

Protein Is an Intelligent Micelle



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Stages Contained in the Biological Dogma
- 1.
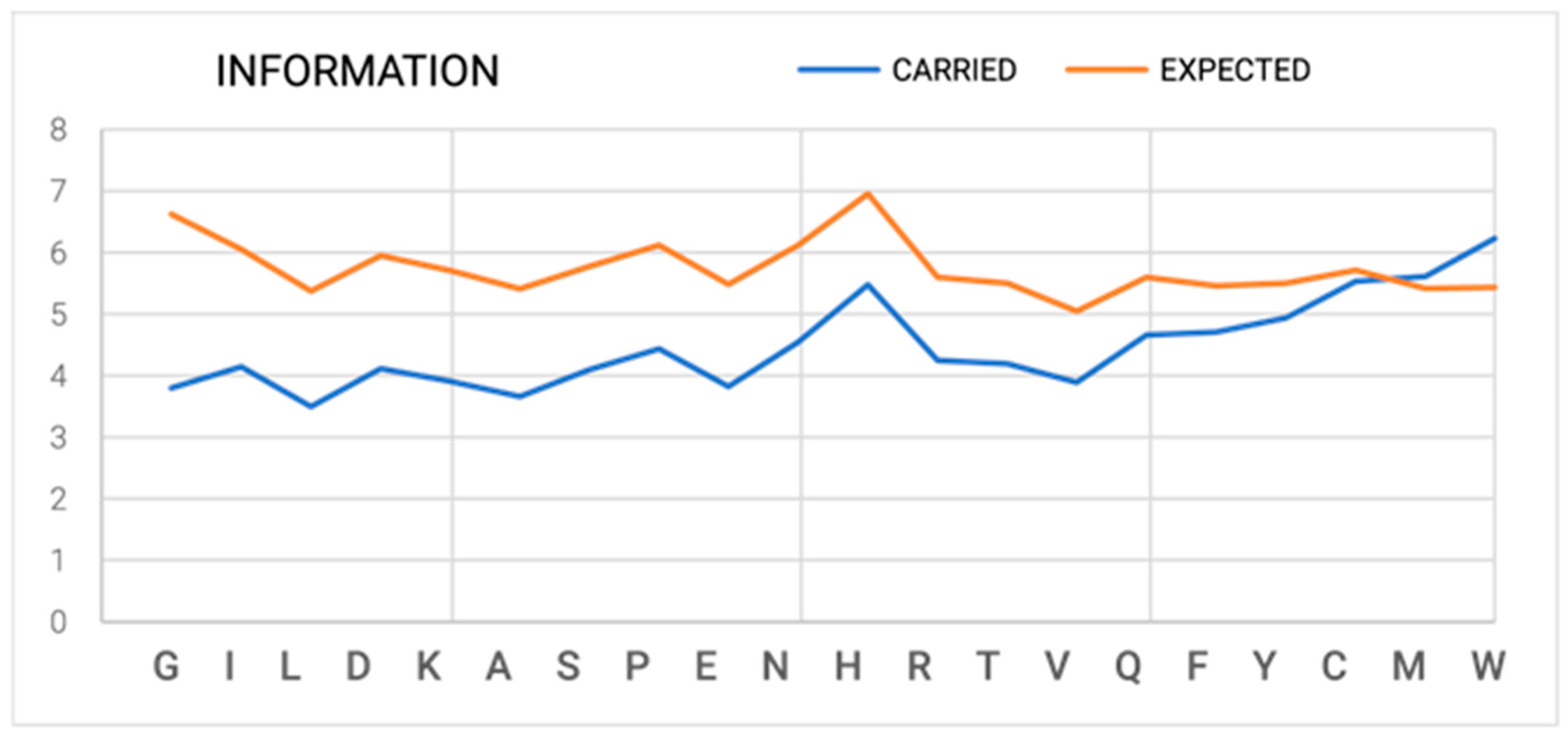
- Step 1—Amount of Information in DNA
- 2.
