Abstract
Mitochondrial dysfunction including deficits of mitophagy is seen in aging and neurodegenerative disorders including Alzheimer’s disease (AD). Apart from traditionally targeting amyloid beta (Aβ), the main culprit in AD brains, other approaches include investigating impaired mitochondrial pathways for potential therapeutic benefits against AD. Thus, a future therapy for AD may focus on novel candidates that enhance optimal mitochondrial integrity and turnover. Bioactive food components, known as nutraceuticals, may serve as such agents to combat AD. Urolithin A is an intestinal microbe-derived metabolite of a class of polyphenols, ellagitannins (ETs). Urolithin A is known to exert many health benefits. Its antioxidant, anti-inflammatory, anti-atherogenic, anti-Aβ, and pro-mitophagy properties are increasingly recognized. However, the underlying mechanisms of urolithin A in inducing mitophagy is poorly understood. This review discusses the mitophagy deficits in AD and examines potential molecular mechanisms of its activation. Moreover, the current knowledge of urolithin A is discussed, focusing on its neuroprotective properties and its potential to induce mitophagy. Specifically, this review proposes potential mechanisms by which urolithin A may activate and promote mitophagy.
1. Introduction
Ageing is the time-dependent deterioration of cellular metabolic functions which has been characterized by several hallmarks. These include genomic instability, attrition in telomeres, loss of proteostasis, deregulated nutrient sensing, altered intercellular communication, cellular senescence, stem cell exhaustion, epigenetic alterations and mitochondrial dysfunction [1]. Ample evidence exists for a role for mitochondrial dysfunction in ageing [2,3,4,5]. In ageing and under stressful conditions, neurons exhibit an increase in oxidative phosphorylation (OXPHOS) by elevating glycolysis in astrocytes to meet the increased neuronal demand for lactate [6,7]. The increased OXPHOS on the other hand, results in an over- production of reactive oxygen species (ROS), namely superoxide (O2−) and hydrogen peroxide (H2O2). As per the mitochondrial free radical theory of ageing (MFRTA) proposed by Harman [8,9], aging is associated with oxidative stress exerted by high levels of ROS [1,10,11]. Mitochondrial nucleoids (mtDNA) located near the inner mitochondrial membrane (IMM), the sites of OXPHOS, are highly vulnerable to ROS-induced damage. Additionally, neurons, glial cells and microvessels in the brain transiently produce nitric oxide (NO) by activating various nitric oxide synthases (NOS) [12,13]. The reaction of NO with O2− produces various reactive nitrogen species (RNS) including peroxynitrites (ONOO-). Oxidative stress occurs when the production of ROS overwhelms the antioxidant defences.
The protection afforded by the brain’s endogenous antioxidant defences are relatively low compared to other vital organs [14,15]. This makes the brain specifically susceptible to oxidative damage, since it also possesses high levels of fatty acids that readily oxidize oxidized transition metal species (Fe3+, Cu2+, Zn2+), and high mitochondrial activity to sustain its comparatively large energy demand [16]. In fact, accumulating mitochondrial bioenergetic abnormalities and mitochondrial damage in neurons may be central to many ROS-induced cellular pathological events in senescence and neurodegeneration, including the initiation and progression of Alzheimer’s disease (AD).
2. Alzheimer’s Disease
Alzheimer’s disease is the most common form of dementia that constitutes approximately 50–75% of clinically diagnosed dementia cases, worldwide [17]. This progressive neurodegenerative disorder is identified as the second major cause of death in Australia [18]. Furthermore, Asian [19] and Asia Pacific countries [17], were reported to have a tremendous increase in AD prevalence. It has been estimated that an 80-year-old has a 30% probability of getting the disease [20]. This higher prevalence rates and the universality of the disease may be dependent on various common conditions that impose a risk towards AD for which old age, female sex, and Apolipoprotein E allele status (APOE ε4) play key roles [21]. Lifestyle factors such as diet, head injury and smoking have also been shown to increase the risk of AD [22].
The key neuropathological features of AD are the accumulation of amyloid beta (Aβ) in extracellular senile plaques and intracellular neurofibrillary tangles made up of hyper-phosphorylated tau protein [23]. The final stage of AD brains also features cerebral atrophy [24]. Having no definite treatment to date, the major obstacle of AD diagnosis is that by the time clinical symptoms of cognitive deficits appear, the brain damage that has occurred is irreversible. Early diagnosis of the disease is critical, as there is a long prodromal/preclinical phase during which neuropathological changes are observed in the brain without clinical presentation [25,26]. It has been reported by Villemagne et al. (2013) that in AD patients, amyloid deposition in the brain occurs approximately two decades before the onset of clinical symptoms [27].
Oxidative stress accompanied by mitochondrial dysfunction is a characteristic feature in AD brains and is thought to be an early feature of the disease [28,29,30]. Amyloid beta directly enters mitochondria and disturbs mitochondrial function. Additionally, there is evidence that mitochondrial dysfunction arising from mitochondrion-derived ROS itself leads to aberrant Aβ production [31]. Therefore, coordinated mitochondrial biogenesis and elimination of dysfunctional mitochondria has the potential to play a pivotal role against AD.
3. Mitophagy Deficits in Alzheimer’s Disease
In living cells, selective degradation of defective mitochondria occurs in a pathway known as mitophagy. Mitophagy is a major mitochondrial quality control mechanism that ensures an efficient turnover to maintain a healthy population of mitochondria in cells. There are three major types of mitophagy: basal mitophagy, PINK1/Parkin-mediated mitophagy and mitochondrial vesicle-derived mitophagy [32]. Briefly, with the aid of various autophagy adaptor proteins, such as P62, Neighbor of BRCA1 gene 1 protein 1 (NBR1), Nuclear dot 10 protein 52 (NDP52/CALCOCO2), and Optineurin (OPTN), mitophagosomes are formed by damaged mitochondria, which are enclosed in double-membranes. These mitophagosomes mature and fuse with lysosomes, leading to enzymatic degradation of the contents and release of recycled materials into the cytosol. However, the efficiency of these mitophagy pathways declines with age [33]. Furthermore, impairment of mitophagy is increasingly recognized as a characteristic feature in AD brains [29].
It has been shown in mAPP mice that a lack of PINK1 promoted Aβ accumulation [34]. On the other hand, it has been reported that N-terminal truncated Tau induces aberrant Parkin recruitment, thus leading to excessive mitophagy and contributing to synaptic failure [35]. Previous studies have reported prominent autophagic accumulations of mitochondria in AD-affected brains [36,37,38,39]. This indicates that autophagic machinery is competent in AD neurons; however, the flux is impaired at the final stages of the turnover process, during the fusion of autophagosomes with lysosomes [40]. Moreover, in vitro studies have shown that hyper-phosphorylated Tau directly localizes and accumulates in mitochondria, leading to an increase in mitochondrial membrane potential and impaired PINK1/Parkin activation, resulting in deficits of mitophagy [41]. Parkin over-expression leads to an enhancement of the autophagic clearance of defective mitochondria [42].
Taken together, mitophagy deficits account for a significant part of the overall mitochondrial dysfunction as seen in AD. Therefore, inducing mitophagy confers neuroprotection against AD, as it ensures the maintenance of mitochondrial homeostasis [43]. Furthermore, an increasing number of review articles report that enhancing mitophagy holds promise as a novel therapeutic strategy for AD [44,45,46,47].
4. Enhancing Longevity: Molecular Pathways Leading to Mitophagy Activation
A build-up of high quantities of ROS in cells directly damage all biomolecules [48]. However, low levels of ROS give rise to an extended lifespan [49,50,51,52]. Whilst this initially seems counterintuitive, it is explained by a pathway termed mitohormesis [53]. Low levels of ROS activate Nuclear factor erythroid 2-related factor (Nrf2) in antioxidant response element (ARE)-mediated transcription of antioxidant proteins and phase I and II detoxification enzymes [54,55]. Moreover, the mitohormetic response promotes stress resistance, systemic antioxidant defence capabilities, and life span extension and is driven by a variety of cellular pathways and lifestyle interventions, which in turn activate the mitophagy process [55,56].
As shown in Figure 1, there are a variety of mitophagy modulatory pathways, namely, adenosine monophosphate (AMP)-dependent kinase (AMPK) signalling, mitochondrial unfolded protein response (UPRmit), Silent information regulator of transcription (Sirtuin) signalling and Mammalian targeting of rapamycin (mTOR). Moreover, caloric restriction and physical exercise are lifestyle interventions that contribute to the induction of mitophagy [57,58,59,60].
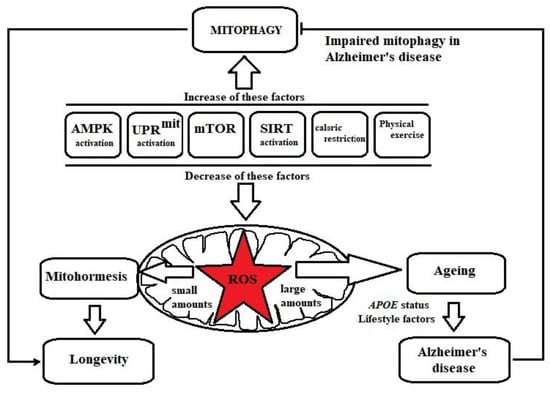
Figure 1.
Factors regulating mitohormesis and mitophagy. Adenosine monophosphate (AMP)-dependent kinase (AMPK) signalling, mitochondrial unfolded protein response (UPRmit), Silent information regulator of transcription (Sirtuin) signalling, inhibition of mammalian target of rapamycin (mTOR), caloric restriction and physical exercise induce low levels of ROS in mitochondria. ROS, at low levels, activate stress resistance mechanisms (mitohormesis), resulting in longevity. However, increased amounts of ROS in mitochondria are responsible for ageing, which is a major risk factor for AD and is influenced by APOE status and a variety of lifestyle factors. AMPK signalling, UPRmit, Sirtuin signalling, mTOR inhibition, caloric restriction and physical exercise also induce the mitophagy process, which is defective in AD.
AMPK is an energy sensor in mitochondria that is regulated by the cellular AMP/ATP ratio. During energy deficits, the ratio of adenosine triphosphate (ATP)/AMP increases, stimulating AMPK activation [61]. The activation of AMPK requires specific phosphorylation events by upstream kinases such as the serine/threonine protein kinase known as liver kinase B1 (LKB1) [62]. AMPK inhibits anabolic pathways and increases catabolic processes to induce ATP generation. Acting as an initiator of mitochondrial biogenesis, AMPK induces mitochondrial metabolism to increase ATP levels via a mitohormetic response, which is based on low levels of ROS [63]. However, it is also reported that AMPK can also be activated by increased ROS levels [64]. The activated AMPK then phosphorylates mitochondrial fission factor (MFF) [65] and unc-51 like autophagy activating kinase 1 (ULK1) [66] to promote mitochondrial fission and subsequent mitophagy, respectively [67].
The UPRmit is a cellular response that occurs when there is an imbalance between nuclear DNA (nDNA)-encoded OXPHOS proteins and mtDNA-encoded OXPHOS proteins. This mitonuclear protein imbalance is characterized by accumulation of misfolded or unfolded proteins in the lumen of the endoplasmic reticulum (ER) [68]. The UPRmit induces mitochondrial protective genes including mitochondrial molecular chaperones and proteases and ROS defence antioxidants to re-establish protein homeostasis within the mitochondrial protein-folding environment [68,69]. Being a stress response mechanism, UPRmit is activated in neurodegenerative diseases including AD [70,71,72]. However, there is evidence that UPRmit drives mitophagy [58], despite the reports that mitophagy is impaired in AD [29]. Moreover, it has been shown in life span extension studies in C. elegans that UPRmit is induced by nicotinamide adenine dinucleotide (NAD+) boosters such as NAD+ precursors and poly ADP ribose polymerase (PARP) inhibitors by acting as co-factors in Sirtuin signalling [59,69].
