
Co-Encapsulation of Methylene Blue and PARP-Inhibitor into Poly(Lactic-Co-Glycolic Acid) Nanoparticles for Enhanced PDT of Cancer



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of PLGA NPs
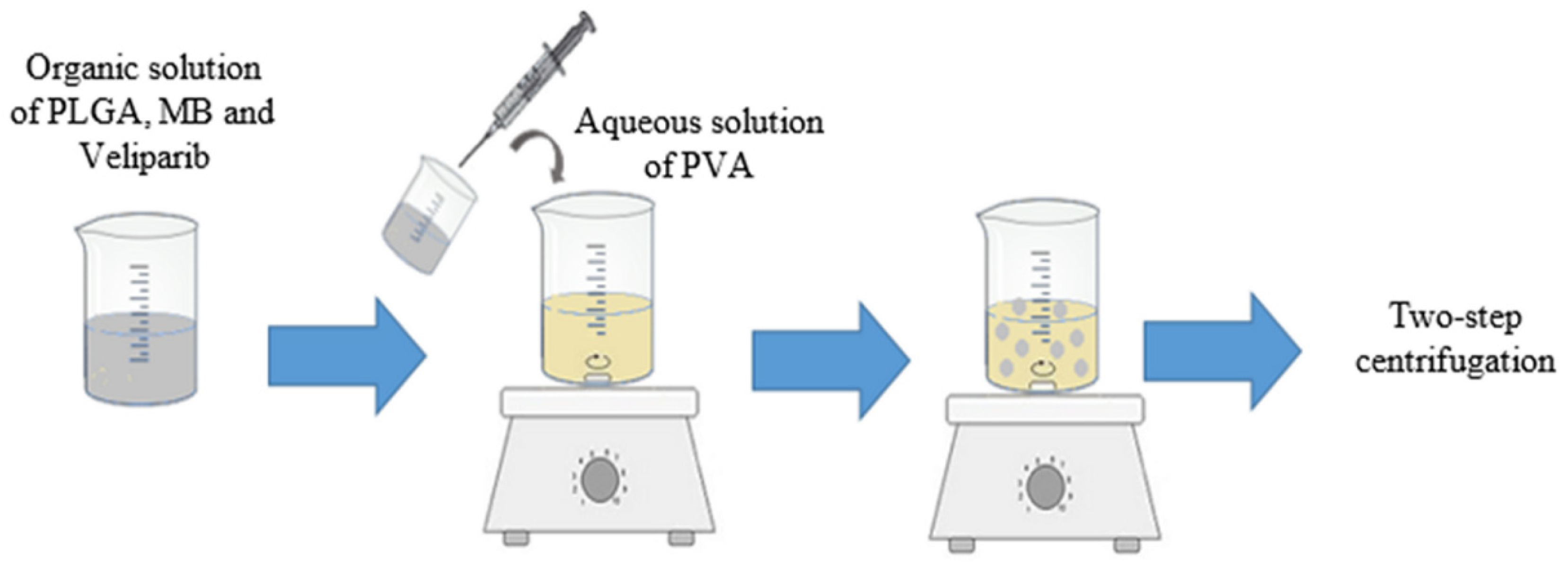
2.3. Synthesis of PLGA NPs with MB and Veliparib Co-Loaded (VMB-NPs)
2.4. Evaluation of NPs Physicochemical Properties
2.5. Determination of Encapsulation Efficiency (%EE)
2.6. In Vitro Drug Release
2.7. Cell Viability Assays
3. Results and Discussion
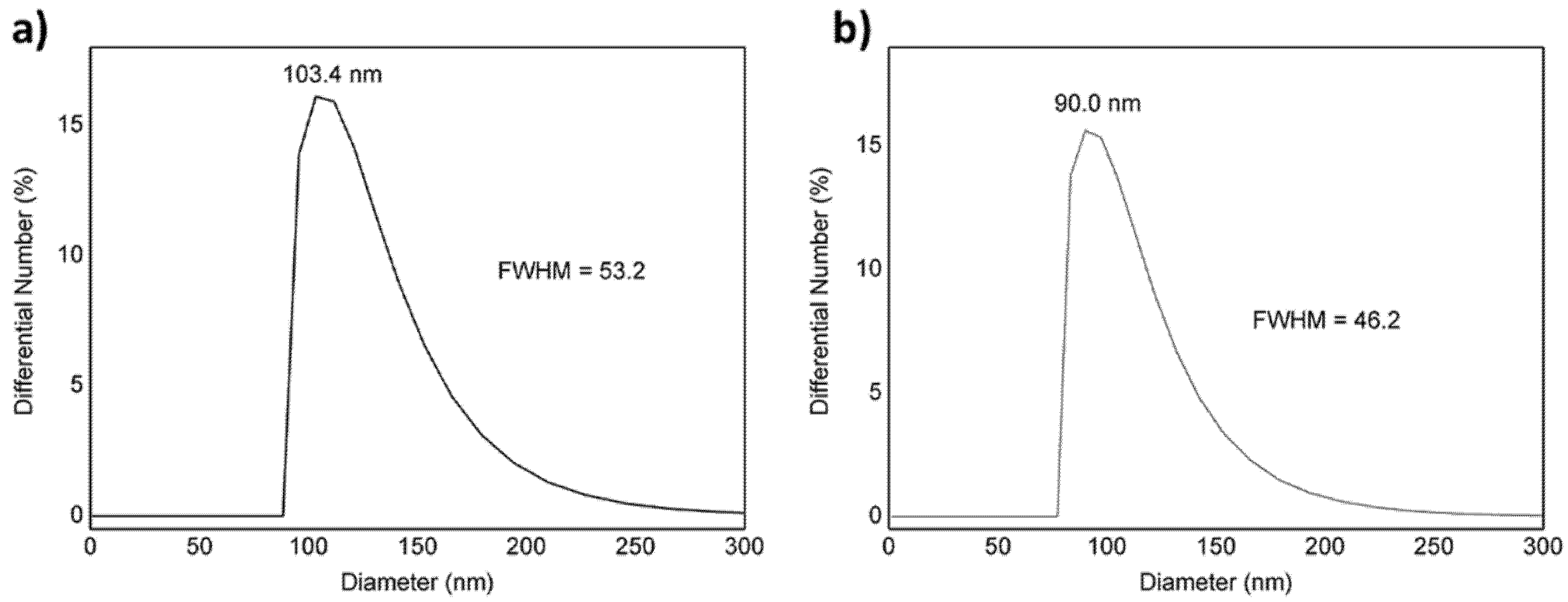
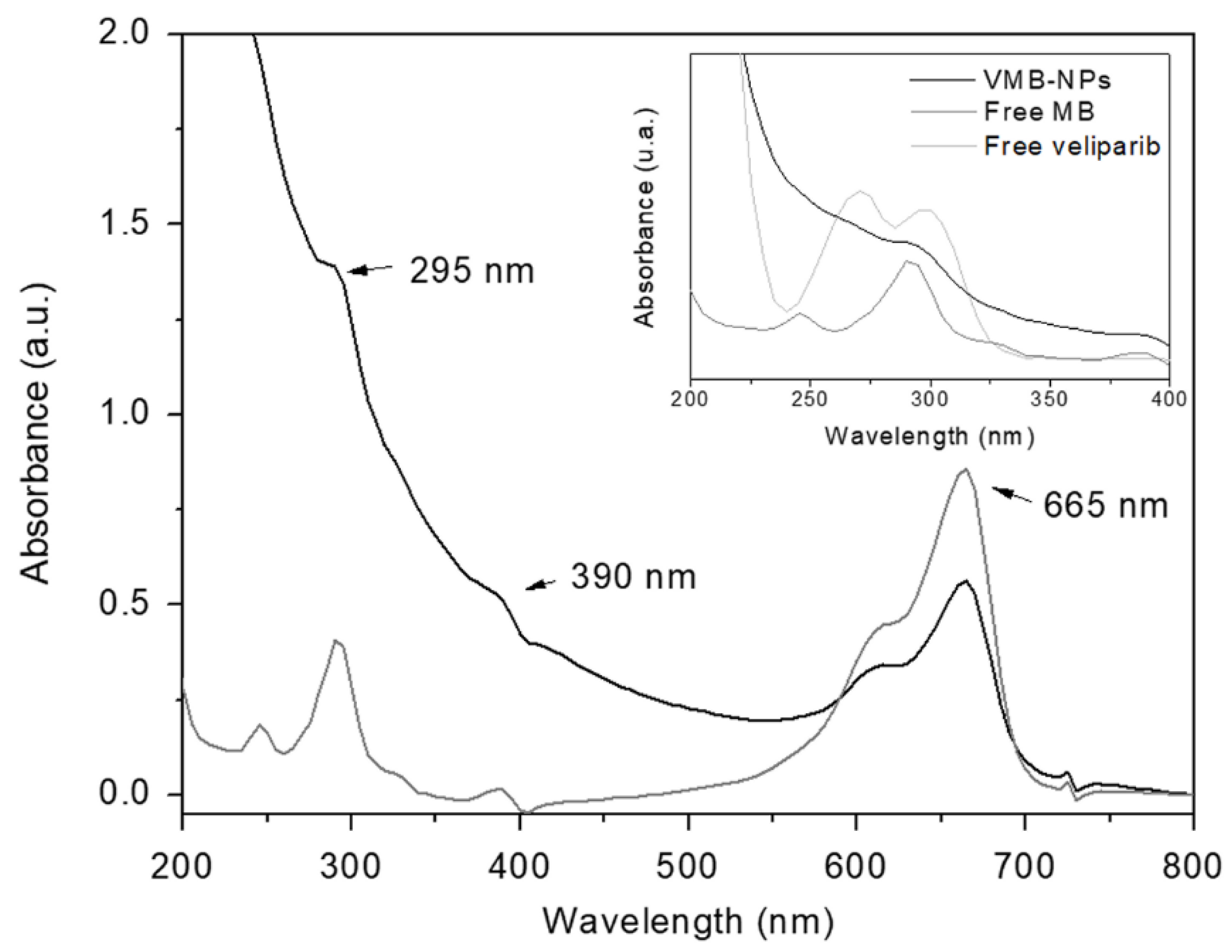
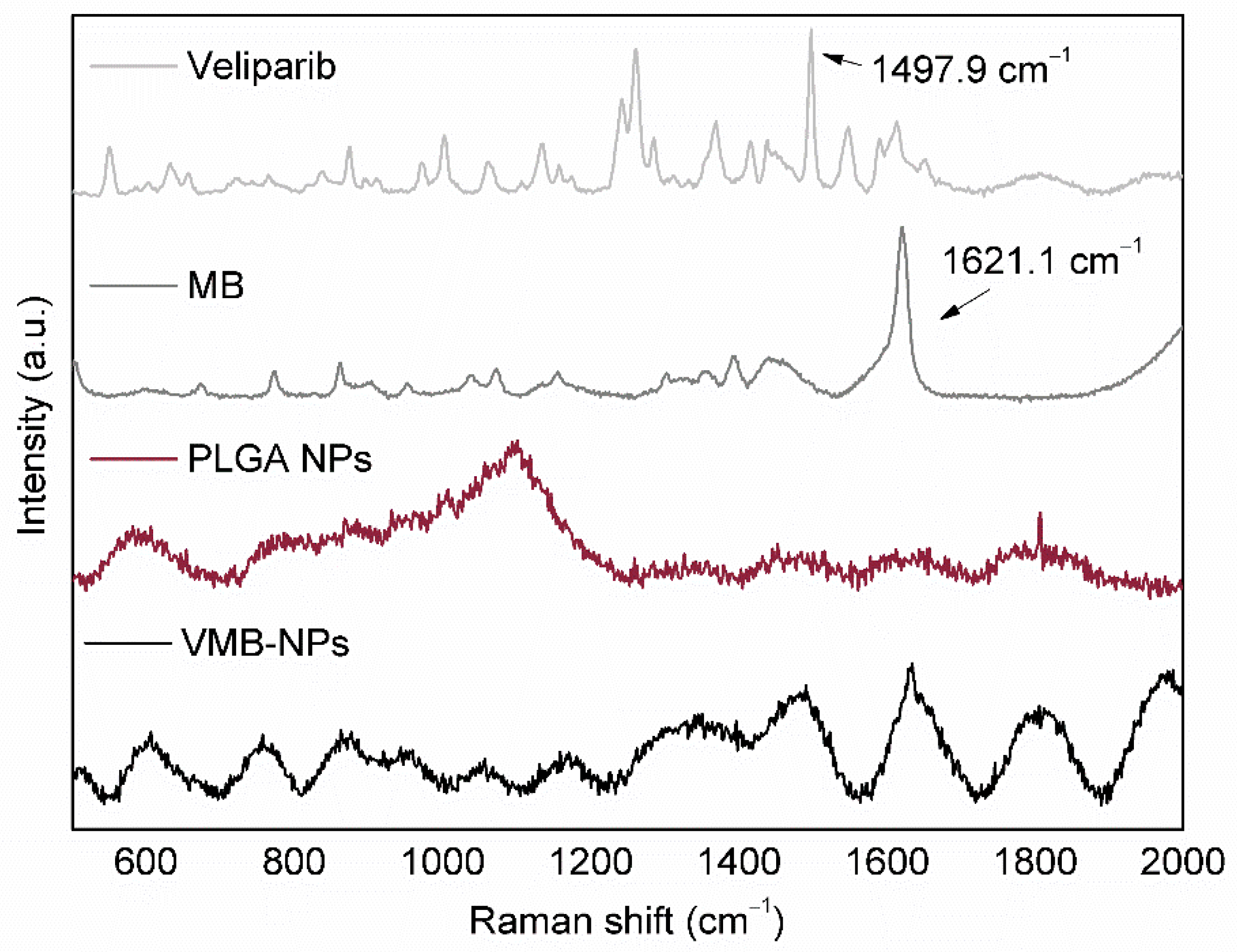
3.1. Physicochemical Properties
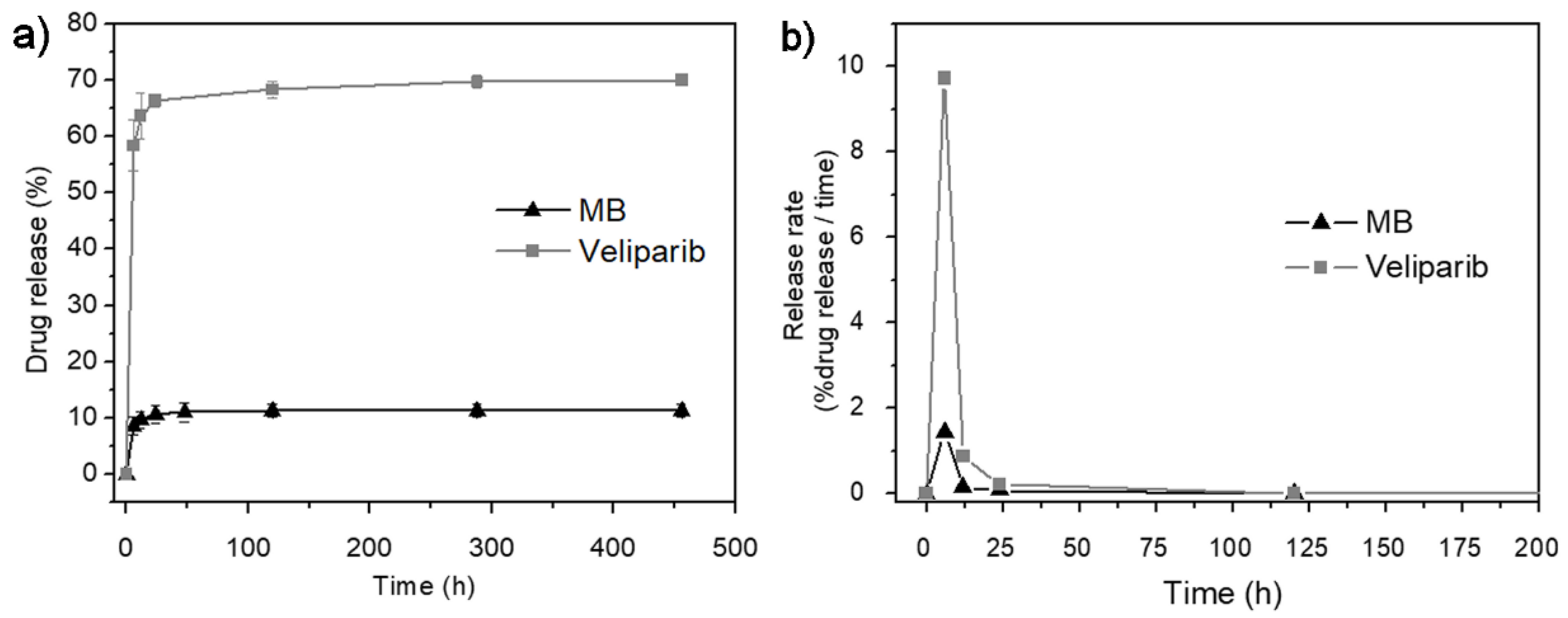
3.2. In Vitro Drug-Release
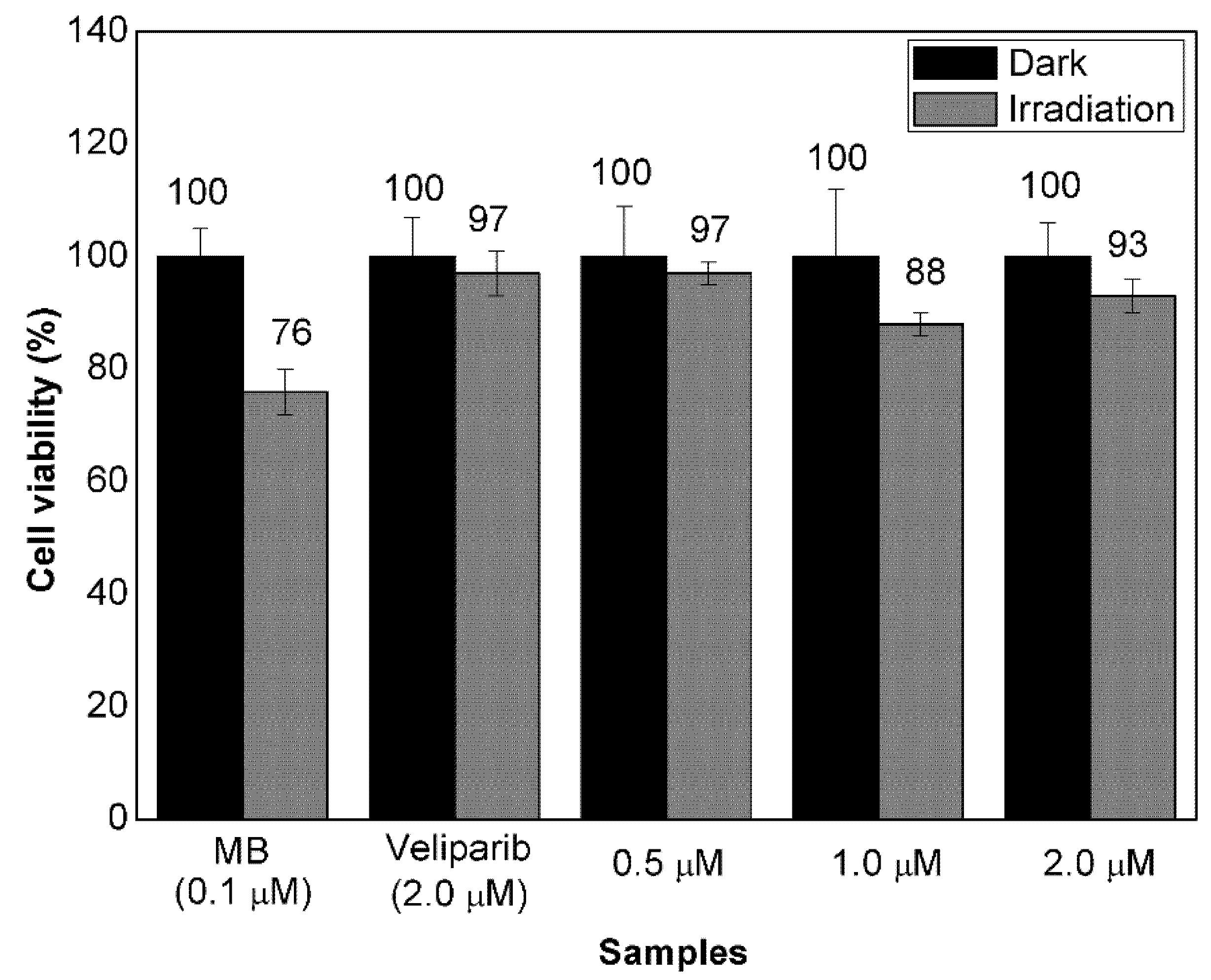
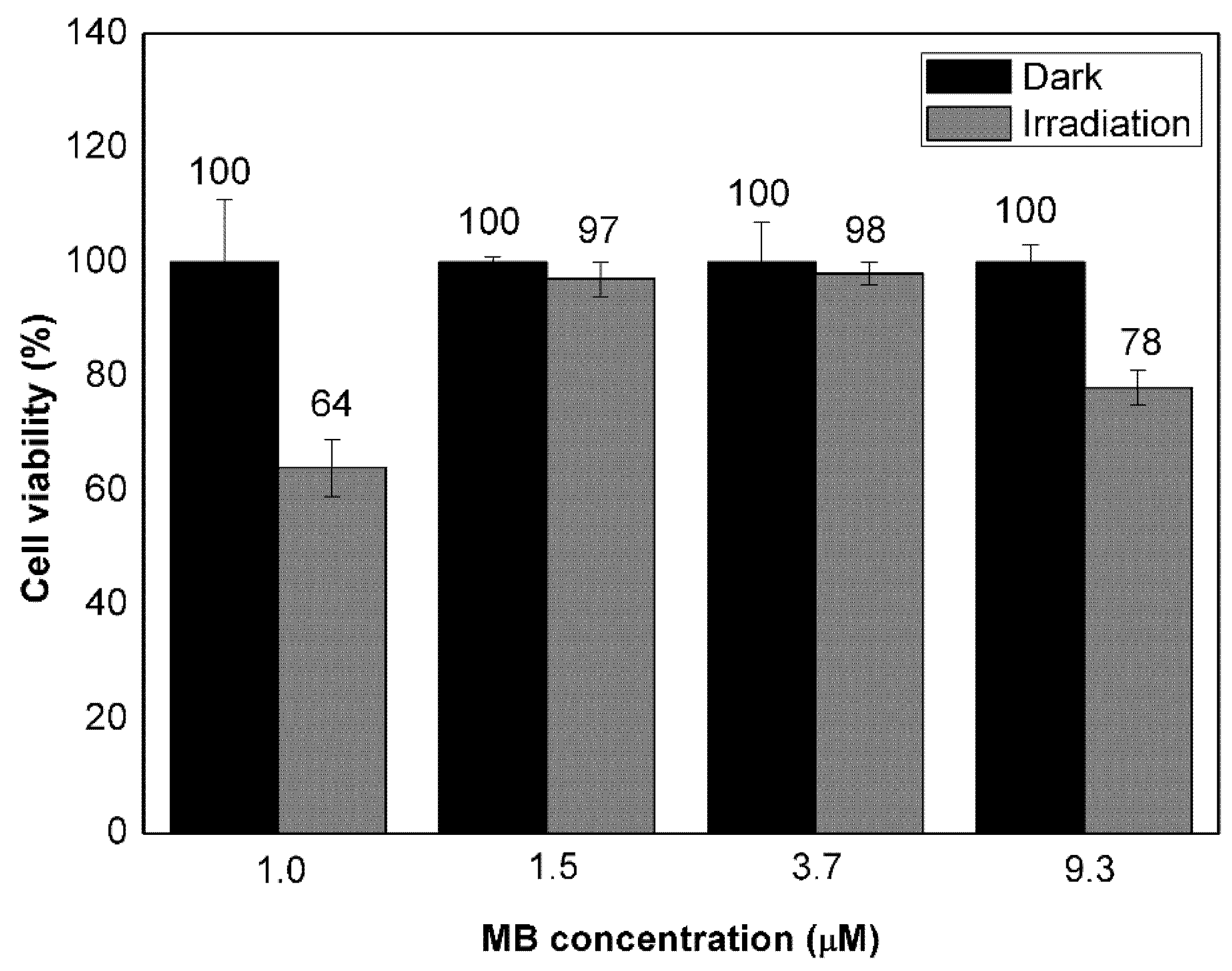
3.3. Evaluation of Cell Response to Non-Encapsulated Molecules and VMB-NPs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McFarland, S.A.; Mandel, A.; Dumoulin-White, R.; Gasser, G. Metal-based photosensitizers for photodynamic therapy: The future of multimodal oncology? Curr. Opin. Chem. Biol. 2020, 56, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Qidwai, A.; Nabi, B.; Kotta, S.; Narang, J.K.; Baboota, S.; Ali, J. Role of nanocarriers in photodynamic therapy. Photodiagn. Photodyn. Ther. 2020, 30, 101782. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gao, Y.; Liu, N.; Suo, Y. Nanoparticles Loading Porphyrin Sensitizers in Improvement of Photodynamic Therapy for Ovarian Cancer. Photodiagn. Photodyn. Ther. 2020, 33, 102156. [Google Scholar] [CrossRef]