- Step 2—mRNA → AA
2.1. Step 3: Amino Acid Sequence (AA) → 3D Structure
2.1.1. Interpretation of Phi, Psi Angles Distribution on Ramachandran Map
2.1.2. Additional Source of Information: The Environment
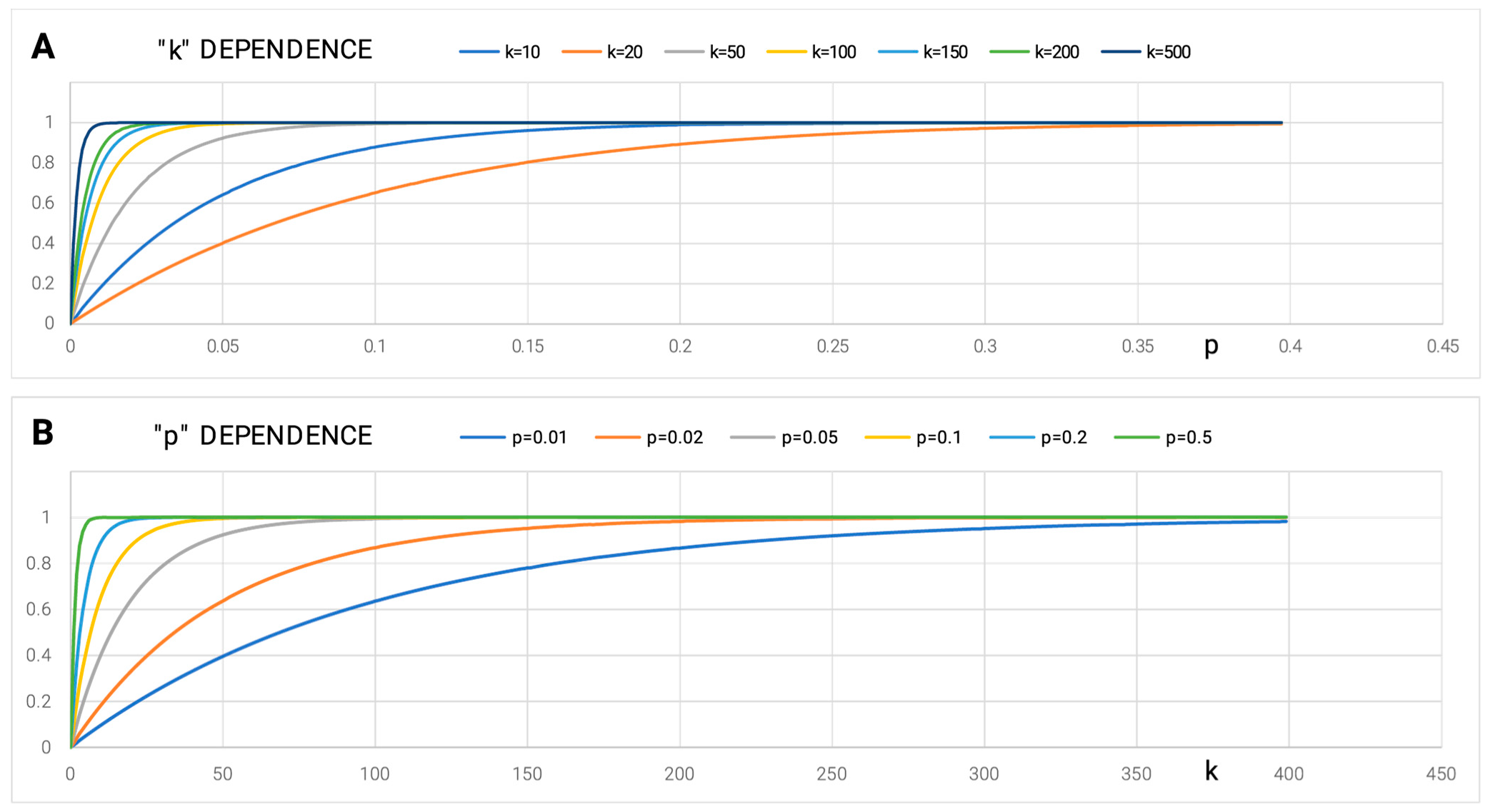
2.1.3. Strategies of Representing Information Deficiency at the Protein Folding Stage
2.2. Environment Participation in the Folding Process
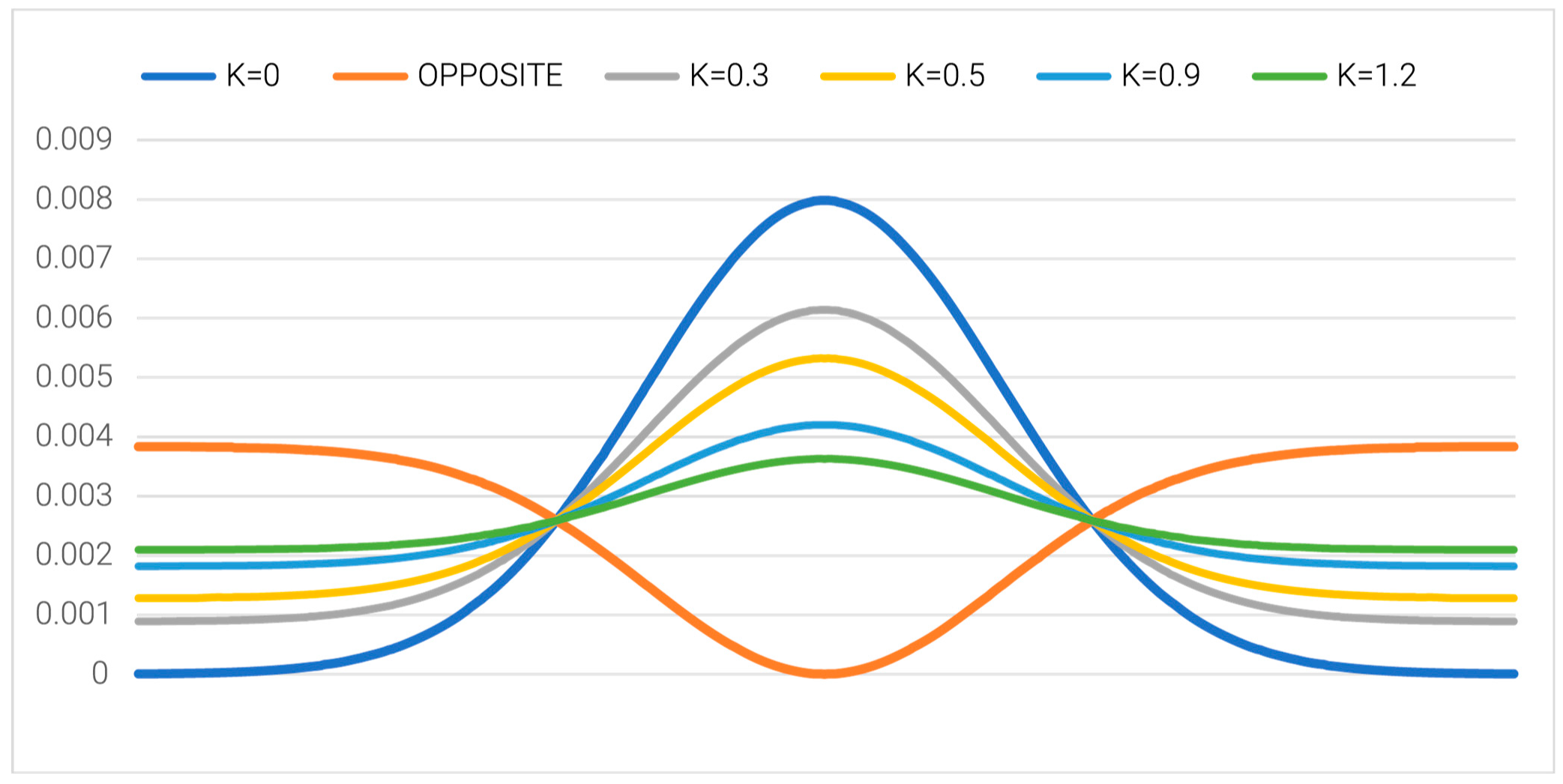
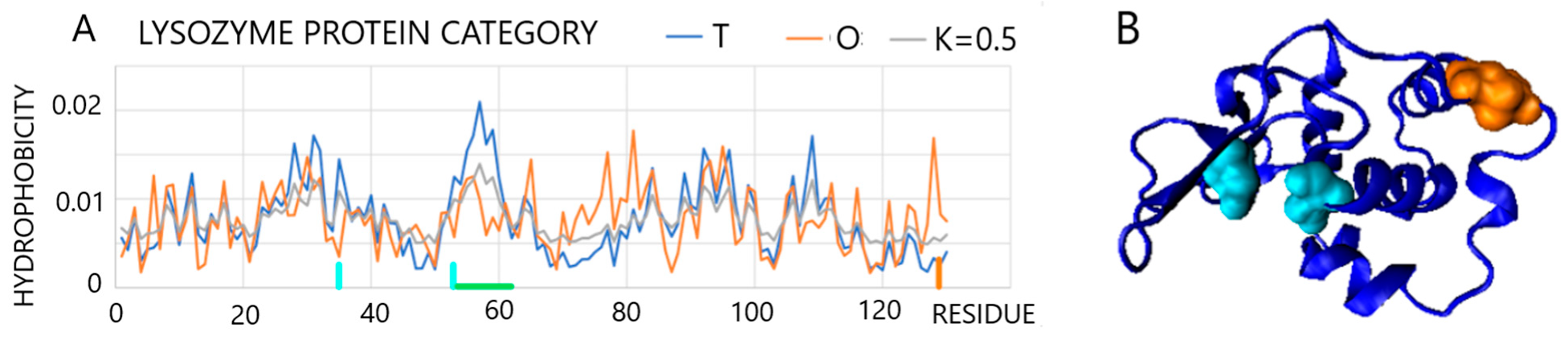
2.2.1. Protein Representing a Structure Consistent with the FOD K = 0 Model
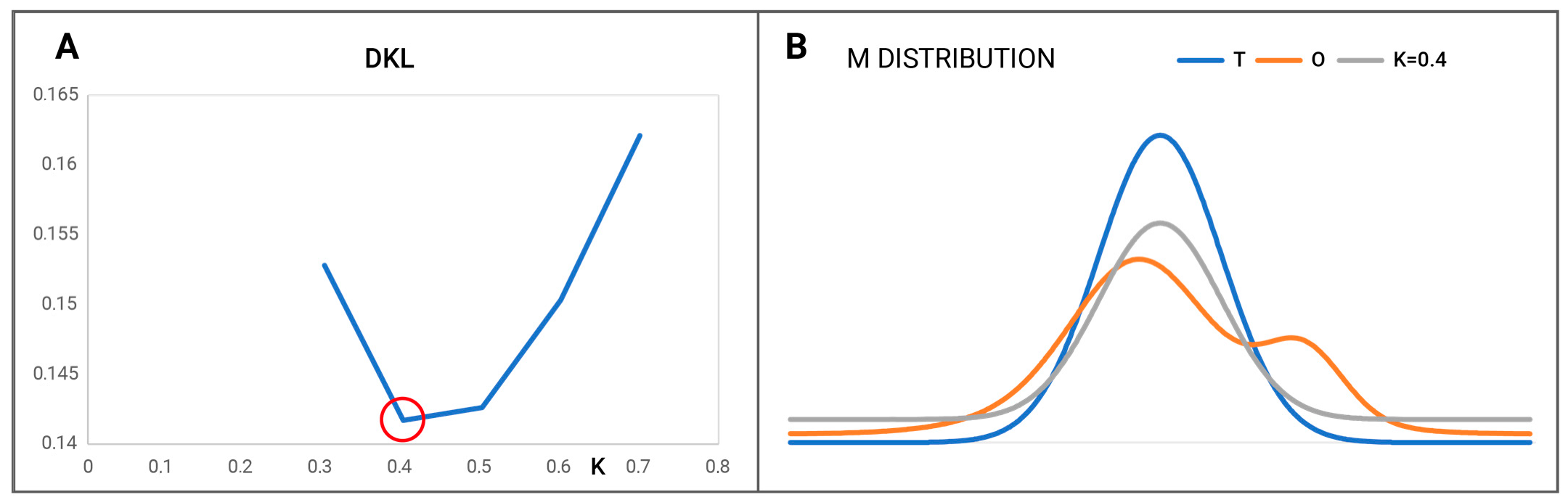
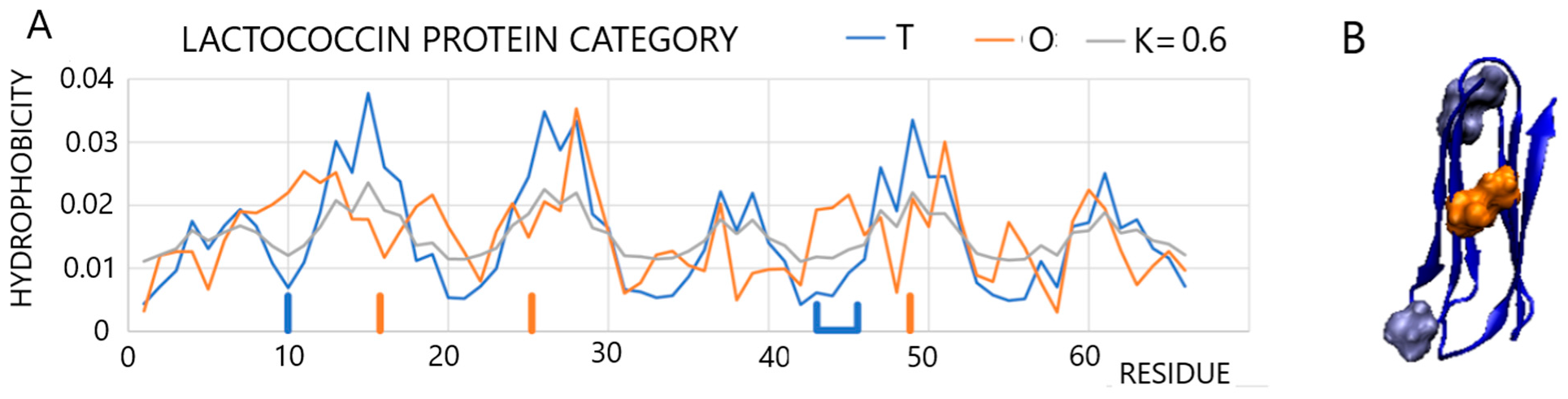
2.2.2. A Protein Representing a Local Maladjustment to the Micelle-Like System
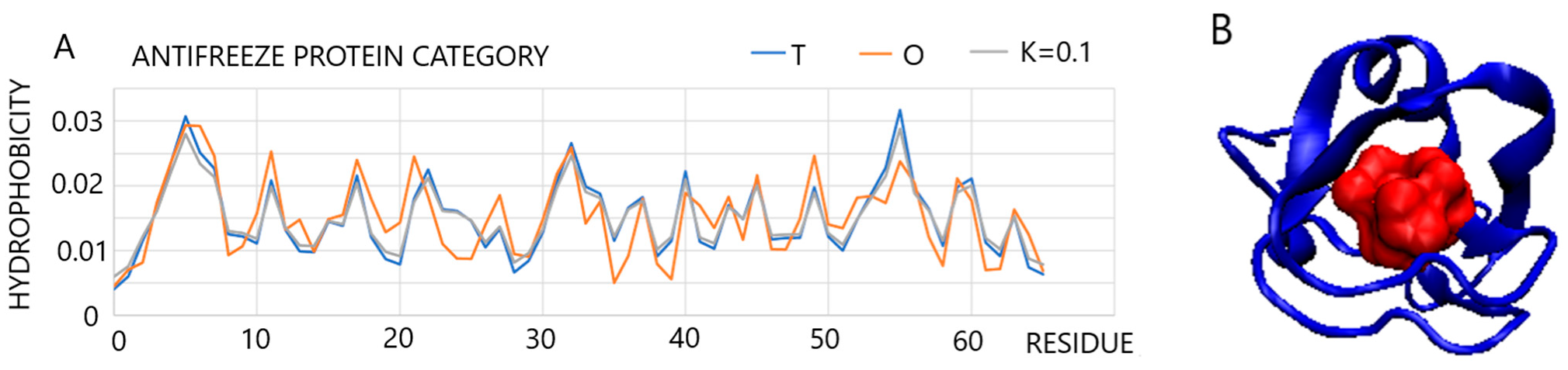
2.2.3. Periplasmic Environment
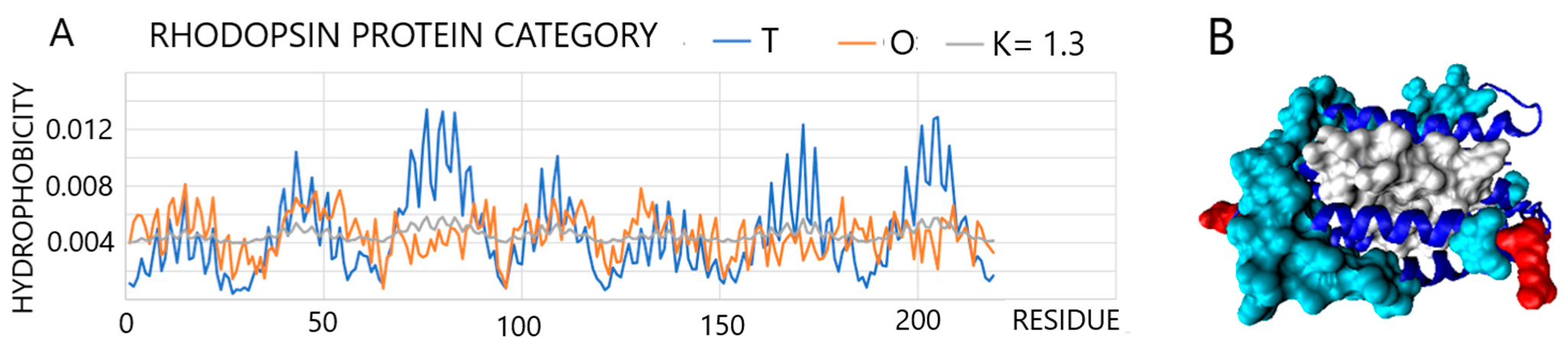
2.2.4. Membrane Environment
2.3. Step 4: 3D Structure → FUNCTION
2.4. Higher Level of Organization
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Oprea, T.I.; May, E.E.; Leitão, A.; Tropsha, A. Computational systems chemical biology. Methods Mol. Biol. 2011, 672, 459–488. [Google Scholar] [CrossRef] [PubMed]
- Perret, N.; Longo, G. Reductionist perspectives and the notion of information. Prog. Biophys. Mol. Biol. 2016, 122, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Youssef, N.; Budd, A.; Bielawski, J.P. Introduction to Genome Biology and Diversity. Methods Mol. Biol. 2019, 1910, 3–31. [Google Scholar] [CrossRef]
