

Identifying Cancer Driver Pathways Based on the Mouth Brooding Fish Algorithm

Abstract
1. Introduction
2. Materials and Methods
2.1. Maximum Weight Submatrix Model
2.2. Correlation Analysis between Covariates and Mutation Rate
2.3. Identifying Driver Pathways Based on the Mouth Brooding Fish Algorithm
2.3.1. Principal Component Analysis of Covariates
2.3.2. Construction of Maximum Weight Submatrix Model with Fusion Covariates
2.3.3. Solving of the Mouth Brooding Fish Algorithm
- (1)
- Search Space and Code of the Mouth Brooding Fish Algorithm
- (2)
- Fitness Function of the Mouth Brooding Fish Algorithm
- (3)
- Setting of Parameters for the Mouth Brooding Fish Algorithm
| Algorithm 1: Identifying the cancer driver pathway based on the Mouth Brooding Fish (MBF) algorithm. |
| Input: The mutation matrix A, the gene expression level vector x, gene replication time vector y, and chromosome state vector z. |
| Output: Optimal driver pathways (that is, a set of driver genes). |
| 1. SP is 0.6, nFish is 50, Dis is 1.8, Pdis is 0.2, and SPdamp is 0.95. |
| 2. Process x,y,z to obtain x′,y′,z′. |
| 3. Analyze principal components and calculate the fusional |
| 4. For : |
| Initialize the mouth brooding fish population randomly . |
| Perform the classic MBF algorithm. |
| Add the variable with the highest fitness in the MBF algorithm to a set of driver genes. |
| 5. Return a set of driver genes. |
2.3.4. Permutation Test
2.4. Datasets
- Lung Adenocarcinoma Dataset
- b.
- Glioblastoma Multiforme Dataset
- c.
- Gene Covariate Data
- d.
- Simulated Data
2.5. Experimental Setup and Evaluation Index
3. Results
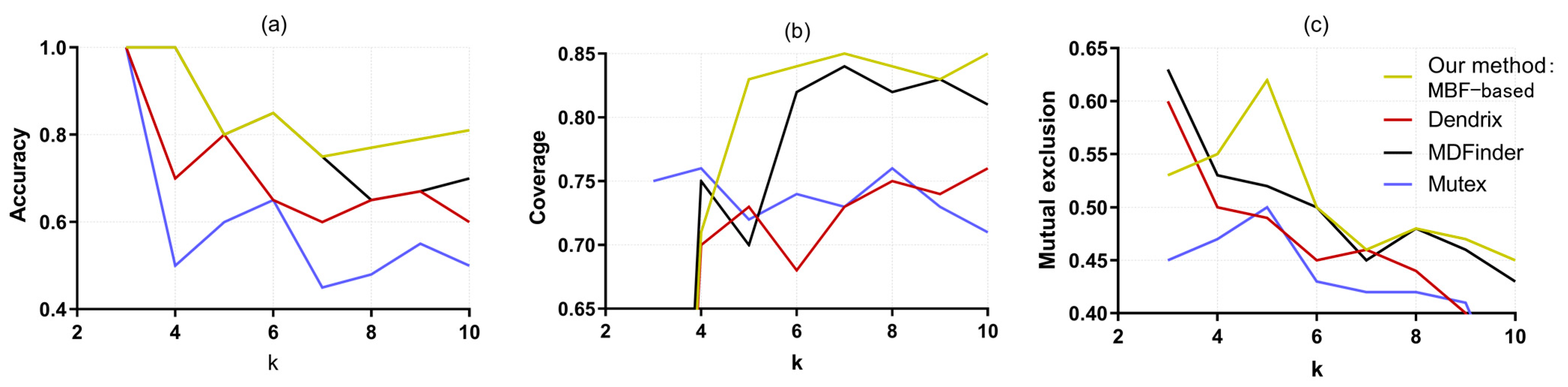
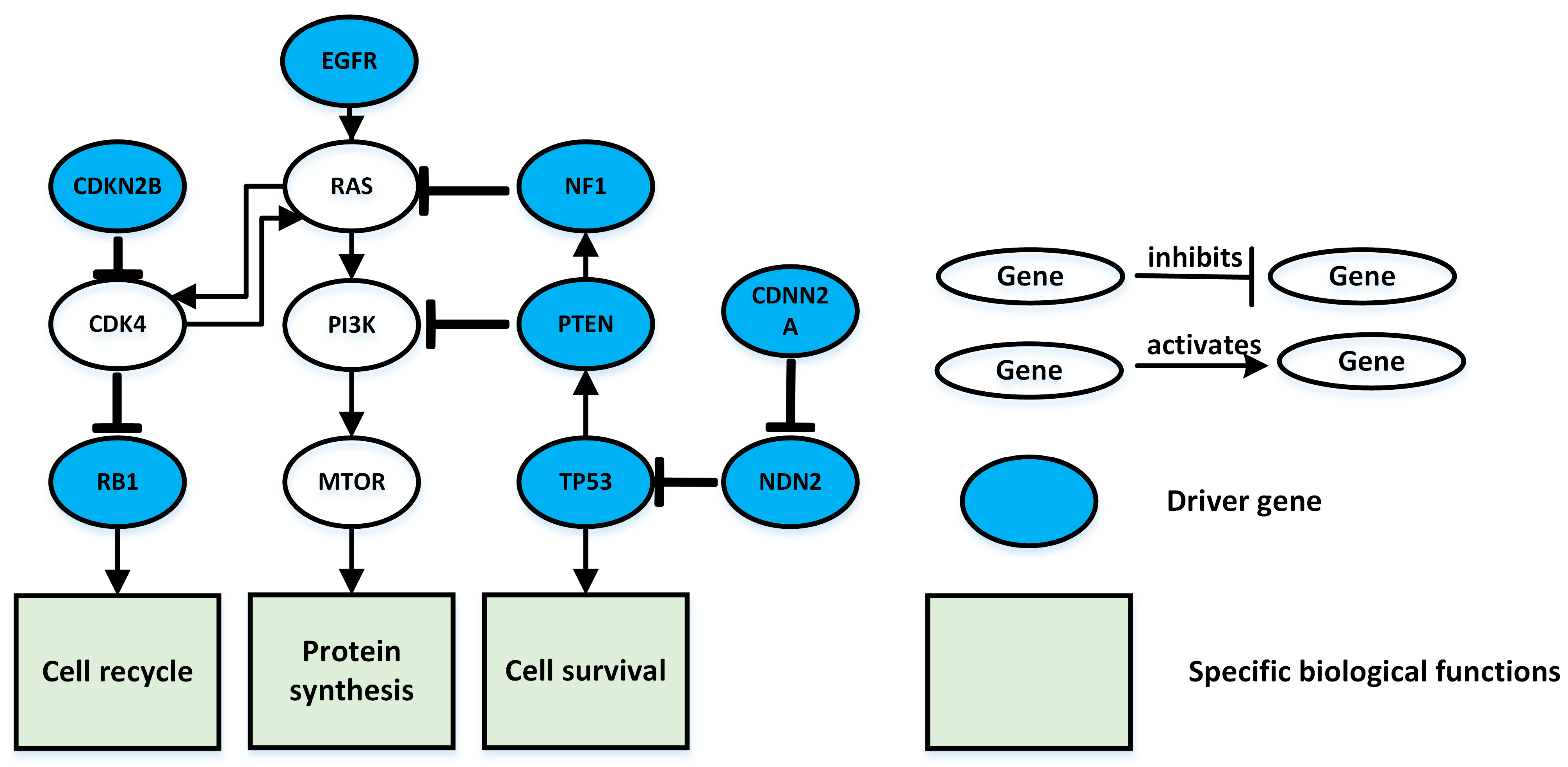
3.1. Lung Adenocarcinoma Test Results
3.2. Glioblastoma Multiforme Test Results
3.3. Simulation Experiment Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, F.; Wang, Y.; Zhang, X.; Fang, J. Five hub genes contributing to the oncogenesis and Trastuzumab-resistant in gastric cancer. Gene 2022, 851, 146942. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; McGray, A.J.R.; Jiang, W.; Lu, B.; Kalinski, P.; Guo, Z.S. Improving cancer immunotherapy by rationally combining oncolytic virus with modulators targeting key signaling pathways. Mol. Cancer 2022, 21, 196. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhou, G.; Liu, X.; Deng, L.; Wu, C.; Zhang, D.; Liu, H. Contrastive learning-based computational histopathology predict differential expression of cancer driver genes. Brief. Bioinform. 2022, 23, bbac294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, Z.; Li, Y.; Sun, Y.; Zhou, X.; Chen, X.; Sun, S. Integrated Bioinformatics Analysis Identifies Robust Biomarkers and Its Correlation With Immune Microenvironment in Nonalcoholic Fatty Liver Disease. Front. Genet. 2022, 13, 942153. [Google Scholar] [CrossRef]
- Northey, J.J.; Barrett, A.S.; Acerbi, I.; Hayward, M.K.; Talamantes, S.; Dean, I.S.; Mouw, J.K.; Ponik, S.M.; Lakins, J.N.; Huang, P.J.; et al. Stiff stroma increases breast cancer risk by inducing the oncogene ZNF217. J. Clin. Investig. 2020, 130, 5721–5737. [Google Scholar] [CrossRef]
- Chen, G.; Li, Y.; Zhang, A.; Gao, L. Circular RNA Circ-BANP Regulates Oxidized Low-density Lipoprotein-induced Endothelial Cell Injury Through Targeting the miR-370/Thioredoxin-interacting Protein Axis. J. Cardiovasc. Pharm. 2021, 77, 349–359. [Google Scholar] [CrossRef]
- Zhao, W.; Gu, X.; Chen, S.; Wu, J.; Zhou, Z. MODIG: Integrating multi-omics and multi-dimensional gene network for cancer driver gene identification based on graph attention network model. Bioinformatics 2022, 38, 4901–4907. [Google Scholar] [CrossRef]
- Vandin, F.; Upfal, E.; Raphael, B.J. De novo discovery of mutated driver pathways in cancer. Genome Res. 2012, 22, 375–385. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, S.L. An Integrated Framework for Identifying Mutated Driver Pathway and Cancer Progression. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 455–464. [Google Scholar] [CrossRef]
- Wu, J.; Chen, X.; Li, G.; Deng, Z.; Zhu, K. A nonlinear model and an algorithm for identifying cancer driver pathways. Appl. Soft Comput. 2022, 129, 109578. [Google Scholar] [CrossRef]
- Ciriello, G.; Cerami, E.; Sander, C.; Schultz, N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012, 22, 398–406. [Google Scholar] [CrossRef]
- Babur, O.; Gonen, M.; Aksoy, B.A.; Schultz, N.; Ciriello, G.; Sander, C.; Demir, E. Systematic identification of cancer driving signaling pathways based on mutual exclusivity of genomic alterations. Genome Biol. 2015, 16, 45. [Google Scholar] [CrossRef]