- Najlah, M.; Ahmed, Z.; Iqbal, M.; Wang, Z.; Tawari, P.; Wang, W.; McConville, C. Development and characterisation of disulfiram-loaded PLGA nanoparticles for the treatment of non-small cell lung cancer. Eur. J. Pharm. Biopharm. 2017, 112, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C 2019, 98, 1252–1276. [Google Scholar] [CrossRef]
- Acharya, S.; Sahoo, S.K. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Fukumori, Y.; Ichikawa, H. Nanoparticles for cancer therapy and diagnosis. Adv. Powder Technol. 2006, 17, 1–28. [Google Scholar] [CrossRef]
- Sanna, V.; Pala, N.; Sechi, M. Targeted therapy using nanotechnology: Focus on cancer. Int. J. Nanomed. 2014, 9, 467–483. [Google Scholar] [CrossRef]
- Nakamura, Y.; Mochida, A.; Choyke, P.L.; Kobayashi, H. Nanodrug delivery: Is the enhanced permeability and retention effect sufficient for curing cancer? Bioconjug. Chem. 2016, 27, 2225–2238. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Tang, K.; Hou, Y.; Yu, J.; Wang, C.; Wang, Y. Fabrication of core/shell/shell structure nanoparticle with anticancer drug and dual-photosensitizer co-loading for synergistic chemotherapy and photodynamic therapy. Microporous Mesoporous Mater. 2020, 297, 110049. [Google Scholar] [CrossRef]