- Wood, C.C. The computational stance in biology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180380. [Google Scholar] [CrossRef]
- Winck, F.V.; Monteiro, L.F.R.; Souza, G.M. Introduction: Advances in Plant Omics and Systems Biology. Adv. Exp. Med. Biol. 2021, 1346, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.R. Information literacy in biology education: An example from an advanced cell biology course. Cell Biol. Educ. 2005, 4, 335–343. [Google Scholar] [CrossRef]
- Tebani, A.; Afonso, C.; Bekri, S. Advances in metabolome information retrieval: Turning chemistry into biology. Part II: Biological information recovery. J. Inherit. Metab. Dis. 2018, 41, 393–406. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Jurkowski, W.; Brylinski, M.; Konieczny, L.; Wiśniowski, Z.; Roterman, I. Conformational subspace in simulation of early stage protein folding. Proteins 2004, 55, 115–127. [Google Scholar] [CrossRef]
- Konieczny, L.; Roterman, I.; Spolnik, P. Information—Its role and meaning in organisms. In Systems Biology—Functional Strategies in Living Organisms; Konieczny, L., Roterman, I., Spolnik, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 65–124. [Google Scholar]
- Konieczny, L.; Brylinski, M.; Roterman, I. Gauss-function-Based model of hydrophobicity density in proteins. Silico Biol. 2006, 6, 15–22. [Google Scholar]
