
Dissipative Photochemical Abiogenesis of the Purines


Abstract
:1. Introduction
2. Thermodynamics of Dissipative Structuring
3. The Dissipative Structuring of the Purines
3.1. The Model
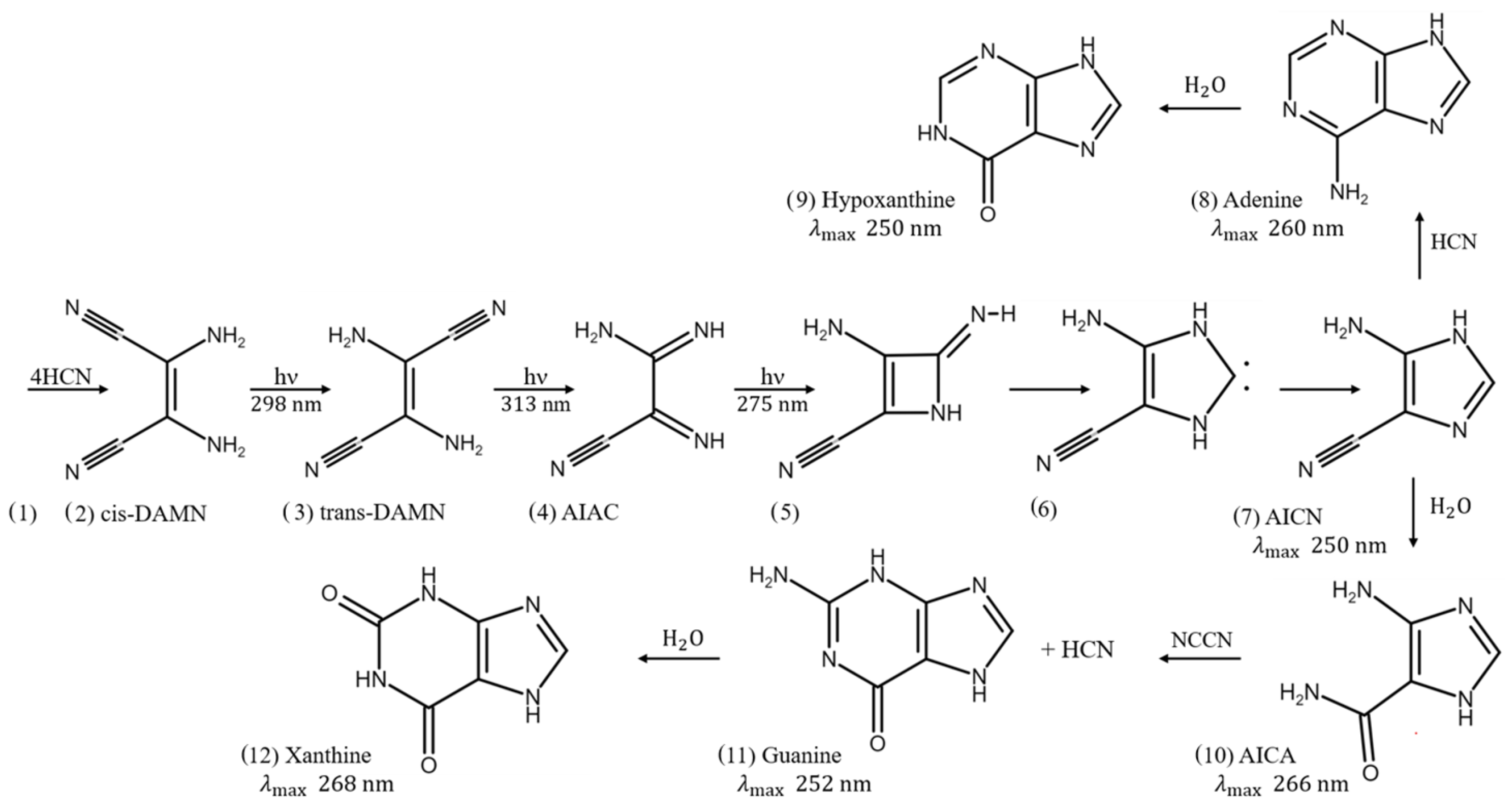
3.2. The Photochemical Route to the Purines
3.3. The Kinetic Equations
- HCN (H) thermally polymerizes into (HCN). Its most stable tetramer () is known as cis-diaminomaleonitrile, cis-DAMN (C) [5];
- HCN (H) can also thermally polymerize into trans-diaminomaleonitrile, trans-DAMN (T) [5];
- Trans-DAMN (T) and cyanogen (Cg) are good catalysts for the polymerization of 4HCN into cis-DAMN (see Table 6 of Sanchez et al. [5]). The catalytic effect of trans-DAMN on the tetramization of HCN was incorporated into the model by reducing the energy of the activation barrier such as to give the same amplification factor of 12 due to the catalytic effect of the inclusion of 0.01 M trans-DAMN in the HCN solution observed in the experiments of Sanchez et al. [5] at a temperature of 20 °C.The catalytic effect of cyanogen (Cg) is taken to be the same as that of trans-DAMN (T) since Sanchez et al. determined both of these to be strong catalysts [5]. As mentioned above, cyanogen is a precursor needed for guanine obtained from the Lyman- line (121.6 nm) on atmospheric HCN giving, through