- Doustvandi, M.A.; Mohammadnejad, F.; Mansoori, B.; Tajalli, H.; Mohammadi, A.; Mokhtarzadeh, A.; Baghbani, E.; Khaze, V.; Hajiasgharzadeh, K.; Moghaddam, M.M.; et al. Photodynamic therapy using zinc phthalocyanine with low dose of diode laser combined with doxorubicin is a synergistic combination therapy for human SK-MEL-3 melanoma cells. Photodiagn. Photodyn.Ther. 2019, 28, 88–97. [Google Scholar] [CrossRef]
- Soriano, J.; Mora-Espí, I.; Alea-Reyes, M.E.; Pérez-García, L.; Barrios, L.; Ibáñez, E.; Nogués, C. Cell Death Mechanisms in Tumoral and Non-Tumoral Human Cell Lines Triggered by Photodynamic Treatments: Apoptosis, Necrosis and Parthanatos. Sci. Rep. 2017, 7, 41340. [Google Scholar] [CrossRef]
- Kessel, D.; Oleinick, N.L. Cell Death Pathways Associated with Photodynamic Therapy: An Update. Photochem. Photobiol. 2018, 94, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Martins, W.K.; Belotto, R.; Silva, M.N.; Grasso, D.; Suriani, M.D.; Lavor, T.S.; Itri, R.; Baptista, M.S.; Tsubone, T.M. Autophagy Regulation and Photodynamic Therapy: Insights to Improve Outcomes of Cancer Treatment. Front. Oncol. 2021, 10, 3121. [Google Scholar] [CrossRef]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part two—Cellular signaling, cell metabolism and modes of cell death. Photodiagn. Photodyn. Ther. 2005, 2, 1–23. [Google Scholar] [CrossRef]
- Martins, W.K.; Santos, N.F.; Rocha, C.S.; Bacellar, I.O.L.; Tsubone, T.M.; Viotto, A.C.; Matsukuma, A.Y.; Abrantes, A.B.P.; Siani, P.; Dias, L.G.; et al. Parallel damage in mitochondria and lysosomes is an efficient way to photoinduce cell death. Autophagy 2019, 15, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D.; Evans, C.L. Promotion of Pro-Apoptotic Signals by Lysosomal Photodamage: Mechanistic Aspects and Influence of Autophagy. Photochem. Photobiol. 2016, 92, 620–623. [Google Scholar] [CrossRef]