- Roterman, I.; Stapor, K.; Fabian, P.; Konieczny, L.; Banach, M. Model of Environmental Membrane Field for Transmembrane Proteins. Int. J. Mol. Sci. 2021, 22, 3619. [Google Scholar] [CrossRef]
- Konieczny, L.; Roterman, I.; Spolnik, P. Regulation in biological systems. In Systems Biology—Functional Strategies in Living Organisms; Konieczny, L., Roterman, I., Spolnik, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 125–166. [Google Scholar]
- Libbrecht, K.G. Physical Dynamics of Ice Crystal Growth. Annu. Rev. Mater. Res. 2017, 47, 271–295. [Google Scholar] [CrossRef]
- Ben-Amotz, D. Electric buzz in a glass of pure water. Science 2022, 376, 800–801. [Google Scholar] [CrossRef]
- Chelli, R.; Pagliai, M.; Procacci, P.; Cardini, G.; Schettino, V. Polarization response of water and methanol investigated by a polarizable force field and density functional theory calculations: Implications for charge transfer. J. Chem. Phys. 2005, 122, 074504. [Google Scholar] [CrossRef]
- Mannfors, B.; Palmo, K.; Krimm, S. Spectroscopically determined force field for water dimer: Physically enhanced treatment of hydrogen bonding in molecular mechanics energy functions. J. Phys. Chem. A 2008, 112, 12667–12678. [Google Scholar] [CrossRef]
- Pullanchery, S.; Kulik, S.; Rehl, B.; Hassanali, A.; Roke, S. Charge transfer across C-H⋅O hydrogen bonds stabilizes oil droplets in water. Science 2021, 374, 1366–1370. [Google Scholar] [CrossRef]