The Sirtuins are a family of deacetylases (SIRT1-7) implicated in longevity [73,74]. Several studies indicate that they promote life span extension through the mitohormetic effect [75,76]. Redox reactivity of Sirtuins with NAD+ produce nicotinamide, which is methylated and is converted into 1-methylnicotinamide. This serves as a substrate for an aldehyde oxidase to produce hydrogen peroxide. The latter acts as a ROS signal to execute Sirtuin effects [77]. SIRT1 also suppresses the inducible nitric oxide synthase (iNOS) and thus, may decrease cellular ROS levels [78]. Primarily localized in the nucleus, SIRT1 translocation into mitochondria deacetylates the Peroxisome proliferation activator receptor gamma-co-activator 1α (PGC-1α), resulting in increased mitochondrial biogenesis [79]. Moreover, it has been demonstrated that SIRT3 promotes mitochondrial metabolic functions [80,81] and mitochondrial dynamics through activation of OPA1 to induce the fusion process [82]. In addition, SIRT2 deacetylation also leads to fork head box O (FOXO) transcriptional factor activation, resulting in the expression of various target genes including p27Kip1, manganese superoxide dismutase (MnSOD) and pro-apoptotic factor, Bcl-2 interacting mediator of cell death (Bim) [83]. FOXO activation by SIRT1 results in transcriptional activation of mitophagy [59].
The mTOR is another pathway involved in longevity [84,85,86]. It is a serine/threonine protein kinase that consists of two complexes, namely mTOR1 and mTOR2. The activity of mTOR1 is dependent on the accessory protein Regulatory-Associated Protein of mTOR (RAPTOR), which is thought to be a major cellular sensor of oxidative stress. Attenuation of mTOR signalling is associated with impaired glucose metabolism [87], resulting in mitohormesis and subsequent tissue-dependent modulation of mitobiogenesis [88]. There is evidence indicating that mTOR inhibition can lead to mitophagy [60,89].
Caloric restriction (CR) is reducing 10–50% ad libitum calorie intake without leading to malnutrition. It has been reported to be beneficial for healthy ageing and extending life span [90,91]. Additionally, agents that mimic the beneficial metabolic effects of CR are known as caloric restriction mimetics (CRMs). As evidenced in Saccharomyces cerevisiae and C. elegans, CR increases cellular OXPHOS levels, giving rise to elevated amounts of ROS and ultimately leading to longevity via hormetic ROS defence pathways [92,93,94]. On the one hand, as shown in rats, CR leads to the activation of SIRT1 and the suppression of mTOR [95]. On the other hand, CR has been shown to preserve the expression of SIRT3 and to increase the expression of AMPK and PGC-1α in skeletal muscle [96]. However, Lanza et al. (2012) [97] have shown that CR only preserves mitochondrial respiratory function rather than increasing mitochondrial abundance. In addition, a study conducted in rat cortical neurons has shown that CR-induced autophagy was stimulated by upregulated levels of neuropeptide Y (NPY) and ghrelin [98], a peripheral gut hormone that increases appetite. Additionally, Cui et al. (2013) [99] have confirmed that CR markedly increases the expression of autophagosome markers in kidneys of Fischer 344 rats. Apart from SIRT1/3 and AMPK activation and mTOR inhibition, which may lead to mitophagy, there is no direct evidence that CR regulates mitophagy. However, CR has been reported to exert neuroprotective effects [100].
An increasing number of studies has indicated the importance of exercise in longevity and healthy ageing [101,102,103]. One of the ways it is thought to exert its actions is through protection against agents that can cause cell death. It has been reported that exercise induces SIRT3 expression in cortical neurons, which deacetylates SOD2 and cyclophilin D in neuronal mitochondria, which provides protection against excitotoxicity [104]. Furthermore, during exercise, mitochondrial biogenesis is induced by increased PGC-1α levels [105,106]. However, a study by Philp et al. (2011) [107] indicates that upregulated PGC-1α levels are not due to the deacetylase activity of SIRT1, but are due to changes in the general control of amino-acid synthesis 5 (GCN5) acetyltransferase activity after exercise. Alternately, the high energy consumption during exercise disrupts the balance between AMP and ATP, leading to AMPK activation followed by induction of PGC-1α [108]. During exercise, PGC-1α promotes ROS generation via increased mitochondrial metabolism, leading to an elevated oxygen consumption in muscle fibres. This ROS generation is dependent on other exercise-induced changes in muscles, including increased CO2 tension, raised temperature and decrease in cellular pH [109]. However, PGC-1α has also been reported to induce ROS-detoxifying enzymes by Nrf2 activation [110]. In addition to the ROS-mediated adaptive responses, there is emerging evidence that exercise induces autophagy/mitophagy in skeletal muscle [111,112,113].
It has been reported that acute exercise induces mitophagy through AMPK-dependent activation of ULK1 in the skeletal muscle [66]. It has been reported that mitophagy occurs only at a later stage and not during or soon after high intensity endurance exercise [114]. The latter observation is consistent with the work of Ogborn et al. (2015) [115], who showed that the mitophagy proteins PINK1 and Parkin are not changed in response to exercise in the muscle from aged subjects immediately after a single bout of resistance training.
Despite the presence of the above-mentioned mitohormetic factors that promote stress resistance at low levels of ROS, the brain remains highly susceptible to oxidative stress induced by high levels of ROS. To counter this, the brain has several endogenous antioxidant defence mechanisms. These encompass the antioxidant enzymes: catalase, superoxide dismutase, superoxide reductase and glutathione peroxidase and antioxidants including glutathione (GSH), vitamin C and vitamin E. For supporting nearby neurons, astrocytes recycle vitamin C [116,117], and also maintain high concentrations of certain antioxidants by expressing Nrf2-dependent antioxidant genes. Moreover, neuronal inhibition of glycolysis stimulates the occurrence of the pentose phosphate pathway, which has significant implications for antioxidant defence due to the generation of the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH), an essential cofactor for the generation of GSH [118]. However, these endogenous antioxidant defence mechanisms get overwhelmed, as ROS is continuously produced by vicious cycles due to Aβ-interactions with transition metal ions in AD brains.
Mitophagy induction has links to oxidative stress, as ROS induces the mitophagy process [119,120]. Therefore, in addition to exercise and caloric restriction, the lifestyle interventions so far known to induce mitophagy through pharmacological or nutraceutical interventions are extremely important.
5. Polyphenols with Potential to Induce Mitophagy
Given the lack of success of Aβ-targeted pharmaceutical approaches to date, the role of naturally occurring bioactive compounds as alternate treatment options for AD has gained considerable interest. Polyphenols in plant extracts are reported to play a role in delaying the onset of AD [121,122]. The most common types of polyphenols are phenolic acids, stilbenes, flavonoids and lignans. A major challenge for the use of these agents for therapy is thought to be their poor bioavailability after ingestion and inability to cross the BBB [123,124]. The most studied polyphenols that may be beneficial for AD include curcumin [125,126], resveratrol [127,128,129], quercetin [130,131], (-)-epigallactocatechin-3-gallate (EGCG) [132], and fisetin [133]. Many polyphenols exert favorable effects on mitochondria in terms of biogenesis and integrity [134]. However, the focus of this review is another group of nutraceutical compounds, urolithins, which have been shown to activate mitophagy [135].
6. Urolithins
Urolithins are 6H-dibenzopyran-6-one derivatives (or aglycons) produced by specific microbial transformations. The polyphenol ellagitannins (ETs) are the precursors of urolithins. ETs are hydrolysable tannins that occur in a wide variety of fruits such as pomegranates, strawberries, raspberries, blackberries and walnuts. Most importantly, pomegranate is a rich source of ETs and the most abundant ET in pomegranate is punicalagin. Its highest concentration is in the pomegranate peel, which is approximately 10.5 g/kg [136,137], while in the juice, it is between 1.5–1.9 g/L [138]. Hydrolysis of ETs to ellagic acid (EA) has been observed in the duodenum due to neutral pH conditions [139,140], whilst both ETs and EAs can undergo biotic changes in the lower gastro-intestinal tract [141].
6.1. Biological Effects of Urolithins
6.1.1. Human Microbiota and Urolithin Metabotypes
Human microbiota is the entire collection of microorganisms living on the surface and the interior of the human body. Most of the microbiota live in the colon, where they exceed a density of 1011 cells per gram of bodyweight [142]. It is known that the human gut is colonized by hundreds of different species, where the species Firmicutes, Bacteroidetes and Actinobacteria are prominent [143]. However, the microbiome is highly dynamic and even within an individual, it is influenced by many factors such as age, diet, hormonal cycles, travel, illnesses, and usage of drugs, especially antibiotics. Most importantly, the gut microbiota composition plays a key role in the maintenance of human health [144].
Modulating the density of various bacterial communities in the gut is linked to both health improvement and deterioration. Beneficial bacteria, commonly known as probiotics (e.g., Bifidobacterium, Lactobacillus), function as a ‘barrier’ against pathogens. However, it is increasingly being observed that the composition of human gut microbiota is prone to changes in various diseases, including certain types of cancer [145] and also AD [146,147]. The role of microbiota in AD could be attributed to diet, which is being recognized to be a risk factor for AD [148,149]. Alterations in the gut microbiome have been found not only in rodent AD models, but recently also in human AD subjects. In one study, faeces of AD participants indicated decreased levels of Firmicutes and Bifidobacterium and increased levels of Bacteroidetes compared with those obtained from age- and gender-matched controls [146]. Interestingly, the different levels of these microbial genera have been observed to correlate with cerebrospinal fluid (CSF) biomarkers of AD [146].
There are specific dietary substrates that modulate the composition and metabolic function of the microbial communities. For instance, dietary phenolic and polyphenolic components of common foods readily contribute to gut bacteria modulation [150]. However, dietary fibre and prebiotics which remain undigested by human enzymes modulate the microbiota into specific gastrointestinal microbes, which generate short chain fatty acids (SCFAs) that are beneficial for human hosts [151]. Similarly, ETs in diet modify the gut microbiota including Clostridium coccoides, Clostridium leptum [152], or Gordonibacter urolithinfaciens [153], which transform the ETs into urolithins [154].
As shown in Figure 2, ETs in ingested foods are converted into EA in the duodenum. EA then converts to urolithin D, retaining four phenolic hydroxyl groups. Subsequent metabolism continues along the gastrointestinal tract with the sequential removal of hydroxyl groups, leading to the production of urolithin C, urolithin A and urolithin B in the distal parts of the colon. Urolithin M-5, urolithin M-6, urolithin M-7, urolithin C and urolithin E are reported to be produced as side metabolites leading to urolithin A [152,155]. Urolithin A is shown to exert anti-ageing effects, increased mitochondrial activity and muscle function, potentially due to its mitophagy-inducing effects [135] and antioxidant effects [156]. Urolithin B is shown to act to reduce muscle atrophy [157], while lipid-lowering effects are exerted by urolithin C [158]. The rate of urolithin production is dependent on the type of microbiota and the ingested type of ET [159]. One study has shown that humans can be categorised into three metabolic phenotypes based on their ability to modulate microbiota, such that different urolithins are produced [160]. For example, after consuming an ET-rich diet, if an individual could excrete only urolithin A, he/she will be of metabotype A. Similarly, the ability to excrete urolithin B and/or isourolithin A defines metabotype B while metabotype O denotes the inability to produce urolithins [161].