- Zhao, J.F.; Zhang, S.H.; Wu, L.Y.; Zhang, X.S. Efficient methods for identifying mutated driver pathways in cancer. Bioinformatics 2012, 28, 2940–2947. [Google Scholar] [CrossRef]
- Leiserson, M.D.M.; Blokh, D.; Sharan, R.; Raphael, B.J. Simultaneous Identification of Multiple Driver Pathways in Cancer. PLoS Comput. Biol. 2013, 9, 23–34. [Google Scholar] [CrossRef]
- Dao, P.; Kim, Y.A.; Wojtowicz, D.; Madan, S.; Sharan, R.; Przytycka, T.M. BeWith: A Between-Within method to discover relationships between cancer modules via integrated analysis of mutual exclusivity, co-occurrence and functional interactions. PLoS Comput. Biol. 2017, 13, e1005695. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, S. Discovery of cancer common and specific driver gene sets. Nucleic Acids Res. 2017, 45, 86. [Google Scholar] [CrossRef]
- Vandin, F.; Upfal, E.; Raphael, B.J. Algorithms for Detecting Significantly Mutated Pathways in Cancer. J. Comput. Biol. 2011, 18, 507–522. [Google Scholar] [CrossRef]
- Leiserson, M.D.; Vandin, F.; Wu, H.T.; Dobson, J.R.; Eldridge, J.V.; Thomas, J.L.; Papoutsaki, A.; Kim, Y.; Niu, B.; McLellan, M.; et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 2015, 47, 106–114. [Google Scholar] [CrossRef]
- Reyna, M.A.; Leiserson, M.D.M.; Raphael, B.J. Hierarchical HotNet: Identifying hierarchies of altered subnetworks. Bioinformatics 2018, 34, 972–980. [Google Scholar] [CrossRef]
- Jahani, E.; Chizari, M. Tackling global optimization problems with a novel algorithm Mouth Brooding Fish algorithm. Appl. Soft Comput. 2018, 62, 987–1002. [Google Scholar] [CrossRef]
- Youn, A.; Simon, R. Identifying cancer driver genes in tumor genome sequencing studies. Bioinformatics 2011, 27, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hao, J.; Jiang, W.; He, T.; Zhang, X.; Jiang, T.; Jiang, R. Identifying potential cancer driver genes by genomic data integration. Sci. Rep. 2013, 3, 3538. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.A.; Cho, D.Y.; Dao, P.; Przytycka, T.M. MEMCover: Integrated analysis of mutual exclusivity and functional network reveals dysregulated pathways across multiple cancer types. Bioinformatics 2015, 31, 84–92. [Google Scholar] [CrossRef]