- Kessel, D. Sub-cellular Targeting as a Determinant of the Efficacy of Photodynamic Therapy. Photochem. Photobiol. 2017, 93, 609–612. [Google Scholar] [CrossRef]
- Kessel, D.; Reiners, J.J., Jr. Enhanced Efficacy of Photodynamic Therapy via a Sequential Targeting Protocol. Photochem. Photobiol. 2014, 90, 889–895. [Google Scholar] [CrossRef]
- Casas, A.; Di Venosa, G.; Hasan, T.; Batlle, A. Mechanisms of resistance to photodynamic therapy. Curr. Med. Chem. 2011, 18, 2486–2515. [Google Scholar] [CrossRef]
- Shanmugapriya, K.; Kim, H.; Kang, H.W. Nanoengineered chlorin e6 conjugated with hydrogel for photodynamic therapy on cancer. Colloids Surf. B Biointerfaces 2019, 181, 778–788. [Google Scholar] [CrossRef]
- Scalfi-Happ, C.; Zhu, Z.; Graefe, S.; Wiehe, A.; Ryabova, A.; Loschenov, V.; Wittig, R.; Steiner, R.W. Chlorin nanoparticles for tissue diagnostics and photodynamic therapy. Photodiagn. Photodyn. 2018, 22, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M.; Sun, J.; Payne, P.W. Activation of poly (adenosine di-phosphate-ribose) polymerase in mouse tumors treated by photodynamic therapy. Photochem. Photobiol. 2003, 78, 400–406. [Google Scholar] [CrossRef]
- Kim, J.; Lim, W.; Kim, S.; Jeon, S.; Hui, Z.; Ni, K.; Kim, C.; Im, Y.; Choi, H.; Kim, O. Photodynamic therapy (PDT) resistance by PARP1 regulation on PDT-induced apoptosis with autophagy in head and neck cancer cells. J. Oral Pathol. Med. 2014, 43, 675–684. [Google Scholar] [CrossRef]