- Poli, E.; Jong, K.H.; Hassanali, A. Charge transfer as a ubiquitous mechanism in determining the negative charge at hydrophobic interfaces. Nat. Commun. 2020, 11, 901. [Google Scholar] [CrossRef]
- Marsalek, O.; Markland, T.E. Quantum Dynamics and Spectroscopy of Ab Initio Liquid Water: The Interplay of Nuclear and Electronic Quantum Effects. J. Phys. Chem. Lett. 2017, 8, 1545–1551. [Google Scholar] [CrossRef]
- Lee, A.J.; Rick, S.W. The effects of charge transfer on the properties of liquid water. J. Chem. Phys. 2011, 134, 184507. [Google Scholar] [CrossRef]
- Agmon, N.; Bakker, H.J.; Campen, R.K.; Henchman, R.H.; Pohl, P.; Roke, S.; Thämer, M.; Hassanali, A. Protons and Hydroxide Ions in Aqueous Systems. Chem. Rev. 2016, 116, 7642–7672. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Li, Y.; Cooks, R.G.; Yan, X. Accelerated Reaction Kinetics in Microdroplets: Overview and Recent Developments. Annu. Rev. Phys. Chem. 2020, 71, 31–51. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Leven, I.; Head-Gordon, T. Can electric fields drive chemistry for an aqueous microdroplet? Nat. Commun. 2022, 13, 280. [Google Scholar] [CrossRef] [PubMed]
- Katsuto, H.; Okamoto, R.; Sumi, T.; Koga, K. Ion Size Dependences of the Salting-Out Effect: Reversed Order of Sodium and Lithium Ions. J. Phys. Chem. B 2021, 125, 6296–6305. [Google Scholar] [CrossRef] [PubMed]
- Chanda, A.; Fokin, V.V. Organic Synthesis “On Water”. Chem. Rev. 2009, 109, 725–748. [Google Scholar] [CrossRef]