- Leiserson, M.D.M.; Wu, H.T.; Vandin, F.; Raphael, B.J. CoMEt: A statistical approach to identify combinations of mutually exclusive alterations in cancer. Genome Biol. 2015, 16, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Deng, Y.L.; Luo, S.Y.; Deng, C.Y.; Luo, T.; Yin, W.K.; Zhang, H.Y.; Zhang, Y.; Zhang, X.X.; Lan, Y.J.; Ping, Y.Y.; et al. Identifying mutual exclusivity across cancer genomes: Computational approaches to discover genetic interaction and reveal tumor vulnerability. Brief. Bioinform. 2019, 20, 254–266. [Google Scholar] [CrossRef]
- Xi, J.N.; Li, A.; Wang, M.H. A novel network regularized matrix decomposition method to detect mutated cancer genes in tumour samples with inter-patient heterogeneity. Sci. Rep. 2017, 7, 2855. [Google Scholar] [CrossRef]
- Cho, A.; Shim, J.E.; Kim, E.; Supek, F.; Lehner, B.; Lee, I. MUFFINN: Cancer gene discovery via network analysis of somatic mutation data. Genome Biol. 2016, 17, 129. [Google Scholar] [CrossRef]
- Kalashnikova, M.; Mattock, K.; Monaghan, P. Mutual exclusivity develops as a consequence of abstract rather than particular vocabulary knowledge. First Lang 2016, 36, 451–464. [Google Scholar] [CrossRef]
- Mularoni, L.; Sabarinathan, R.; Deu-Pons, J.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveFML: A general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 2016, 17, 128. [Google Scholar] [CrossRef]
- Yi, S.; He, Z.; Jing, X.Y.; Li, Y.; Cheung, Y.M.; Nie, F. Adaptive Weighted Sparse Principal Component Analysis for Robust Unsupervised Feature Selection. IEEE Trans. Neural Netw. Learn. Syst. 2020, 31, 2153–2163. [Google Scholar] [CrossRef]
- Kamath, V.P.; Torres-Roca, J.F.; Eschrich, S.A. Integrating Biological Covariates into Gene Expression-Based Predictors of Radiation Sensitivity. Int. J. Genom. 2017, 2017, 6576840. [Google Scholar] [CrossRef]
- DeCordova, S.; Shastri, A.; Tsolaki, A.G.; Yasmin, H.; Klein, L.; Singh, S.K.; Kishore, U. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front. Immunol. 2020, 11, 1402. [Google Scholar] [CrossRef]
- Palmieri, A.; Valentinis, L.; Zanchin, G. Update on headache and brain tumors. Cephalalgia Int. J. Headache 2021, 41, 431–437. [Google Scholar] [CrossRef]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Song, J.; Peng, W.; Wang, F. Identifying cancer patient subgroups by finding co-modules from the driver mutation profiles and downstream gene expression profiles. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 19, 2863–2872. [Google Scholar] [CrossRef]