- Kästle, M.; Grimm, S.; Nagel, R.; Breusing, N.; Grune, T. Combination of PDT and inhibitor treatment affects melanoma cells and spares keratinocytes. Free Radic. Biol. Med. 2011, 50, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.J.; Oak, C.H.; Heo, J.; Kim, Y.H. Methylene blue-mediated photodynamic therapy enhances apoptosis in lung cancer cells. Oncol. Rep. 2013, 30, 856–862. [Google Scholar] [CrossRef]
- Arruda, D.C.; de Oliveira, T.D.; Cursino, P.H.F.; Maia, V.S.C.; Berzaghi, R.; Travassos, L.R.; Tada, D.B. Inhibition of melanoma metastasis by dual-peptide PLGA NPs. Biopolymers 2017, 108, e23029. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; LeBreton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Almoustafa, H.A.; Alshawsh, M.A.; Chik, Z. Technical aspects of preparing PEG-PLGA nanoparticles as carrier for chemotherapeutic agents by nanoprecipitation method. Int. J. Pharm. 2017, 533, 275–284. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Groshev, A.; Zhang, J.; Grover, A.; Sutariya, V.B. Preparation and characterization of methylene blue nanoparticles for Alzheimer’s disease and other tauopathies. Curr. Drug Deliv. 2014, 11, 541–550. [Google Scholar] [CrossRef]
- Bergmann, K.; O’Konski, C.T. A spectroscopy study of methylene blue monomer, dimer, and complexes with montmorillonite. J. Phys. Chem. 1963, 67, 2169–2177. [Google Scholar] [CrossRef]
