −24.19 ± 1.77 |
| Samples | %EE |
|---|---|
| NPPOS2 | 91.2 ± 3.5 |
| CaP-NPPOS2B | 75.6 ± 2.7 |
| NPNEG1 | 97.4 ± 3.3 |
| CaP-NPNEG1B | 77.5 ± 2.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sapino, S.; Chindamo, G.; Chirio, D.; Manzoli, M.; Peira, E.; Riganti, C.; Gallarate, M. Calcium Phosphate-Coated Lipid Nanoparticles as a Potential Tool in Bone Diseases Therapy. Nanomaterials 2021, 11, 2983. https://doi.org/10.3390/nano11112983
Sapino S, Chindamo G, Chirio D, Manzoli M, Peira E, Riganti C, Gallarate M. Calcium Phosphate-Coated Lipid Nanoparticles as a Potential Tool in Bone Diseases Therapy. Nanomaterials. 2021; 11(11):2983. https://doi.org/10.3390/nano11112983
Chicago/Turabian StyleSapino, Simona, Giulia Chindamo, Daniela Chirio, Maela Manzoli, Elena Peira, Chiara Riganti, and Marina Gallarate. 2021. "Calcium Phosphate-Coated Lipid Nanoparticles as a Potential Tool in Bone Diseases Therapy" Nanomaterials 11, no. 11: 2983. https://doi.org/10.3390/nano11112983
APA StyleSapino, S., Chindamo, G., Chirio, D., Manzoli, M., Peira, E., Riganti, C., & Gallarate, M. (2021). Calcium Phosphate-Coated Lipid Nanoparticles as a Potential Tool in Bone Diseases Therapy. Nanomaterials, 11(11), 2983. https://doi.org/10.3390/nano11112983