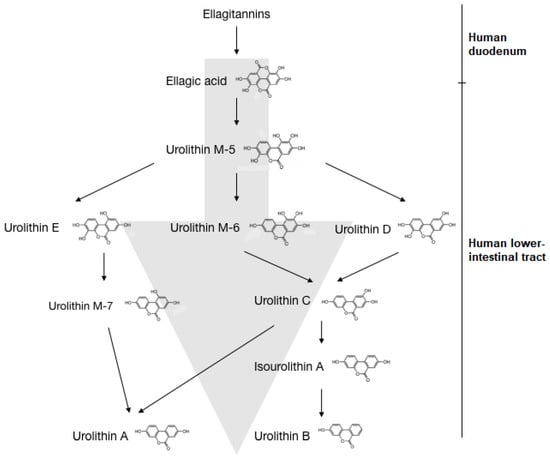
Figure 2.
Bacterial transformation of ellagitannins into urolithin A and B in humans. The ETs in ingested food are converted to EA in the human duodenum. In the lower intestinal tract, EA is converted to urolithin M-5, which can be converted into the intermediates urolithin E, urolithin M-6 and urolithin D. Urolithin E converts to urolithin M-7, while urolithin M-6 and urolithin D give rise to urolithin C. Urolithin C converts to isourolithin A and urolithin B respectively. Alternatively, urolithin M-7 and urolithin C are converted to urolithin A.
Urolithin metabotypes have been reported to be potential cardiometabolic risk biomarkers [162]. The gut microbiota associated with obesity have been shown to have a link with a differential metabolism of EA and the urolithin-producing bacterial genus Gordonibacter [163]. However, further studies for urolithin metabotypes are required, as many of the basic facts including their induction, stability over time, diet preference and genetic and epigenetic influence remain largely unknown. Moreover, there are four new urolithin metabolites (urolithin M6R, M7R, CR, AR) which have been recently identified in human faeces and urine following intake of pomegranate extract, leading to further studies of unravelling gut microbial action [164].
6.1.2. Bioavailability and Toxicity of Urolithins
Urolithins appear in the human circulation within a few hours of consumption of ET-containing foods, reaching maximum concentrations after 24–48 h and complete excretion in urine/faeces within 72 h. They are present in the plasma at very low concentrations ranging from 0.2–20 μM [165] and undergo further biotransformation, such as methylation, sulfation and glucuronidation, upon entering the enterohepatic circulation [166,167]. For oral and intravenous administration urolithin A, only glucuronidated and sulfonated phase II metabolites have been reported to be the prevalent metabolites from Absorption, Distribution, Metabolism and Excretion (ADME) studies [168]. It has been reported very recently that a phase II metabolism can also negatively affect certain pharmacological properties of urolithins [169].
Furthermore, liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI–MS/MS) has been recently utilised for the determination of urolithin C in rat plasma. In this study, glucuronyl and sulphate conjugates of urolithin C were the main metabolites detected in plasma [170]. In silico studies have predicted that urolithins can permeate the blood–brain barrier (BBB) [171]. Taken together with a report in which urolithin B has been detected in rat brains upon intravenous administration, this implies that urolithin A can enter the brain [172]. Furthermore, it has been reported very recently that urolithin A was detected in brains of a Parkinson’s disease rat model [173].
Urolithins are relatively non-toxic, as shown by studies in rats [172,173], as well as humans [174]. Furthermore, Urolithin A has been shown to be non-genotoxic by a battery of assays [168]. The lethal dose 50 (LD50) has been found to be greater than 5 g/kg body weight in rats for a pomegranate fruit extract standardized to consist of 30% punicalagins [175]. Another study has reported the effect of the pomegranate fruit extract in Wistar rats, at levels up to 600 mg/kg body weight for 90 days with no signs of toxicity [176]. In another toxicological study, rats were fed with chow diets with 6% punicalagin, where the daily intake of punicalagin ranged from 0.6 to 1.2 g. No evidence of toxicity was observed after feeding the rats with the punicalagin-enriched diet for 37 days [166]. Overall, their non-toxicity and relatively short half-life until excretion from the body are favorable characteristics for urolithins to be considered for use as potential therapeutic agents. However, a recent report hints at the possibility of urolithin A as a potential thyroid disruptor [177].
6.1.3. Therapeutic Effects of Urolithins
Urolithins are rapidly being identified as possible therapeutic agents for many types of diseases, including cardiac dysfunction [178] and various cancers [179,180,181]. Urolithin C has been shown to exert triglyceride-lowering effects in both adipocytes and hepatocytes [158], as well as to induce apoptosis in PC12 cells through a mitochondria-mediated pathway [182]. Moreover, urolithin A, B, C and D have exerted different antiproliferative effects on human colon cancer cell lines [183].
Pomegranate has been shown to ameliorate inflammation in the gastrointestinal tract, owing to the presence of ETs, EA and the urolithins [184]. Furthermore, the gut health of the host can determine the effectiveness of pomegranate-derived compounds against inflammation. In a study of Fisher rats [185], urolithin A was found to be the most effective anti-inflammatory compound derived from pomegranate consumption. However, in rats with colon inflammation, the nonmetabolized ET-related fraction was more effective. The compounds in the peel of the fruit have also been shown to be effective. In a study conducted using a mouse model of obesity associated with hypercholesterolemia and inflammatory disorders, it was found that the symptoms were alleviated by pomegranate peel extract [186]. Moreover, urolithin A has been studied in a randomized, placebo-controlled, double blind clinical trial, which concluded that urolithin A is safe and bioavailable in humans and is effective against age-related muscle decline [187]. Another clinical trial has shown that UA at doses of 500 mg and 1,000 mg for 4 weeks modulated plasma acylcarnitines and skeletal muscle mitochondrial gene expression in elders [174].
6.2. Considerations for Urolithin A as a Therapeutic Agent for Alzheimer’s Disease
Due to their prominent roles in the disease process, Aβ accumulation, oxidative stress and inflammation are key therapeutic targets in AD. Evidence, primarily from in vitro studies, suggest a neuroprotective potential for urolithins via mechanisms that act upon these targets. The evidence presented in Table 1 indicates that not only can EA and punicalagin attenuate the neurodegenerative process, but it can also act upstream to reduce the accumulation of Aβ. There is evidence suggesting that these molecules inhibit BACE1 activity, leading to reduced Aβ production. A study based on activity-guided purification has revealed that EA and punicalagin have an inhibitory action on BACE1 [188]. Furthermore, other ETs, geraniin and corilagin from the plant Geranium thunbergii have also been shown to selectively inhibit BACE1 without modulating the activity of other common serine proteases [189].

Table 1.
Pomegranate/ellagitannins/urolithins-based studies in relation to neuroprotection.
No study has evaluated the potential of downstream ET metabolites, particularly urolithins, in the inhibition of Aβ production. Additionally, urolithins have been shown to inhibit Aβ fibrillization dose-dependently, as indicated by the Thioflavin T assay [171].
ETs and urolithins possess antioxidant properties due to the presence of phenolic hydroxyl groups in their chemical structure. Compared to urolithin A, EA, urolithin C and D have been reported to have a higher antioxidant potency, which was even higher than for vitamin C [200]. Moreover, the effect of punicalagin on oxidative stress has been investigated in rat intestinal epithelial cells (IEC-6) [201]. Its antioxidative properties are exerted by up-regulating the expression of HO-1 via a mechanism that involves PI3K/Akt activation and Nrf2 translocation [156].
As neuroinflammation is a key feature in AD [202,203], the compounds that show anti-inflammatory activity can be beneficial against AD. Pomegranate constituents have been reported to inhibit the enzymes cyclooxygenase (COX) and lipoxygenase (LOX) [184] that inhibit the formation of prostaglandins, which are the key modulators of inflammation. Furthermore, it has been demonstrated that pomegranate juice inhibits the p38-mitogen-activated protein kinase (p38 MAPK) pathway, subsequently decreasing the levels of transcription factor NF-κB and down-regulating the production of the proinflammatory mediators TNF-α, IL-1β and MCP1 [204]. These observations were confirmed by Olajide et al. (2014) [191] for punicalagin, including the inhibition of tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF-6) mediated neuroinflammation. Histone deacetylation also plays an important role in inflammation, as it is implicated in the deactivation of inflammation-related transcription factors NF-κB and AP-1. Inhibition of histone acetyltransferases (HATs) is another mechanism by which the urolithins exhibit anti-inflammatory properties [165].
ETs and urolithins can also promote neuroprotection via estrogen receptors, potentially through the activation of nuclear estrogen receptors (ERs) in the brain [205,206]. Due to their plant-based origin, ETs can be considered as phytoestrogens and the urolithin metabolites generated within the gut are referred to as enterophytoestrogens. Urolithins exhibit estrogenic or antiestrogenic properties, depending on the availability of endogenous estrogen (estradiol) and thus may be considered as selective estrogen receptor modulators (SERMs) [167,177,207]. Even among urolithins, urolithin A has been shown to exhibit a high affinity to ERs, specifically to ERα, compared to urolithin B [167]. Consistently, a more recent study has reported the expression of estrogen-regulated genes by urolithin A in an ERα-dependent pathway [208].
Hypercholesterolemia has a central role in AD [209,210]; therefore, modulation of cholesterol or enhancing anti-atherogenic effects may be beneficial against AD. Studies have shown that urolithins act as activation ligands for liver X receptors (LXRs) [190], which are involved in uptake, transport, efflux and excretion of cholesterol in a tissue-dependent manner [211]. EA also promotes cholesterol efflux in oxidized LDL-induced foam cells, inhibiting macrophage lipid uptake [212]. However, Mele et al. (2016) [190] showed that EA and urolithin C decrease the accumulation of cholesterol but do not promote cholesterol efflux in THP-1-derived macrophages.
As mitophagy deficits are key features of mitochondrial dysfunction in AD, safeguarding mitochondrial integrity by inducing mitophagy may potentially be an effective intervention for AD. In this respect, urolithins have been shown to restore mitochondrial homeostasis by improving mitochondrial function and biogenesis [213], and inducing mitophagy [135,214]. Furthermore, Webb et al. (2017) have reported that urolithin dose-dependently induced mitophagy in C2C12 myotubes [215]. However, research directed at an understanding of the underpinning mechanisms of urolithin-induced mitophagy is still in its infancy.
6.3. Insights on How Urolithin A May Trigger Mitophagy
As shown in Figure 3 and discussed further below, diverse evidence suggests that urolithins may modulate key signalling molecules in mitophagy and thereby regulate the activity of this pathway.
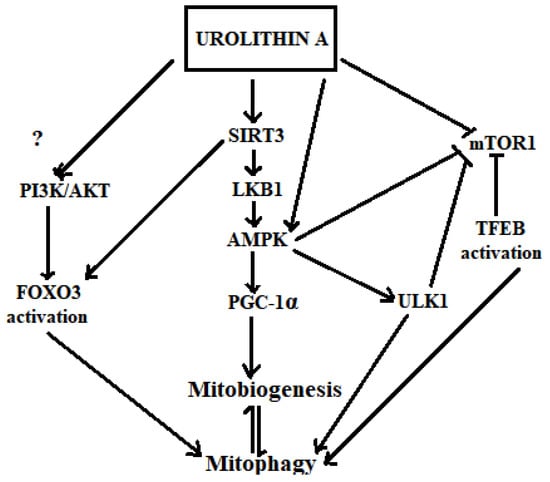
Figure 3.
Hypothetical mechanisms of urolithin A in inducing mitophagy. Urolithin A activates SIRT1, SIRT3 [216] and AMPK. Activated AMPK increases PGC-1α levels [108] that directly increases mitochondrial biogenesis, which is in equilibrium with the mitophagy process. Activated AMPK also increases the activation of ULK1 [66]. Inhibition of mTOR1 is triggered by both urolithin A [217] and AMPK, which then activates ULK1 towards inducing mitophagy. The mTOR1 inhibition by Urolithin A may also induce the transcriptional activation of mitophagy via TFEBs. Urolithin A transcriptionally activates mitophagy via SIRT3-dependent FOXO activation [218,219].