photolysis, the CN radical with high quantum yield (reaction R177 Table 1 of Zahnle [51]), and this can form cyanogen (CN) by interacting with a second HCN molecule (reaction R199, Table 1 of Zahnle);
- Trans-DAMN also acts as an auto-catalyst for its own thermal production from 4HCN. Cyanogen is also an effective catalyst for this reaction (Table 6 of Sanchez et al. [5]);
- A photon at 313 nm electronically excites trans-DAMN (T) which then transforms into 2-amino-3-iminoacrylimidoyl cyanide, AIAC (J), through proton transfer from one of the amino groups [60];
- AIAC (J), on absorbing a photon at 275 nm, transforms through photon-induced cyclicization (ring closure) into an azetene intermediate ((5) of Figure 3) in an electronic excited state, which then transforms to the N-heterocyclic carbene ((6) of Figure 3) and finally this tautomerizes to give the imidazole, 4-aminoimidazole-5-carbonitrle, AICN (I) ((7) of Figure 3) [60];
- The imidazole AICN (I), created in the previous photochemical reaction #11, is converted through hydrolysis to 4-aminoimidazole-5-carboxamide, AICA (L) [6];
- Adenine (A) can also be obtained through the attachment of HCN (H) to AICN (I) to form amidine (Am), which is a formamide (F) catalyzed thermal reaction involving formimidic acid (Fa) [100];