- Hong, E.J.; Choi, D.G.; Shim, M.S. Targeted and effective photodynamic therapy for cancer using functionalized nanomaterials. Acta Pharm. Sin. B 2016, 6, 297–307. [Google Scholar] [CrossRef]
- Rui, L.L.; Cao, H.L.; Xue, Y.D.; Liu, L.C.; Xu, L.; Gao, Y.; Zhang, W.A. Functional organic nanoparticles for photodynamic therapy. Chin. Chem. Lett. 2016, 27, 1412–1420. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, C. Tuning the size of poly (lactic-co-glycolic acid) (PLGA) nanoparticles fabricated by nanoprecipitation. Biotechnol. J. 2018, 13, 1700203. [Google Scholar] [CrossRef]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef]
- Sadat, S.M.A.; Jahan, S.; Haddadi, A. Effects of size and surface charge of polymeric nanoparticles on in vitro and in vivo applications. J. Biomater. Nanobiotechnol. 2016, 7, 91–108. [Google Scholar] [CrossRef]
- Klepac-Ceraj, V.; Patel, N.; Song, X.; Holewa, C.; Patel, C.; Kent, R.; Amiji, M.M.; Soukos, N.S. Photodynamic effects of methylene blue-loaded polymeric nanoparticles on dental plaque bacteria. Lasers Surg. Med. 2011, 43, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Valenzuela, C.A.; Esquivel, R.; Guerrero-Germán, P.; Zavala-Rivera, P.; Rodríguez-Figueroa, J.C.; Guzmán-Z, R.; Lucero-Acuña, A. Evaluation of a combined emulsion process to encapsulate methylene blue into PLGA nanoparticles. RSC Adv. 2018, 8, 414–422. [Google Scholar] [CrossRef]