6.3.1. Activation of SIRT1/3, AMPK, PGC1-α and Inhibition of mTOR1
Activation of SIRT, AMPK and PGC1-α and inhibition of mTOR tends to induce mitophagy and mitochondrial biogenesis to maintain a healthy mitochondrial population [220]. These key signalling pathways have been shown to be modulated by urolithins. SIRT deacetylates and activates LKB1, which is an upstream kinase that activates AMPK [221]. Zhao et al. (2016), have reported enhanced SIRT3 promoter activity in Caco-2 cells by urolithin A [216]. Urolithin A also increases ATP and NAD+ levels, leading to the activation of SIRT1 promoters, effecting the SIRT1-PGC-1α pathway. [222]. The SIRT1 activity modulates mitofusin 2 (Mfn2) expression and subsequent mitophagy [223]. It has also been shown that urolithin A increases Mfn2 in its pathway of inducing mitophagy [174]. Furthermore, activation of SIRT1 increases Parkin levels [224] and urolithin A (1000 mg) has been shown to transcriptionally increase Parkin and BECN1 levels after 28 days of treatment in humans [174].
Activation of AMPK is a broad checkpoint for many cell signalling pathways, including the induction of downstream mitophagy effectors such as ULK1 through the inhibition of mTOR1 [225,226]. Furthermore, urolithins’ ability to activate AMPK [227], and impair mTOR signalling [217], is reported. Therefore, these studies provide support that urolithins may induce AMPK-mediated activation of ULK1, which may then phosphorylate the LIR motif of FUNDC1 to promote mitophagy [174].
6.3.2. Transcriptional Activation by TFEB and FOXO3
Transcription factor EB (TFEB) and FOXO3 are activated in response to low nutrient or energy status to upregulate the expression of autophagic and lysosomal genes [228,229,230]. In addition to their role in general autophagy, they have also been reported as key players in mitophagy [218].
The TFEB belongs to the helix-loop-helix-leucine-zipper (bHLH-Zip) class of microphthalmia-associated transcription factors (MiTFs) and regulates lysosomal biogenesis. The levels of TFEB in the nucleus reportedly corresponds to varying cellular demands for autophagosome-lysosome function [231]. TFEB binds to the coordinated lysosomal expression and regulation (CLEAR) motif. In the de-activated state, TFEB resides in the cytoplasm, recruited to the mTOR complex, which is localized in lysosomes to the cytoplasmic end [232]. Alternatively, mTOR1 inhibition promotes the translocation of TFEB to the nucleus and the downstream effect of mitophagy through transcriptional activity in a PINK1 and a Parkin-dependent manner [233,234]. There is no direct evidence to date that urolithins activate TFEB; however, pomegranate extract has been reported to activate TFEB independently of ERK1/2, mTORC1 and calcineurin [234]. This hints that urolithin A may also give rise to TFEB activation. This vision is further reinforced as TFEB activation also occurs independent of mTORC1 via Akt inhibition [235], and for the reports showing that urolithin A attenuates Akt signalling [165,199,236,237]. However, one exception is a report that urolithin A alleviates myocardial ischemia/reperfusion injury through activating the PI3K/Akt pathway [238].
On the other hand, SIRT3 deacetylates and activates FOXO3 to promote its nuclear translocation for transcriptional activation of PINK [239,240]. Furthermore, the activity of FOXO3 during the process of mitophagy has been evaluated in SH-SY5Y cells [219]. In this study, the levels of the mitophagy marker proteins Beclin 1, PINK and Parkin were shown to be increased, following the induction of mitophagy by MnCl2 and this was accompanied by FOXO3 nuclear retention. A more recent study carried out in SH-SY5Y and induced pluripotent stem cell (iPSC)-derived neurons, has also shown that Akt signalling increases mitophagy via regulating PINK 1 levels [241]. Additionally, FOXO3 is activated by the Phosphoinositide-3-kinase/ Protein kinase B (PI3K/Akt) pathway depending on the mTORC2 activation [242]. Therefore, due to controversy on the effect of urolithin A on the PI3K/Akt pathway, whether FOXO3 might be activated by urolithin A as a mode of transcriptional activation of mitophagy needs further evaluation.
7. Conclusions
The neurodegenerative processes that lead to AD remain to be fully understood and although the accumulation of Aβ and hyper-phosphorylated Tau protein have a key role, investigating the relevant cellular pathways that are impaired in the early stages of the disease process is required. Mitochondrial dysfunction is an early feature of the disease and although it is yet to be established as a cause or consequence of accumulating pathology, it is increasingly being recognized as a key contributor to the disease process. Avenues to attenuate or restore mitochondrial function could offer potential targets; however, an alternative is to promote removal of damaged/dysfunctional organelles. In this regard, enhancing the process of mitophagy is considered to be beneficial.
In addition to a greater understanding of disease mechanisms, it is increasingly apparent that any intervention for AD needs to be initiated as early as possible before the onset of irreversible neurodegeneration is to be effective. Due to their relative safety and efficacy compared to many other drugs in development, nutraceuticals are more attractive alternatives for the prevention and treatment of AD. Urolithins are particularly promising, as they represent microbial metabolites of ingested polyphenols and initial studies suggest that they may have a multifaceted therapeutic value for AD by their actions to reduce BACE1 activity, Aβ fibrillation, ROS damage, inflammation, and atherogenesis and most importantly, their ability to restore/induce mitophagy, which is impaired in AD. Furthermore, urolithin metabotypes warrant investigation as their impact on the microbiome may be an additional contribution to reducing AD risk.
Author Contributions
Conceptualization, R.N.M., D.P.W.J. and H.K.; writing—original draft preparation, D.P.W.J.; writing—review and editing, E.H., G.V., M.L.G., H.S., T.L., B.F., G.J.G. and R.N.M.; visualization, D.P.W.J. and supervision, R.N.M., E.H., G.V., M.L.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors wish to dedicate this review article to Harjot Khaira (H.K.) an outstanding PhD student with considerable passion, talent and motivation who suddenly passed away in 2017.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-C.; Wei, Y.-H. Role of mitochondria in human aging. J. Biomed. Sci. 1997, 4, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K. Mitochondria damage checkpoint, aging, and cancer. Ann. N. Y. Acad. Sci. 2006, 1067, 182–190. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellerin, L.; Bouzier-Sore, A.K.; Aubert, A.; Serres, S.; Merle, M.; Costalat, R.; Magistretti, P.J. Activity-dependent regulation of energy metabolism by astrocytes: An update. Glia 2007, 55, 1251–1262. [Google Scholar] [CrossRef]
- Riske, L.; Thomas, R.K.; Baker, G.B.; Dursun, S.M. Lactate in the brain: An update on its relevance to brain energy, neurons, glia and panic disorder. Ther. Adv. Psychopharmacol. 2017, 7, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Lenaz, G. Role of mitochondria in oxidative stress and ageing. Biochim. Biophys. Acta (BBA) Bioenerg. 1998, 1366, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef] [Green Version]
- Moncada, S.; Bolanos, J.P. Nitric oxide, cell bioenergetics and neurodegeneration. J. Neurochem. 2006, 97, 1676–1689. [Google Scholar] [CrossRef]
- Contestabile, A.; Monti, B.; Polazzi, E. Neuronal-glial Interactions Define the Role of Nitric Oxide in Neural Functional Processes. Curr. Neuropharmacol. 2012, 10, 303–310. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef]
- Shohami, E.; Beit-Yannai, E.; Horowitz, M.; Kohen, R. Oxidative Stress in Closed-Head Injury: Brain Antioxidant Capacity as an Indicator of Functional Outcome. J. Cereb. Blood Flow Metab. 1997, 17, 1007–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Disease International. Dementia and Risk Reduction: An Analysis of Protective and Modifiable Risk Factors; Alzheimer’s Disease International: London, UK, 2014. [Google Scholar]
- Australian Institute of Health and Welfare. Dementia in Australia. Available online: https://www.aihw.gov.au/reports-data/health-conditions-disability-deaths/dementia/overview (accessed on 21 June 2021).
- Yang, Y.H.; Meguro, K.; Kim, S.Y.; Shim, Y.S.; Yu, X.; Chen, C.L.H.; Wang, H.; Lam, L.; Senanarong, V.; Dominguez, J.; et al. Impact of Alzheimer’s Disease in Nine Asian Countries. Gerontology 2016, 62, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, P.B. Risk factors for vascular dementia and Alzheimer disease. Stroke 2004, 35, 2620–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedel, B.C.; Thompson, P.M.; Brinton, R.D. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2016, 160, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Edwards Iii, G.A.; Gamez, N.; Escobedo, G., Jr.; Calderon, O.; Moreno-Gonzalez, I. Modifiable Risk Factors for Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, H.J.; Graeber, M.B. The case described by Alois Alzheimer in 1911. Historical and conceptual perspectives based on the clinical record and neurohistological sections. Euro. Arch. Psychiatry Clin. Neurosci. 1998, 248, 111–122. [Google Scholar] [CrossRef]
- Fox, N.C.; Schott, J.M. Imaging cerebral atrophy: Normal ageing to Alzheimer’s disease. Lancet 2004, 363, 392–394. [Google Scholar] [CrossRef]
- Kern, S.; Zetterberg, H.; Kern, J.; Zettergren, A.; Waern, M.; Höglund, K.; Andreasson, U.; Wetterberg, H.; Börjesson-Hanson, A.; Blennow, K.; et al. Prevalence of preclinical Alzheimer disease. Neurology 2018, 90, e1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayan, A.D. Quantitative histological studies on the aged human brain. I. Senile plaques and neurofibrillary tangles in “normal” patients. Acta Neuropathol. 1970, 16, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017. [Google Scholar] [CrossRef] [Green Version]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Leuner, K.; Schütt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-Derived Reactive Oxygen Species Lead to Enhanced Amyloid Beta Formation. Antioxid. Redox Sgnal. 2012, 16, 1421–1433. [Google Scholar] [CrossRef] [Green Version]