- Van Daele, D.; Weytjens, B.; De Raedt, L.; Marchal, K. OMEN: Network-based driver gene identification using mutual exclusivity. Bioinformatics 2022, 38, 3245–3251. [Google Scholar] [CrossRef]
- Wang, C.; Shi, J.; Cai, J.; Zhang, Y.; Zheng, X.; Zhang, N. DriverRWH: Discovering cancer driver genes by random walk on a gene mutation hypergraph. BMC Bioinform. 2022, 23, 277. [Google Scholar] [CrossRef]
- Bozic, I.; Gerold, J.M.; Nowak, M.A. Quantifying Clonal and Subclonal Passenger Mutations in Cancer Evolution. PLoS Comput. Biol. 2016, 12, e1004731. [Google Scholar] [CrossRef]
- Seisenova, A.; Daniyarov, A.; Molkenov, A.; Sharip, A.; Zinovyev, A.; Kairov, U. Meta-Analysis of Esophageal Cancer Transcriptomes Using Independent Component Analysis. Front. Genet. 2021, 12, 683632. [Google Scholar] [CrossRef] [PubMed]
- Chanda, P.; Costa, E.; Hu, J.; Sukumar, S.; Van Hemert, J.; Walia, R. Information Theory in Computational Biology: Where We Stand Today. Entropy 2020, 22, 627. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| k (Number of Genes in Pathway) | The Driver Pathway |
|---|---|
| 3 | EGFR KRAS TP53 |
| 4 | EGFR KRAS NF1 STK11 |
| 5 | EGFR KRAS NF1 STK11 LRP1B |
| 6 | EGFR KRAS NF1 STK11 LRP1B ERBB4 |
| 7 | EGFR KRAS NF1 STK11 LRP1B ERBB4 CDKN2A |
| 8 | EGFR KRAS NF1 STK11 LRP1B ERBB4 CDKN2A TERT |
| 9 | EGFR KRAS NF1 STK11 LRP1B ERBB4 CDKN2A TERT CYSLTR2 |
| 10 | EGFR KRAS NF1 STK11 ATM TP53 APC LRP1B ERBB4 AKT1 |
| (a) | ||||
| Method | Pathway | Submatrix Weight | Accuracy | |
| Our method: MBF-based | EGFR KRAS NF1 STK11 ATM TP53 APC LRP1B ERBB4, AKT1 PAK6 ABL1 CYSLTR2 EGFR | 1.7 | 0.8 | |
| Dendrix [8] | SRC MAP3K15 MAST1 STK11 WT1 YES1 STK11 EGFR KRAS MKNK2 | 1.436 | 0.6 | |
| MDPFinder [13] | FES KRAS NF1 MAP3K3 STK11 TFDP1 KRAS EGFR BUB1 MAP3K3 | 1.682 | 0.8 | |
| Mutex [12] | MAP3K3 NF1 ERBB4 MAST1 ABL1 PAK6 | 1.393 | 0.5 | |
| (b) | ||||
| Method | Coverage | Mutex | Enrichment Pathway (q Value) | |
| Our method: MBF-based | 0.870 | 0.840 | Non-small cell Lung Cancer (8.58 × 10−8) MAPK Signaling Pathway (8 × 10−8) | |
| Dendrix [8] | 0.740 | 0.620 | GnRH Signaling Pathway (1.43 × 10−6) Adherens Junction (5.94 × 10−5) | |
| MDPFinder [13] | 0.850 | 0.830 | Neurotrophin Signaling Pathway (1.1 × 10−4) ErbB Signaling Pathway (1 × 10−4) | |