- Castañeda-Gill, J.M.; Ranjan, A.P.; Vishwanatha, J.K. Development and Characterization of Methylene Blue Oleate Salt-Loaded Polymeric Nanoparticles and their Potential Application as a Treatment for Glioblastoma. J. Nanomed. Nanotechnol. 2017, 8, 449. [Google Scholar] [CrossRef]
- Muñoz-Gamez, J.A.; Viota, J.L.; Barrientos, A.; Carazo, Á.; Sanjuán-Nuñez, L.; Quiles-Perez, R.; Muñoz-de-Rueda, P.; Delgado, Á.; Ruiz-Extremera, A.; Salmerón, J. Synergistic cytotoxicity of the poly (ADP-ribose) polymerase inhibitor ABT-888 and temozolomide in dual-drug targeted magnetic nanoparticles. Liver Int. 2015, 35, 1430–1441. [Google Scholar] [CrossRef]
- Hines, D.J.; Kaplan, D.L. Poly (lactic-co-glycolic acid) controlled release systems: Experimental and modeling insights. Crit. Rev. Ther. Drug Carr. Syst. 2013, 30, 257–276. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.L. Mathematical models of drug release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Woodhead Publishing: Cambridge, UK, 2015; pp. 63–86. [Google Scholar]
- Chiarelli-Neto, O.; Pavani, C.; Ferreira, A.S.; Uchoa, A.F.; Severino, D.; Baptista, M.S. Generation and suppression of singlet oxygen in hair by photosensitization of melanin. Free Radic. Biol. Med. 2011, 51, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.M. Profile of veliparib and its potential in the treatment of solid tumors. Onco Targets Ther. 2015, 8, 1931–1939. [Google Scholar] [CrossRef]