- Lemasters, J.J. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Diot, A.; Morten, K.; Poulton, J. Mitophagy plays a central role in mitochondrial ageing. Mamm. Genome 2016, 27, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.F.; Walker, D.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 2017, 140, 3233–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsetti, V.; Florenzano, F.; Atlante, A.; Bobba, A.; Ciotti, M.T.; Natale, F.; Della Valle, F.; Borreca, A.; Manca, A.; Meli, G.; et al. NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: Implications in Alzheimer’s disease. Hum. Mol. Genet. 2015, 24, 3058–3081. [Google Scholar] [CrossRef] [Green Version]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [Green Version]
- Moreira, P.I.; Siedlak, S.L.; Wang, X.; Santos, M.S.; Oliveira, C.R.; Tabaton, M.; Nunomura, A.; Szweda, L.I.; Aliev, G.; Smith, M.A.; et al. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy 2007, 3, 614–615. [Google Scholar] [CrossRef] [Green Version]
- Baloyannis, S.J. Mitochondrial alterations in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 119–126. [Google Scholar] [CrossRef]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef]
- Hu, Y.; Xia-Chun, L.; Zhi-Hao, W.; Yu, L.; Xiangnan, Z.; Xiu-Ping, L.; Qiong Feng, Q.W.; Zhenyu, Y.; Zhong, C.; Keqiang, Y.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Jayatunga, D.P.W.; Hone, E.; Bharadwaj, P.; Garg, M.; Verdile, G.; Guillemin, G.J.; Martins, R.N. Targeting Mitophagy in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 78, 1273–1297. [Google Scholar] [CrossRef]
- Li, W.; Kui, L.; Demetrios, T.; Gong, X.; Tang, M. A Glimmer of Hope: Maintain Mitochondrial Homeostasis to Mitigate Alzheimer’s Disease. Aging Dis. 2020, 11. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Xie, C.; Aman, Y.; Adriaanse, B.A.; Cader, M.Z.; Plun-Favreau, H.; Xiao, J.; Fang, E.F. Culprit or Bystander: Defective Mitophagy in Alzheimer’s Disease. Front. Cell Dev. Biol. 2020, 7, 391. [Google Scholar] [CrossRef]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabiola, S.; Tsitsilonis, O.E.; Athanassios, K.; Ioannis, P.T. Oxidative Stress-mediated Biomolecular Damage and Inflammation in Tumorigenesis. In Vivo 2012, 26, 395–402. [Google Scholar]
- Melov, S.; Ravenscroft, J.; Malik, S.; Gill, M.S.; Walker, D.W.; Clayton, P.E.; Wallace, D.C.; Malfroy, B.; Doctrow, S.R.; Lithgow, G.J. Extension of life-span with superoxide dismutase/catalase mimetics. Science 2000, 289, 1567–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, H.; Tang, X.D.; Chen, M.L.; Joiner, M.L.; Sun, G.; Brot, N.; Weissbach, H.; Heinemann, S.H.; Iverson, L.; Wu, C.F.; et al. High-quality life extension by the enzyme peptide methionine sulfoxide reductase. Proc. Nat. Acad. Sci. USA 2002, 99, 2748–2753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.F.; Santana, L.F.; Vermulst, M.; Tomazela, D.M.; Emond, M.J.; MacCoss, M.J.; Gollahon, K.; Martin, G.M.; Loeb, L.A.; Ladiges, W.C.; et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009, 119, 2789–2797. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaiahgari, S.; Zhang, Q.; Kleeberger, S.R.; Cho, H.Y.; Reddy, S.P. Hyperoxia stimulates an Nrf2-ARE transcriptional response via ROS-EGFR-PI3K-Akt/ERK MAP kinase signaling in pulmonary epithelial cells. Antioxid. Redox Sgnal. 2006, 8, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 2014, 12, 288–341. [Google Scholar] [CrossRef]
- Markaki, M.; Palikaras, K.; Tavernarakis, N. Novel Insights into the Anti-aging Role of Mitophagy. Int. Rev. Cell Mol. Biol. 2018, 340, 169–208. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, B.; Li, T.; Zhu, Y.; Luo, G.; Jiang, Y.; Tang, F.; Jian, Z.; Xiao, Y. AMPK activation serves a critical role in mitochondria quality control via modulating mitophagy in the heart under chronic hypoxia. Int. J. Mol. Med. 2018, 41, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrino, M.W.; Haynes, C.M. Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol. 2015, 13, 22. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy: Event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef] [Green Version]
- Gilkerson, R.W.; De Vries, R.L.; Lebot, P.; Wikstrom, J.D.; Torgyekes, E.; Shirihai, O.S.; Przedborski, S.; Schon, E.A. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum. Mol. Genet. 2012, 21, 978–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Scott, J.W.; Pan, D.A.; Hudson, E.R. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003, 546, 113–120. [Google Scholar] [CrossRef]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.D.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase Cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [Green Version]
- Zong, H.; Ren, J.M.; Young, L.H.; Pypaert, M.; Mu, J.; Birnbaum, M.J.; Shulman, G.I. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Nat. Acad. Sci. USA 2002, 99, 15983–15987. [Google Scholar] [CrossRef] [Green Version]
- Song, S.B.; Hwang, E.S. A Rise in ATP, ROS, and Mitochondrial Content upon Glucose Withdrawal Correlates with a Dysregulated Mitochondria Turnover Mediated by the Activation of the Protein Deacetylase SIRT1. Cells 2018, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.S.; Lin, S.C. AMPK Promotes Autophagy by Facilitating Mitochondrial Fission. Cell Metab. 2016, 23, 399–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liang, B.; Shirwany, N.A.; Zou, M.-H. 2-Deoxy-D-Glucose Treatment of Endothelial Cells Induces Autophagy by Reactive Oxygen Species-Mediated Activation of the AMP-Activated Protein Kinase. PLoS ONE 2011, 6, e17234. [Google Scholar] [CrossRef] [Green Version]
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014, 217, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.-S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Imaizumi, K.; Miyoshi, K.; Katayama, T.; Yoneda, T.; Taniguchi, M.; Kudo, T.; Tohyama, M. The unfolded protein response and Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2001, 1536, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Scheper, W.; Hoozemans, J.J. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2017, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Rose, G.; Dato, S.; Altomare, K.; Bellizzi, D.; Garasto, S.; Greco, V.; Passarino, G.; Feraco, E.; Mari, V.; Barbi, C.; et al. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp. Gerontol. 2003, 38, 1065–1070. [Google Scholar] [CrossRef]
- Bellizzi, D.; Rose, G.; Cavalcante, P.; Covello, G.; Dato, S.; De Rango, F.; Greco, V.; Maggiolini, M.; Feraco, E.; Mari, V.; et al. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics 2005, 85, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.R.; Lu, Z.; Sack, M.N.; Scott, I. The role of sirtuins in modulating redox stressors. Free Radic. Biol. Med. 2012, 52, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Merksamer, P.I.; Liu, Y.; He, W.; Hirschey, M.D.; Chen, D.; Verdin, E. The sirtuins, oxidative stress and aging: An emerging link. Aging 2013, 5, 144–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmeisser, K.; Mansfeld, J.; Kuhlow, D.; Weimer, S.; Priebe, S.; Heiland, I.; Birringer, M.; Groth, M.; Segref, A.; Kanfi, Y.; et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat. Chem. Biol. 2013, 9, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Song, M.Y.; Song, E.K.; Kim, E.K.; Moon, W.S.; Han, M.K.; Park, J.W.; Kwon, K.B.; Park, B.H. Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes 2009, 58, 344–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Kim, H.S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [Green Version]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 Deacetylates and Activates OPA1 To Regulate Mitochondrial Dynamics during Stress. Mol. Cell. Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Nguyen, M.; Qin, F.X.; Tong, Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M. Targeting mTOR signaling to promote healthy longevity. FASEB J. 2017, 31, 256.4. [Google Scholar] [CrossRef]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [Green Version]
- Polak, P.; Cybulski, N.; Feige, J.N.; Auwerx, J.; Ruegg, M.A.; Hall, M.N. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008, 8, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, A.; García-Aguilar, A.; Asahara, S.-I.; Kido, Y.; Guillén, C.; Pajvani, U.B.; Benito, M. MTORC1 Regulates both General Autophagy and Mitophagy Induction after Oxidative Phosphorylation Uncoupling. Mol. Cell. Biol. 2017, 37, e00441-17. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—From yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Vendelbo, M.H.; Nair, K.S. Mitochondrial longevity pathways. Biochim. Biophys. Acta 2011, 1813, 634–644. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Sharma, S.; Agrawal, V.; Roy, N. Caloric restriction augments ROS defense in S. cerevisiae, by a Sir2p independent mechanism. Free Rad. Res. 2005, 39, 55–62. [Google Scholar] [CrossRef]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.K.; Agrawal, V.; Roy, N. Mitochondria-mediated hormetic response in life span extension of calorie-restricted Saccharomyces cerevisiae. Age 2011, 33, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Dong, W.; Wang, R.; Li, Y.; Xu, B.; Zhang, J.; Zhao, Z.; Wang, Y. Effect of caloric restriction on the SIRT1/mTOR signaling pathways in senile mice. Brain Res. Bull. 2015, 116, 67–72. [Google Scholar] [CrossRef]
- Palacios, O.M.; Carmona, J.J.; Michan, S.; Chen, K.Y.; Manabe, Y.; Ward, J.L., 3rd; Goodyear, L.J.; Tong, Q. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging 2009, 1, 771–783. [Google Scholar] [CrossRef]
- Lanza, I.R.; Zabielski, P.; Klaus, K.A.; Morse, D.M.; Heppelmann, C.J.; Bergen, H.R.; Dasari, S.; Walrand, S.; Short, K.R.; Johnson, M.L.; et al. Chronic Caloric Restriction Preserves Mitochondrial Function in Senescence Without Increasing Mitochondrial Biogenesis. Cell Metab. 2012, 16, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Ferreira-Marques, M.; Aveleira, C.A.; Carmo-Silva, S.; Botelho, M.; Pereira de Almeida, L.; Cavadas, C. Caloric restriction stimulates autophagy in rat cortical neurons through neuropeptide Y and ghrelin receptors activation. Aging 2016, 8, 1470–1484. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Shi, S.; Sun, X.; Cai, G.; Cui, S.; Hong, Q.; Chen, X.; Bai, X.-Y. Mitochondrial Autophagy Involving Renal Injury and Aging Is Modulated by Caloric Intake in Aged Rat Kidneys. PLoS ONE 2013, 8, e69720. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, T.V.; Gleichmann, M.; Tang, S.C.; Mattson, M.P. Hormesis/preconditioning mechanisms, the nervous system and aging. Ageing Res. Rev. 2006, 5, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D. Physical activity is medicine for older adults. Postgrad. Med. J. 2014, 90, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLeod, M.; Breen, L.; Hamilton, D.L.; Philp, A. Live strong and prosper: The importance of skeletal muscle strength for healthy ageing. Biogerontology 2016, 17, 497–510. [Google Scholar] [CrossRef] [Green Version]