- Jung, Y.; Marcus, R.A. On the nature of organic catalysis “on water”. J. Am. Chem. Soc. 2007, 129, 5492–5502. [Google Scholar] [CrossRef]
- Wise, P.K.; Ben-Amotz, D. Interfacial Adsorption of Neutral and Ionic Solutes in a Water Droplet. J. Phys. Chem. B 2018, 122, 3447–3453. [Google Scholar] [CrossRef]
- Qiu, L.; Wei, Z.; Nie, H.; Cooks, R.G. Reaction Acceleration Promoted by Partial Solvation at the Gas/Solution Interface. Chempluschem 2021, 86, 1362–1365. [Google Scholar] [CrossRef]
- Rogers, B.A.; Okur, H.I.; Yan, C.; Yang, T.; Heyda, J.; Cremer, P.S. Weakly hydrated anions bind to polymers but not monomers in aqueous solutions. Nat. Chem. 2022, 14, 40–45. [Google Scholar] [CrossRef]
- Bredt, A.J.; Ben-Amotz, D. Influence of crowding on hydrophobic hydration-shell structure. Phys. Chem. Chem. Phys. 2020, 22, 11724–11730. [Google Scholar] [CrossRef]
- Sugimoto, Y. Seeing how ice breaks the rule. Science 2022, 377, 264–265. [Google Scholar] [CrossRef]
- Tian, Y.; Hong, J.; Cao, D.; You, S.; Song, Y.; Cheng, B.; Wang, Z.; Guan, D.; Liu, X.; Zhao, Z.; et al. Visualizing Eigen/Zundel cations and their interconversion in monolayer water on metal surfaces. Science 2022, 377, 315–319. [Google Scholar] [CrossRef]