| Mutex [12] | 0.730 | 0.650 | ErbB Signaling Pathway (1.94 × 10−9) MAPK Signaling Pathway (2.8 × 10−7) | |
| k (Number of Genes in Pathway) | The Driver Pathway |
|---|---|
| 3 | CDK4 CDKN2B RB1 |
| 4 | CDK4 CDKN2B NF1 RB1 |
| 5 | CDK4 CDKN2B EMP3 NF1 RB1 |
| 6 | CDK4 CDKN2B EMP3 NF1 PRNP RB1 |
| 7 | CDK4 CDKN2B NF1 FGFR TP53 RB1 CDKN2A |
| 8 | CDK4 CDKN2B NF1 FGFR TP53 RB1 CDKN2A SPHK2 |
| 9 | FGFR NOTCH1 TP53 RB1 CDKN2A ZNF175 CDK4 CDKN2B NF1 |
| 10 | CDK4 CDKN2B PTEN NF1 FGFR CDKN2A TP53 PRF1 RB1 SIGLEC9 |
| (a) | ||||
| Method | Pathway | Submatrix Weight | Accuracy | |
| Our Method: MBF-Based | CDK4 CDKN2B PTEN NF1 FGFR CDKN2A TP53 PRF1 RB1 SIGLEC9 | 1.890 | 0.800 | |
| Dendrix [8] | CDKN2B ERBB2 SHH PI15 FGFR | 1.540 | 0.600 | |
| MDPFinder [13] | CDK4 CDKN2B CSF1R ERBB2 FGFR3 HGF NTRK3 PRF1 RB1 SIGLEC9 CYP27B1 RB1 TP53 CDK4 CDKN2A EGFR CDKN2A TP53 MDM2 | 1.786 | 0.700 | |
| Mutex [12] | CDKN2B PTEN TRPV4 MYH1 RHOC HGF | 1.393 | 0.5 | |
| (b) | ||||
| Method | Coverage | Mutual Exclusion | Enrichment Pathway (q Value) | |
| Our Method: MBF-Based | 0.850 | 0.450 | Glioma (1.83 × 10−8) Cell Cycle (2.73 × 10−9) | |
| Dendrix [8] | 0.820 | 0.430 | Pathways in Cancer (4.54 × 10−11) Bladder Cancer (1.98 × 10−8) | |
| MDPFinder [13] | 0.760 | 0.370 | Pathways in Cancer (1.12 × 10−11) Bladder Cancer (1.12 e× 10−11) | |
| Mutex [12] | 0.730 | 0.380 | Melanoma (9.34 × 10−13) P53 Signaling Pathway (5.74 × 10−8) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Xiang, X.; Zhao, B.; Huang, J.; Yang, L.; Zeng, Y. Identifying Cancer Driver Pathways Based on the Mouth Brooding Fish Algorithm. Entropy 2023, 25, 841. https://doi.org/10.3390/e25060841
Zhang W, Xiang X, Zhao B, Huang J, Yang L, Zeng Y. Identifying Cancer Driver Pathways Based on the Mouth Brooding Fish Algorithm. Entropy. 2023; 25(6):841. https://doi.org/10.3390/e25060841
Chicago/Turabian StyleZhang, Wei, Xiaowen Xiang, Bihai Zhao, Jianlin Huang, Lan Yang, and Yifu Zeng. 2023. "Identifying Cancer Driver Pathways Based on the Mouth Brooding Fish Algorithm" Entropy 25, no. 6: 841. https://doi.org/10.3390/e25060841
APA StyleZhang, W., Xiang, X., Zhao, B., Huang, J., Yang, L., & Zeng, Y. (2023). Identifying Cancer Driver Pathways Based on the Mouth Brooding Fish Algorithm. Entropy, 25(6), 841. https://doi.org/10.3390/e25060841