- McPhee, J.S.; French, D.P.; Jackson, D.; Nazroo, J.; Pendleton, N.; Degens, H. Physical activity in older age: Perspectives for healthy ageing and frailty. Biogerontology 2016, 17, 567–580. [Google Scholar] [CrossRef]
- Cheng, A.; Yang, Y.; Zhou, Y.; Maharana, C.; Lu, D.; Peng, W.; Liu, Y.; Wan, R.; Marosi, K.; Misiak, M.; et al. Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise, and Metabolic and Excitatory Challenges. Cell Metab. 2016, 23, 128–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baar, K. Involvement of PPAR gamma co-activator-1, nuclear respiratory factors 1 and 2, and PPAR alpha in the adaptive response to endurance exercise. Proc. Nutr. Soc. 2004, 63, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef] [Green Version]
- Philp, A.; Chen, A.; Lan, D.; Meyer, G.A.; Murphy, A.N.; Knapp, A.E.; Olfert, I.M.; McCurdy, C.E.; Marcotte, G.R.; Hogan, M.C.; et al. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation following endurance exercise. J. Biol. Chem. 2011, 286, 30561–30570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atherton, P.J.; Babraj, J.; Smith, K.; Singh, J.; Rennie, M.J.; Wackerhage, H. Selective activation of AMPK-PGC-1alpha or PKB-TSC2-mTOR signaling can explain specific adaptive responses to endurance or resistance training-like electrical muscle stimulation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 786–788. [Google Scholar] [CrossRef]
- Arbogast, S.; Reid, M.B. Oxidant activity in skeletal muscle fibers is influenced by temperature, CO2 level, and muscle-derived nitric oxide. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R698–R705. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Tam, B.T.; Siu, P.M. Autophagic cellular responses to physical exercise in skeletal muscle. Sports Med. 2014, 44, 625–640. [Google Scholar] [CrossRef]
- Saleem, A.; Carter, H.N.; Hood, D.A. p53 is necessary for the adaptive changes in cellular milieu subsequent to an acute bout of endurance exercise. American journal of physiology. Cell Physiol. 2014, 306, C241–C249. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.C.; Wilson, R.J.; Yan, Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, C.; Deldicque, L.; Francaux, M. Lack of Activation of Mitophagy during Endurance Exercise in Human. Med. Sci. Sports Exerc. 2017, 49, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Ogborn, D.I.; McKay, B.R.; Crane, J.D.; Safdar, A.; Akhtar, M.; Parise, G.; Tarnopolsky, M.A. Effects of age and unaccustomed resistance exercise on mitochondrial transcript and protein abundance in skeletal muscle of men. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R734–R741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, J.M. Vitamin C transport and its role in the central nervous system. Sub Cell. Biochem. 2012, 56, 85–103. [Google Scholar] [CrossRef] [Green Version]
- Nualart, F.; Mack, L.; García, A.; Cisternas, P.; Bongarzone, E.R.; Heitzer, M.; Jara, N.; Martínez, F.; Ferrada, L.; Espinoza, F.; et al. Vitamin C Transporters, Recycling and the Bystander Effect in the Nervous System: SVCT2 versus Gluts. J. Stem Cell Res. Ther. 2014, 4, 209. [Google Scholar] [CrossRef] [Green Version]
- Bolanos, J.P.; Almeida, A. The pentose-phosphate pathway in neuronal survival against nitrosative stress. IUBMB Life 2010, 62, 14–18. [Google Scholar] [CrossRef]
- Shefa, U.; Jeong, N.Y.; Song, I.O.; Chung, H.-J.; Kim, D.; Jung, J.; Huh, Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural. Regen. Res. 2019, 14, 749–756. [Google Scholar] [CrossRef]
- Fan, P.; Xie, X.-H.; Chen, C.-H.; Peng, X.; Zhang, P.; Yang, C.; Wang, Y.-T. Molecular Regulation Mechanisms and Interactions Between Reactive Oxygen Species and Mitophagy. DNA Cell Biol. 2018, 38, 10–22. [Google Scholar] [CrossRef]
- Apetz, N.; Munch, G.; Govindaraghavan, S.; Gyengesi, E. Natural compounds and plant extracts as therapeutics against chronic inflammation in Alzheimer’s disease—A translational perspective. CNS Neurol. Dis. Drug Targets 2014, 13, 1175–1191. [Google Scholar] [CrossRef]
- Reddy, V.P.; Aryal, P.; Robinson, S.; Rafiu, R.; Obrenovich, M.; Perry, G. Polyphenols in Alzheimer’s Disease and in the Gut-Brain Axis. Microorganisms 2020, 8, 199. [Google Scholar] [CrossRef] [Green Version]
- Pandareesh, M.D.; Mythri, R.B.; Srinivas Bharath, M.M. Bioavailability of dietary polyphenols: Factors contributing to their clinical application in CNS diseases. Neurochem. Int. 2015, 89, 198–208. [Google Scholar] [CrossRef]
- Figueira, I.; Garcia, G.; Pimpão, R.C.; Terrasso, A.P.; Costa, I.; Almeida, A.F.; Tavares, L.; Pais, T.F.; Pinto, P.; Ventura, M.R.; et al. Polyphenols journey through blood-brain barrier towards neuronal protection. Sci. Rep. 2017, 7, 11456. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Dunbar, G.L. Comparative Neuroprotective Effects of Dietary Curcumin and Solid Lipid Curcumin Particles in Cultured Mouse Neuroblastoma Cells after Exposure to Abeta42. Int. J. Alzheimer’s Dis. 2017, 2017, 4164872. [Google Scholar] [CrossRef] [Green Version]
- Draczynska-Lusiak, B.; Doung, A.; Sun, A.Y. Oxidized lipoproteins may play a role in neuronal cell death in Alzheimer disease. Mol. Chem. Neuropathol. 1998, 33, 139–148. [Google Scholar] [CrossRef]
- Ma, T.; Tan, M.-S.; Yu, J.-T.; Tan, L. Resveratrol as a Therapeutic Agent for Alzheimer’s Disease. BioMed. Res. Int. 2014, 2014, 13. [Google Scholar] [CrossRef]
- Sawda, C.; Moussa, C.; Turner, R.S. Resveratrol for Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2017, 1403, 142–149. [Google Scholar] [CrossRef]
- Jung, S.H.; Murphy, E.A.; McClellan, J.L.; Carmichael, M.D.; Davis, J.M. The dietary flavonoid quercetin decreases neuroinflammation in a mouse model of Alzheimer’s disease. FASEB J. 2010, 24, 604.17. [Google Scholar] [CrossRef]
- Maria, S.-G.A.; Ignacio, M.-M.J.; Ramírez-Pineda Jose, R.; Marisol, L.-R.; Edison, O.; Patricia, C.-G.G. The flavonoid quercetin ameliorates Alzheimer’s disease pathology and protects cognitive and emotional function in aged triple transgenic Alzheimer’s disease model mice. Neuropharmacology 2015, 93, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.A.; Mandal, A.K.; Khan, Z.A. Potential neuroprotective properties of epigallocatechin-3-gallate (EGCG). Nutr. J. 2016, 15, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currais, A.; Prior, M.; Dargusch, R.; Armando, A.; Ehren, J.; Schubert, D.; Maher, P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell 2014, 13, 379–390. [Google Scholar] [CrossRef]
- Sandoval-Acuna, C.; Ferreira, J.; Speisky, H. Polyphenols and mitochondria: An update on their increasingly emerging ROS-scavenging independent actions. Arch. Biochem. Biophys. 2014, 559, 75–90. [Google Scholar] [CrossRef]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22. [Google Scholar] [CrossRef] [PubMed]
- Amakura, Y.; Okada, M.; Tsuji, S.; Tonogai, Y. Determination of phenolic acids in fruit juices by isocratic column liquid chromatography. J. Chromatogr. A 2000, 891, 183–188. [Google Scholar] [CrossRef]
- Fischer, U.A.; Carle, R.; Kammerer, D.R. Identification and quantification of phenolic compounds from pomegranate (Punica granatum L.) peel, mesocarp, aril and differently produced juices by HPLC-DAD–ESI/MSn. Food Chem. 2011, 127, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.I.; Tomas-Barberan, F.A.; Hess-Pierce, B.; Holcroft, D.M.; Kader, A.A. Antioxidant activity of pomegranate juice and its relationship with phenolic composition and processing. J. Agric. Food Chem. 2000, 48, 4581–4589. [Google Scholar] [CrossRef]
- Gil-Izquierdo, A.; Zafrilla, P.; Tomás-Barberán, F.A. An in vitro method to simulate phenolic compound release from the food matrix in the gastrointestinal tract. Euro. Food Res. Technol. 2002, 214, 155–159. [Google Scholar] [CrossRef]
- Larrosa, M.; Tomas-Barberan, F.A.; Espin, J.C. The dietary hydrolysable tannin punicalagin releases ellagic acid that induces apoptosis in human colon adenocarcinoma Caco-2 cells by using the mitochondrial pathway. J. Nutr. Biochem. 2006, 17, 611–625. [Google Scholar] [CrossRef]
- Espin, J.C.; Gonzalez-Barrio, R.; Cerda, B.; Lopez-Bote, C.; Rey, A.I.; Tomas-Barberan, F.A. Iberian pig as a model to clarify obscure points in the bioavailability and metabolism of ellagitannins in humans. J. Agric. Food Chem. 2007, 55, 10476–10485. [Google Scholar] [CrossRef]
- Walker, A.W.; Duncan, S.H.; Louis, P.; Flint, H.J. Phylogeny, culturing, and metagenomics of the human gut microbiota. Trends Microbiol. 2014, 22, 267–274. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- D’Argenio, V.; Salvatore, F. The role of the gut microbiome in the healthy adult status. Clin. Chim. Acta 2015, 451, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Onoue, M.; Kado, S.; Sakaitani, Y.; Uchida, K.; Morotomi, M. Specific species of intestinal bacteria influence the induction of aberrant crypt foci by 1,2-dimethylhydrazine in rats. Cancer Lett. 1997, 113, 179–186. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Hu, X.; Wang, T.; Jin, F. Alzheimer’s disease and gut microbiota. Sci. China Life Sci. 2016, 59, 1006–1023. [Google Scholar] [CrossRef] [Green Version]
- Solfrizzi, V.; Panza, F.; Frisardi, V.; Seripa, D.; Logroscino, G.; Imbimbo, B.P.; Pilotto, A. Diet and Alzheimer’s disease risk factors or prevention: The current evidence. Exp. Rev. Neurother. 2011, 11, 677–708. [Google Scholar] [CrossRef]
- Hu, N.; Yu, J.-T.; Tan, L.; Wang, Y.-L.; Sun, L.; Tan, L. Nutrition and the Risk of Alzheimer’s Disease. BioMed Res. Int. 2013, 2013, 524820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.C.; Jenner, A.M.; Low, C.S.; Lee, Y.K. Effect of tea phenolics and their aromatic fecal bacterial metabolites on intestinal microbiota. Res. Microbiol. 2006, 157, 876–884. [Google Scholar] [CrossRef] [PubMed]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-Generated Metabolites Promote Metabolic Benefits via Gut-Brain Neural Circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Villalba, R.; Beltran, D.; Espin, J.C.; Selma, M.V.; Tomas-Barberan, F.A. Time course production of urolithins from ellagic acid by human gut microbiota. J. Agric. Food Chem. 2013, 61, 8797–8806. [Google Scholar] [CrossRef] [PubMed]