- Stewart, E.; Goshorn, R.; Bradley, C.; Griffiths, L.M.; Benavente, C.; Twarog, N.R.; Miller, G.M.; Caufield, W.; Freeman, B.B.; Bahrami, A.; et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep. 2014, 9, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Hydrodynamic Diameter (nm) | PDI | Zeta Potential (mV) | Encapsulation Efficiency (%) | |
|---|---|---|---|---|---|
| PLGA NPs | 103.4 | 0.07 ± 0.03 | −6.8 ± 0.6 | ||
| VMB-NPs | 90.0 | 0.08 ± 0.03 | −3.7 ± 0.2 | MB 23 | Veliparib 58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magalhães, J.A.; Arruda, D.C.; Baptista, M.S.; Tada, D.B. Co-Encapsulation of Methylene Blue and PARP-Inhibitor into Poly(Lactic-Co-Glycolic Acid) Nanoparticles for Enhanced PDT of Cancer. Nanomaterials 2021, 11, 1514. https://doi.org/10.3390/nano11061514
Magalhães JA, Arruda DC, Baptista MS, Tada DB. Co-Encapsulation of Methylene Blue and PARP-Inhibitor into Poly(Lactic-Co-Glycolic Acid) Nanoparticles for Enhanced PDT of Cancer. Nanomaterials. 2021; 11(6):1514. https://doi.org/10.3390/nano11061514
Chicago/Turabian StyleMagalhães, Jéssica A., Denise C. Arruda, Maurício S. Baptista, and Dayane B. Tada. 2021. "Co-Encapsulation of Methylene Blue and PARP-Inhibitor into Poly(Lactic-Co-Glycolic Acid) Nanoparticles for Enhanced PDT of Cancer" Nanomaterials 11, no. 6: 1514. https://doi.org/10.3390/nano11061514
APA StyleMagalhães, J. A., Arruda, D. C., Baptista, M. S., & Tada, D. B. (2021). Co-Encapsulation of Methylene Blue and PARP-Inhibitor into Poly(Lactic-Co-Glycolic Acid) Nanoparticles for Enhanced PDT of Cancer. Nanomaterials, 11(6), 1514. https://doi.org/10.3390/nano11061514