- Levitt, M.A. A simplified representation of protein conformations for rapid simulation of protein folding. J. Mol. Biol. 1976, 104, 59–107. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On information and sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Banach, M.; Stapor, K.; Konieczny, L.; Fabian, P.; Roterman, I. Downhill, Ultrafast and Fast Folding Proteins Revised. Int. J. Mol. Sci. 2020, 21, 7632. [Google Scholar] [CrossRef]
- Graether, S.P.; DeLuca, C.I.; Baardsnes, J.; Hill, G.A.; Davies, P.L.; Jia, Z. Quantitative and qualitative analysis of type III antifreeze protein structure and function. J. Biol. Chem. 1999, 274, 11842–11847. [Google Scholar] [CrossRef]
- Choi, S.R.; Lee, J.; Seo, Y.J.; Kong, H.S.; Kim, M.; Jin, E.; Lee, J.R.; Lee, J.H. Molecular basis of ice-binding and cryopreservation activities of type III antifreeze proteins. Comput. Struct. Biotechnol. J. 2021, 19, 897–909. [Google Scholar] [CrossRef]
- Patel, S.N.; Graether, S.P. Structures and ice-binding faces of the alanine-rich type I antifreeze proteins. Biochem. Cell Biol. 2010, 88, 223–229. [Google Scholar] [CrossRef]
- Bredow, M.; Walker, V.K. Ice-Binding Proteins in Plants. Front. Plant Sci. 2017, 8, 2153. [Google Scholar] [CrossRef]
- Artymiuk, J.; Blake, C.C. Refinement of human lysozyme at 1.5 A resolution analysis of non-bonded and hydrogen-bond interactions. J. Mol. Biol. 1981, 152, 737–762. [Google Scholar] [CrossRef]
- Turner, D.L.; Lamosa, P.; Martinez, B. Structure and Properties of Lactococcin 972 from Lactococcus lactis. PDB 2LGN. Available online: https://www.rcsb.org/structure/2LGN (accessed on 1 August 2012).