- Selma, M.V.; Beltran, D.; Garcia-Villalba, R.; Espin, J.C.; Tomas-Barberan, F.A. Description of urolithin production capacity from ellagic acid of two human intestinal Gordonibacter species. Food Funct. 2014, 5, 1779–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bialonska, D.; Ramnani, P.; Kasimsetty, S.G.; Muntha, K.R.; Gibson, G.R.; Ferreira, D. The influence of pomegranate by-product and punicalagins on selected groups of human intestinal microbiota. Int. J. Food Microbiol. 2010, 140, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Iguchi, A.; Hatano, T. Identification of urinary and intestinal bacterial metabolites of ellagitannin geraniin in rats. J. Agric. Food Chem. 2008, 56, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-F.; Li, X.-L.; Zhang, Z.-L.; Qiu, L.; Ding, S.-X.; Xue, J.-X.; Zhao, G.-P.; Li, J. Antiaging Effects of Urolithin A on Replicative Senescent Human Skin Fibroblasts. Rejuvenation Res. 2018, 22, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.; Pierre, N.; Naslain, D.; Bontemps, F.; Ferreira, D.; Priem, F.; Deldicque, L.; Francaux, M. Urolithin B, a newly identified regulator of skeletal muscle mass. J. Cachexia Sarcopenia Muscle 2017. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Tomás-Barberán, F.; Carlos Espín, J.; Chung, S. Urolithin C, a Gut Microbiota Metabolite Derived from Ellagic Acid, Attenuates Triglyceride Accumulation in Human Adipocytes and Hepatoma Huh7 Cells. FASEB J. 2015, 29, 130.1. [Google Scholar] [CrossRef]
- Piwowarski, J.P.; Granica, S.; Stefanska, J.; Kiss, A.K. Differences in Metabolism of Ellagitannins by Human Gut Microbiota ex Vivo Cultures. J. Nat. Prod. 2016, 79, 3022–3030. [Google Scholar] [CrossRef]
- Li, Z.; Henning, S.M.; Lee, R.P.; Lu, Q.Y.; Summanen, P.H.; Thames, G.; Corbett, K.; Downes, J.; Tseng, C.H.; Finegold, S.M.; et al. Pomegranate extract induces ellagitannin metabolite formation and changes stool microbiota in healthy volunteers. Food Funct. 2015, 6, 2487–2495. [Google Scholar] [CrossRef]
- Tomas-Barberan, F.A.; Garcia-Villalba, R.; Gonzalez-Sarrias, A.; Selma, M.V.; Espin, J.C. Ellagic acid metabolism by human gut microbiota: Consistent observation of three urolithin phenotypes in intervention trials, independent of food source, age, and health status. J. Agric. Food Chem. 2014, 62, 6535–6538. [Google Scholar] [CrossRef]
- Romo-Vaquero, M.; Cortés-Martín, A.; Loria-Kohen, V.; Ramírez-de-Molina, A.; García-Mantrana, I.; Collado, M.C.; Espín, J.C.; Selma, M.V. Deciphering the Human Gut Microbiome of Urolithin Metabotypes: Association with Enterotypes and Potential Cardiometabolic Health Implications. Mol. Nutr. Food Res. 2019, 63, 1800958. [Google Scholar] [CrossRef]
- Selma, M.V.; Gonzalez-Sarrias, A.; Salas-Salvado, J.; Andres-Lacueva, C.; Alasalvar, C.; Orem, A.; Tomas-Barberan, F.A.; Espin, J.C. The gut microbiota metabolism of pomegranate or walnut ellagitannins yields two urolithin-metabotypes that correlate with cardiometabolic risk biomarkers: Comparison between normoweight, overweight-obesity and metabolic syndrome. Clin. Nutr. 2017. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Villalba, R.; Selma, M.V.; Espin, J.C.; Tomas-Barberan, F.A. Identification of Novel Urolithin Metabolites in Human Feces and Urine after the Intake of a Pomegranate Extract. J. Agric. Food Chem. 2019, 67, 11099–11107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espín, J.C.; Larrosa, M.; García-Conesa, M.T.; Tomás-Barberán, F. Biological significance of urolithins, the gut microbial ellagic acid-derived metabolites: The evidence so far. Evid.-Based Complement. Altern. Med. 2013, 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerda, B.; Llorach, R.; Ceron, J.J.; Espin, J.C.; Tomas-Barberan, F.A. Evaluation of the bioavailability and metabolism in the rat of punicalagin, an antioxidant polyphenol from pomegranate juice. Eur. J. Nutr. 2003, 42, 18–28. [Google Scholar] [CrossRef]
- Larrosa, M.; Gonzalez-Sarrias, A.; Garcia-Conesa, M.T.; Tomas-Barberan, F.A.; Espin, J.C. Urolithins, ellagic acid-derived metabolites produced by human colonic microflora, exhibit estrogenic and antiestrogenic activities. J. Agric. Food Chem. 2006, 54, 1611–1620. [Google Scholar] [CrossRef]
- Heilman, J.; Andreux, P.; Tran, N.; Rinsch, C.; Blanco-Bose, W. Safety assessment of Urolithin A, a metabolite produced by the human gut microbiota upon dietary intake of plant derived ellagitannins and ellagic acid. Food Chem. Toxicol. Biol. Res. Assoc. 2017, 108, 289–297. [Google Scholar] [CrossRef]
- Bobowska, A.; Granica, S.; Filipek, A.; Melzig, M.F.; Moeslinger, T.; Zentek, J.; Kruk, A.; Piwowarski, J.P. Comparative studies of urolithins and their phase II metabolites on macrophage and neutrophil functions. Eur. J. Nutr. 2021, 60, 1957–1972. [Google Scholar] [CrossRef]
- Bayle, M.; Roques, C.; Marion, B.; Audran, M.; Oiry, C.; Bressolle-Gomeni, F.M.; Cros, G. Development and validation of a liquid chromatography-electrospray ionization-tandem mass spectrometry method for the determination of urolithin C in rat plasma and its application to a pharmacokinetic study. J. Pharm. Biomed. Anal. 2016, 131, 33–39. [Google Scholar] [CrossRef]
- Yuan, T.; Ma, H.; Liu, W.; Niesen, D.B.; Shah, N.; Crews, R.; Rose, K.N.; Vattem, D.A.; Seeram, N.P. Pomegranate’s Neuroprotective Effects against Alzheimer’s Disease Are Mediated by Urolithins, Its Ellagitannin-Gut Microbial Derived Metabolites. ACS Chem. Neurosci. 2016, 7, 26–33. [Google Scholar] [CrossRef]
- Gasperotti, M.; Passamonti, S.; Tramer, F.; Masuero, D.; Guella, G.; Mattivi, F.; Vrhovsek, U. Fate of microbial metabolites of dietary polyphenols in rats: Is the brain their target destination? ACS Chem. Neurosci. 2015, 6, 1341–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujawska, M.; Jourdes, M.; Kurpik, M.; Szulc, M.; Szaefer, H.; Chmielarz, P.; Kreiner, G.; Krajka-Kuźniak, V.; Mikołajczak, P.; Teissedre, P.L.; et al. Neuroprotective Effects of Pomegranate Juice against Parkinson’s Disease and Presence of Ellagitannins-Derived Metabolite-Urolithin A-In the Brain. Int. J. Mol. Sci. 2020, 21, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Dadhaniya, P.; Hingorani, L.; Soni, M.G. Safety assessment of pomegranate fruit extract: Acute and subchronic toxicity studies. Food Chem. Toxicol. 2008, 46, 2728–2735. [Google Scholar] [CrossRef] [PubMed]
- Johanningsmeier, S.D.; Harris, G.K. Pomegranate as a functional food and nutraceutical source. Annu. Rev. Food Sci. Technol. 2011, 2, 181–201. [Google Scholar] [CrossRef] [PubMed]
- Gramec Skledar, D.; Tomasic, T.; Sollner Dolenc, M.; Peterlin Masic, L.; Zega, A. Evaluation of endocrine activities of ellagic acid and urolithins using reporter gene assays. Chemosphere 2019, 220, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Savi, M.; Bocchi, L.; Mena, P.; Dall’Asta, M.; Crozier, A.; Brighenti, F.; Stilli, D.; Del Rio, D. In vivo administration of urolithin A and B prevents the occurrence of cardiac dysfunction in streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2017, 16, 80. [Google Scholar] [CrossRef] [PubMed]
- Kasimsetty, S.G.; Bialonska, D.; Reddy, K.K.; Ma, G.; Khan, S.I.; Ferreira, D. Colon Cancer Chemopreventive Activities of Pomegranate Ellagitannins and Urolithins. J. Agric. Food Chem. 2010, 58, 2180–2187. [Google Scholar] [CrossRef]
- Qiu, Z.; Zhou, B.; Jin, L.; Yu, H.; Liu, L.; Liu, Y.; Zhu, F. In vitro antioxidant and antiproliferative effects of ellagic acid and its colonic metabolite, urolithins, on human bladder cancer T24 cells. Food Chem. Toxicol. 2013, 59, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Shi, F.; Guo, Z.; Zhao, J.; Song, X.; Yang, H. Metabolite of ellagitannins, urolithin A induces autophagy and inhibits metastasis in human sw620 colorectal cancer cells. Mol. Carcinog. 2018, 57, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Yin, P.; Zhang, J.; Yan, L.; Yang, L.; Sun, L.; Shi, L.; Ma, C.; Liu, Y. Urolithin C, a gut metabolite of ellagic acid, induces apoptosis in PC12 cells through a mitochondria-mediated pathway. RSC Adv. 2017, 7, 17254–17263. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Sarrias, A.; Gimenez-Bastida, J.A.; Nunez-Sanchez, M.A.; Larrosa, M.; Garcia-Conesa, M.T.; Tomas-Barberan, F.A.; Espin, J.C. Phase-II metabolism limits the antiproliferative activity of urolithins in human colon cancer cells. Eur. J. Nutr. 2014, 53, 853–864. [Google Scholar] [CrossRef]
- Colombo, E.; Sangiovanni, E.; Dell’agli, M. A review on the anti-inflammatory activity of pomegranate in the gastrointestinal tract. Evid. Based Complement. Altern. Med. 2013, 2013, 247145. [Google Scholar] [CrossRef] [Green Version]
- Larrosa, M.; Gonzalez-Sarrias, A.; Yanez-Gascon, M.J.; Selma, M.V.; Azorin-Ortuno, M.; Toti, S.; Tomas-Barberan, F.; Dolara, P.; Espin, J.C. Anti-inflammatory properties of a pomegranate extract and its metabolite urolithin-A in a colitis rat model and the effect of colon inflammation on phenolic metabolism. J. Nutr. Biochem. 2010, 21, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Neyrinck, A.M.; Van Hee, V.F.; Bindels, L.B.; De Backer, F.; Cani, P.D.; Delzenne, N.M. Polyphenol-rich extract of pomegranate peel alleviates tissue inflammation and hypercholesterolaemia in high-fat diet-induced obese mice: Potential implication of the gut microbiota. Br. J. Nutr. 2013, 109, 802–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Andreux, P.; Blanco, W.; Auwerx, J.; Rinsch, C. Urolithin A, a Gut Microbiome Derived Metabolite Improves Mitochondrial and Cellular Health: Results from a Randomized, Placebo-controlled, Double-blind Clinical Trial (FS09-06-19). Curr. Dev. Nutr. 2019, 3, FS09-06-19. [Google Scholar] [CrossRef]
- Kwak, H.M.; Jeon, S.Y.; Sohng, B.H.; Kim, J.G.; Lee, J.M.; Lee, K.B.; Jeong, H.H.; Hur, J.M.; Kang, Y.H.; Song, K.S. J3-Secretase (BACE1) Inhibitors from Pomegranate (Punica granatum) Husk. Arch. Pharm. Res. 2005, 28, 1328–1332. [Google Scholar] [CrossRef] [PubMed]
- Youn, K.; Jun, M. In vitro BACE1 inhibitory activity of geraniin and corilagin from Geranium thunbergii. Planta Med. 2013, 79, 1038–1042. [Google Scholar] [CrossRef] [Green Version]
- Mele, L.; Mena, P.; Piemontese, A.; Marino, V.; Lopez-Gutierrez, N.; Bernini, F.; Brighenti, F.; Zanotti, I.; Del Rio, D. Antiatherogenic effects of ellagic acid and urolithins in vitro. Arch. Biochem. Biophys. 2016, 599, 42–50. [Google Scholar] [CrossRef]
- Olajide, O.A.; Kumar, A.; Velagapudi, R.; Okorji, U.P.; Fiebich, B.L. Punicalagin inhibits neuroinflammation in LPS-activated rat primary microglia. Mol. Nutr. Food Res. 2014, 58, 1843–1851. [Google Scholar] [CrossRef]
- Velagapudi, R.; Baco, G.; Khela, S.; Okorji, U.; Olajide, O. Pomegranate inhibits neuroinflammation and amyloidogenesis in IL-1beta-stimulated SK-N-SH cells. Eur. J. Nutr. 2016, 55, 1653–1660. [Google Scholar] [CrossRef] [Green Version]
- Forouzanfar, F.; Afkhami Goli, A.; Asadpour, E.; Ghorbani, A.; Sadeghnia, H.R. Protective Effect of Punica granatum L. against Serum/Glucose Deprivation-Induced PC12 Cells Injury. Evid. Based Complement. Altern. Med. 2013, 2013, 716730. [Google Scholar] [CrossRef] [PubMed]
- Morzelle, M.C.; Salgado, J.M.; Telles, M.; Mourelle, D.; Bachiega, P.; Buck, H.S.; Viel, T.A. Neuroprotective Effects of Pomegranate Peel Extract after Chronic Infusion with Amyloid-beta Peptide in Mice. PLoS ONE 2016, 11, e0166123. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Lee, J.H.; Heo, H.J.; Cho, H.Y.; Kim, H.K.; Kim, C.J.; Kim, M.O.; Suh, S.H.; Shin, D.H. Punica granatum protects against oxidative stress in PC12 cells and oxidative stress-induced Alzheimer’s symptoms in mice. J. Med. Food 2011, 14, 695–701. [Google Scholar] [CrossRef]
- Subash, S.; Essa, M.M.; Al-Asmi, A.; Al-Adawi, S.; Vaishnav, R.; Braidy, N.; Manivasagam, T.; Guillemin, G.J. Pomegranate from Oman Alleviates the Brain Oxidative Damage in Transgenic Mouse Model of Alzheimer’s disease. J. Trad. Complement. Med. 2014, 4, 232–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verzelloni, E.; Pellacani, C.; Tagliazucchi, D.; Tagliaferri, S.; Calani, L.; Costa, L.G.; Brighenti, F.; Borges, G.; Crozier, A.; Conte, A.; et al. Antiglycative and neuroprotective activity of colon-derived polyphenol catabolites. Mol. Nutr. Food Res. 2011, 55, S35–S43. [Google Scholar] [CrossRef]
- DaSilva, N.A.; Nahar, P.P.; Ma, H.; Eid, A.; Wei, Z.; Meschwitz, S.; Zawia, N.H.; Slitt, A.L.; Seeram, N.P. Pomegranate ellagitannin-gut microbial-derived metabolites, urolithins, inhibit neuroinflammation in vitro. Nutr. Neurosci. 2017, 1–11. [Google Scholar] [CrossRef]
- Xu, J.; Yuan, C.; Wang, G.; Luo, J.; Ma, H.; Xu, L.; Mu, Y.; Li, Y.; Seeram, N.P.; Huang, X.; et al. Urolithins Attenuate LPS-Induced Neuroinflammation in BV2Microglia via MAPK, Akt, and NF-κB Signaling Pathways. J. Agric. Food Chem. 2018, 66, 571–580. [Google Scholar] [CrossRef]
- Bialonska, D.; Kasimsetty, S.G.; Khan, S.I.; Ferreira, D. Urolithins, intestinal microbial metabolites of Pomegranate ellagitannins, exhibit potent antioxidant activity in a cell-based assay. J. Agric. Food Chem. 2009, 57, 10181–10186. [Google Scholar] [CrossRef]
- Xu, L.; He, S.; Yin, P.; Li, D.; Mei, C.; Yu, X.; Shi, Y.; Jiang, L.; Liu, F. Punicalagin induces Nrf2 translocation and HO-1 expression via PI3K/Akt, protecting rat intestinal epithelial cells from oxidative stress. Int. J. Hyperth. 2016, 32, 465–473. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer Disease—A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef] [PubMed]
- Hamid Reza Rahimia, M.A.a.S.N.O. A Comprehensive Review of Punica granatum (Pomegranate) Properties in Toxicological, Pharmacological, Cellular and Molecular Biology Researches. Iran. J. Pharm. Res. 2012, 11, 385–400. [Google Scholar]
- Green, P.S.; Simpkins, J.W. Neuroprotective effects of estrogens: Potential mechanisms of action. Int. J. Dev. Neurosci. Off. J. Int. Soc. Develop. Neurosci. 2000, 18, 347–358. [Google Scholar] [CrossRef]
- Brann, D.W.; Dhandapani, K.; Wakade, C.; Mahesh, V.B.; Khan, M.M. Neurotrophic and Neuroprotective Actions of Estrogen: Basic Mechanisms and Clinical Implications. Steroids 2007, 72, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papoutsi, Z.; Kassi, E.; Tsiapara, A.; Fokialakis, N.; Chrousos, G.P.; Moutsatsou, P. Evaluation of estrogenic/antiestrogenic activity of ellagic acid via the estrogen receptor subtypes ERalpha and ERbeta. J. Agric. Food Chem. 2005, 53, 7715–7720. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, J.H.; Aguilera-Barrantes, I.; Shiau, C.W.; Sheng, X.; Wang, L.S.; Stoner, G.D.; Huang, Y.W. Urolithin A suppresses the proliferation of endometrial cancer cells by mediating estrogen receptor-alpha-dependent gene expression. Mol. Nutr. Food Res. 2016, 60, 2387–2395. [Google Scholar] [CrossRef] [PubMed]
- Rojo, L.; Sjöberg, M.K.; Hernández, P.; Zambrano, C.; Maccioni, R.B. Roles of Cholesterol and Lipids in the Etiopathogenesis of Alzheimer’s Disease. J. Biomed. Biotechnol. 2006, 2006, 73976. [Google Scholar] [CrossRef] [Green Version]
- Ghribi, O. Potential Mechanisms Linking Cholesterol to Alzheimer’s Disease-like Pathology in Rabbit Brain, Hippocampal Organotypic Slices, and Skeletal Muscle. J. Alzheimer’s Dis. JAD 2008, 15, 673–684. [Google Scholar] [CrossRef]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, J.L.; Lee, E.S.; Han, S.Y.; Gong, J.H.; Kang, M.K.; Kang, Y.H. Dietary ellagic acid attenuates oxidized LDL uptake and stimulates cholesterol efflux in murine macrophages. J. Nutr. 2011, 141, 1931–1937. [Google Scholar] [CrossRef] [Green Version]
- Toney, A.M.; Fan, R.; Xian, Y.; Chaidez, V.; Ramer-Tait, A.E.; Chung, S. Urolithin A, a Gut Metabolite, Improves Insulin Sensitivity Through Augmentation of Mitochondrial Function and Biogenesis. Obesity 2019, 27, 612–620. [Google Scholar] [CrossRef]
- D’Amico, D.; Andreux, P.A.; Valdés, P.; Singh, A.; Rinsch, C.; Auwerx, J. Impact of the Natural Compound Urolithin A on Health, Disease, and Aging. Trends Mol. Med. 2021, 27, 687–699. [Google Scholar] [CrossRef]
- Webb, M. A Novel Mitophagy Assay for Skeletal Myotubes. Open Access J. Neurol. Neurosurg. 2017, 4. [Google Scholar] [CrossRef]
- Zhao, C.; Sakaguchi, T.; Fujita, K.; Ito, H.; Nishida, N.; Nagatomo, A.; Tanaka-Azuma, Y.; Katakura, Y. Pomegranate-Derived Polyphenols Reduce Reactive Oxygen Species Production via SIRT3-Mediated SOD2 Activation. Oxid. Med. Cell. Longev. 2016, 2016, 2927131. [Google Scholar] [CrossRef] [Green Version]
- Boakye, Y.D.; Groyer, L.; Heiss, E.H. An increased autophagic flux contributes to the anti-inflammatory potential of urolithin A in macrophages. Biochim. Biophys. Acta (BBA) Gen. Subj. 2018, 1862, 61–70. [Google Scholar] [CrossRef]
- Tan, S.; Wong, E. Mitophagy Transcriptome: Mechanistic Insights into Polyphenol-Mediated Mitophagy. Oxid. Med. Cell. Longev. 2017, 2017, 9028435. [Google Scholar] [CrossRef]
- Song, D.; Ma, J.; Chen, L.; Guo, C.; Zhang, Y.; Chen, T.; Zhang, S.; Zhu, Z.; Tian, L.; Niu, P. FOXO3 promoted mitophagy via nuclear retention induced by manganese chloride in SH-SY5Y cells. Met. Integr. Biometal Sci. 2017, 9, 1251–1259. [Google Scholar] [CrossRef]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef]
- Pillai, V.B.; Sundaresan, N.R.; Kim, G.; Gupta, M.; Rajamohan, S.B.; Pillai, J.B.; Samant, S.; Ravindra, P.V.; Isbatan, A.; Gupta, M.P. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J. Bol. Chem. 2010, 285, 3133–3144. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, N.; Das, A.; Biswas, N.; Gnyawali, S.; Singh, K.; Gorain, M.; Polcyn, C.; Khanna, S.; Roy, S.; Sen, C.K. Urolithin A augments angiogenic pathways in skeletal muscle by bolstering NAD+ and SIRT1. Sci. Rep. 2020, 10, 20184. [Google Scholar] [CrossRef]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Ren, H.; Du, H.; Zhang, M.; Xiong, X.; Lv, R. Liraglutide repairs the infarcted heart: The role of the SIRT1/Parkin/mitophagy pathway. Mol. Med. Rep. 2018, 17, 3722–3734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2011, 331, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Z.; Huang, J.; Xu, B.; Ou, Z.; Zhang, L.; Lin, X.; Ye, X.; Kong, X.; Long, D.; Sun, X.; et al. Urolithin A attenuates memory impairment and neuroinflammation in APP/PS1 mice. J. Neuroinflamm. 2019, 16, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raben, N.; Puertollano, R. TFEB and TFE3: Linking Lysosomes to Cellular Adaptation to Stress. Annu. Rev. Cell Dev. Biol. 2016, 32, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; Kumsta, C.; Sandri, M.; Ballabio, A.; Hansen, M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 2015, 11, 867–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Füllgrabe, J.; Klionsky, D.J.; Joseph, B. The return of the nucleus: Transcriptional and epigenetic control of autophagy. Nat. Rev. Mol. Cell Biol. 2014, 15, 65–74. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The Transcription Factor TFEB Links mTORC1 Signaling to Transcriptional Control of Lysosome Homeostasis. Sci. Sign. 2012, 5, ra42. [Google Scholar] [CrossRef] [Green Version]
- Nezich, C.L.; Wang, C.; Fogel, A.I.; Youle, R.J. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J. Cell Biol. 2015, 210, 435–450. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Yu, C.Y.; Sim, Z.W.; Low, Z.S.; Lee, B.; See, F.; Min, N.; Gautam, A.; Chu, J.J.H.; Ng, K.W.; et al. Pomegranate activates TFEB to promote autophagy-lysosomal fitness and mitophagy. Sci. Rep. 2019, 9, 727. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef]
- Totiger, T.M.; Srinivasan, S.; Jala, V.R.; Lamichhane, P.; Dosch, A.R.; Gaidarski, A.A.; Joshi, C.; Rangappa, S.; Castellanos, J.; Vemula, P.K.; et al. Urolithin A, a novel natural compound to target PI3K/AKT/mTOR pathway in pancreatic cancer. Mol. Cancer Ther. 2018. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, W.; Kishi, H.; Yagasaki, K.; Ohhira, S. Urolithin A attenuates pro-inflammatory mediator production by suppressing PI3-K/Akt/NF-κB and JNK/AP-1 signaling pathways in lipopolysaccharide-stimulated RAW264 macrophages: Possible involvement of NADPH oxidase-derived reactive oxygen species. Eur. J. Pharmacol. 2018, 833, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Mo, Y.; Li, Y.; Zhong, Y.; He, S.; Zhang, Y.; Tang, Y.; Fu, S.; Wang, X.; Chen, A. Urolithin A alleviates myocardial ischemia/reperfusion injury via PI3K/Akt pathway. Biochem. Biophys. Res. Commun. 2017, 486, 774–780. [Google Scholar] [CrossRef]
- Das, S.; Mitrovsky, G.; Vasanthi, H.R.; Das, D.K. Antiaging properties of a grape-derived antioxidant are regulated by mitochondrial balance of fusion and fission leading to mitophagy triggered by a signaling network of Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid. Med. Cell. Longev. 2014, 2014, 345105. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.M.; Pennington, J.D.; Bisht, K.S.; Aykin-Burns, N.; Kim, H.S.; Mishra, M.; Sun, L.; Nguyen, P.; Ahn, B.H.; Leclerc, J.; et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int. J. Biol. Sci. 2008, 4, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soutar, M.P.M.; Kempthorne, L.; Miyakawa, S.; Annuario, E.; Melandri, D.; Harley, J.; O’Sullivan, G.A.; Wray, S.; Hancock, D.C.; Cookson, M.R.; et al. AKT signalling selectively regulates PINK1 mitophagy in SHSY5Y cells and human iPSC-derived neurons. Sci. Rep. 2018, 8, 8855. [Google Scholar] [CrossRef] [Green Version]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).