- Roterman, I.; Stapor, K.; Fabian, P.; Konieczny, L. The Functional Significance of Hydrophobic Residue Distribution in Bacterial Beta-Barrel Transmembrane Proteins. Membranes 2021, 11, 580. [Google Scholar] [CrossRef] [PubMed]
- Roterman, I.; Stapor, K.; Gądek, K.; Gubała, T.; Nowakowski, P.; Fabian, P.; Konieczny, L. Dependence of Protein Structure on Environment: FOD Model Applied to Membrane Proteins. Membranes 2021, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Gushchin, I.; Reshetnyak, A.; Borshchevskiy, V.; Ishchenko, A.; Round, E.; Grudinin, S.; Engelhard, M.; Büldt, G.; Gordeliy, V. Active state of sensory rhodopsin II: Structural determinants for signal transfer and proton pumping. J. Mol. Biol. 2011, 412, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Wach, J.; Bubak, M.; Nowakowski, P.; Roterman, I.; Konieczny, L.; Chłopaś, K. Negative feedback inhibition—Fundamental biological regulation in cells and organisms. In Simulation in Medicine—Pre-Clinical and Clinical Applications; Roterman-Konieczna, I., Ed.; Walter de Gruyter GmbH: Berlin, Germany; Boston, MA, USA, 2015; pp. 31–56. [Google Scholar]
- Konieczny, L.; Roterman-Konieczna, I.; Spólnik, P. Systems Biology—Functional Strategies of Living Organisms; Springer Science + Business Media: Dordrecht, The Netherlands, 2014; p. 104. [Google Scholar]
- Mikuta, K.; Konieczny, L.; Roterman, I. System to simulate the activity of living organism—Construction of proteome. J. Comput. Sci. 2020, 45, 101195. [Google Scholar]
- Roterman, I.; Sieradzan, A.; Stapor, K.; Fabian, P.; Wesołowski, P.; Konieczny, L. On the need to introduce environmental characteristics in ab initio protein structure prediction using a coarse-grained UNRES force field. J. Mol. Graph. Model. 2022, 114, 108166. [Google Scholar] [CrossRef]
- CASP. Available online: https://predictioncenter.org/ (accessed on 11 May 2023).
- Roterman-Konieczna, I. Information—A tool to interpret the biological phenomena. In Simulation in Medicine—Pre-Clinical and Clinical Applications; Roterman-Konieczna, I., Ed.; Walter de Gruyter GmbH: Berlin, Germany; Boston, MA, USA, 2015; pp. 57–64. [Google Scholar]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roterman, I.; Konieczny, L. Protein Is an Intelligent Micelle. Entropy 2023, 25, 850. https://doi.org/10.3390/e25060850
Roterman I, Konieczny L. Protein Is an Intelligent Micelle. Entropy. 2023; 25(6):850. https://doi.org/10.3390/e25060850
Chicago/Turabian StyleRoterman, Irena, and Leszek Konieczny. 2023. "Protein Is an Intelligent Micelle" Entropy 25, no. 6: 850. https://doi.org/10.3390/e25060850
APA StyleRoterman, I., & Konieczny, L. (2023). Protein Is an Intelligent Micelle. Entropy, 25(6), 850. https://doi.org/10.3390/e25060850