- A subsequent tautomerization of amidine (Am) is required (calculated to have a high barrier of about 50 kcal mol) which, once overcome by absorbing a photon at 250 nm, allows the system to proceed through a subsequent barrier-less cyclicization to form adenine (A) [61];
- Hydrolysis of adenine (A) gives hypoxathine (Hy), determined by Zheng and Meng to have a transition state barrier of 23.4 kcal mol [96];
- The combination of AIAC (L) with cyanogen (Cg) (a precursor produced in the middle atmosphere from HCN—see reaction # 7) through a thermal reaction leads to guanine (G) and HCN (H) [6];
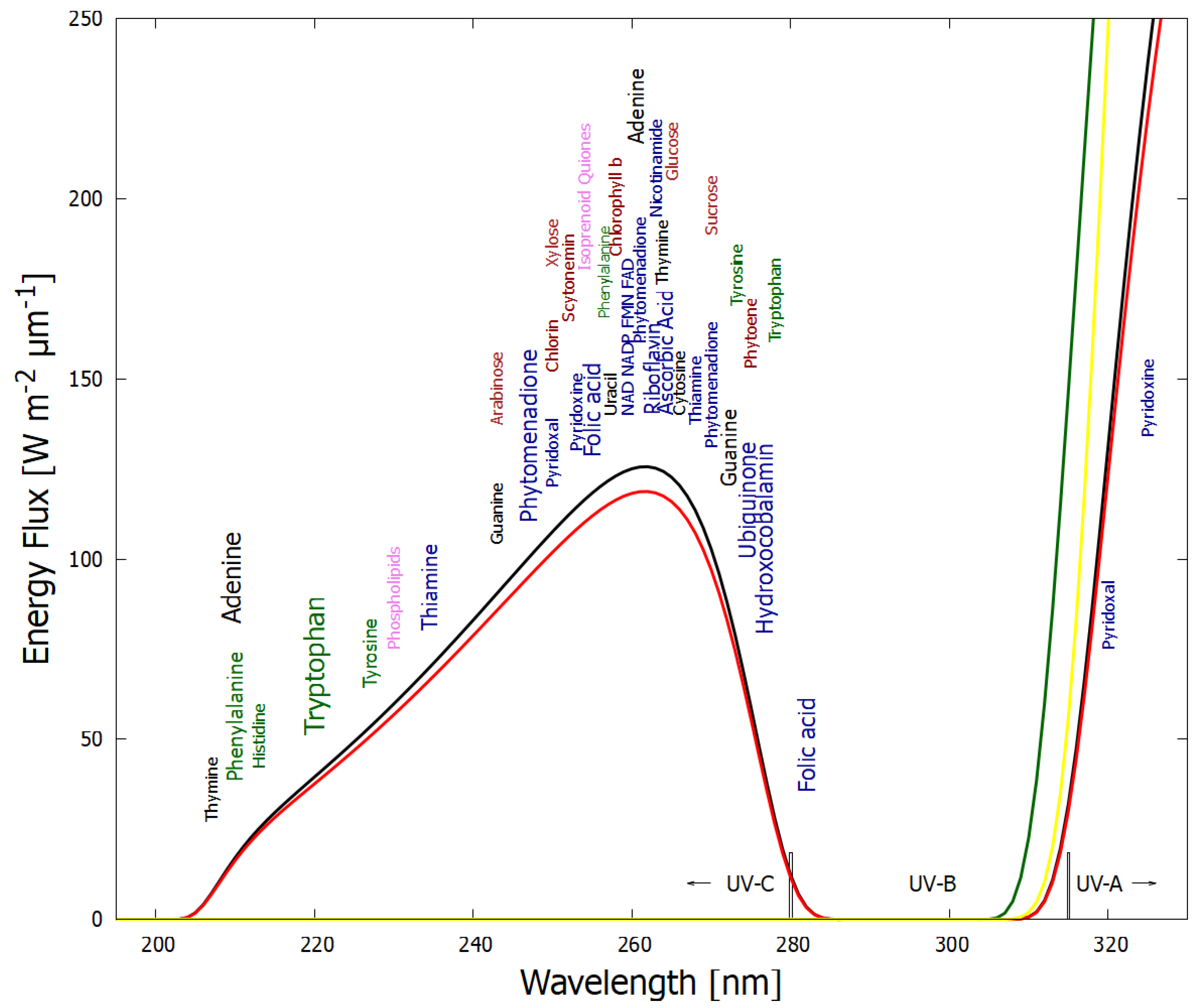
- Photochemical reactions 19 to 28. These represent the absorption (within in a 20 nm region centered on the wavelength of peak absorption) and dissipation through internal conversion at a conical intersection to the ground state on a sub-picosecond time scale. All molecules listed in this set of photo-reactions are photo-stable because of this peaked conical intersection connecting the electronic excited state to the ground state. These reactions, with large quantum efficiencies, represent the bulk of the flow of energy from the incident UV-C spectrum to the emitted outgoing infrared ocean surface spectrum and therefore contribute most to photon dissipation, or entropy production.
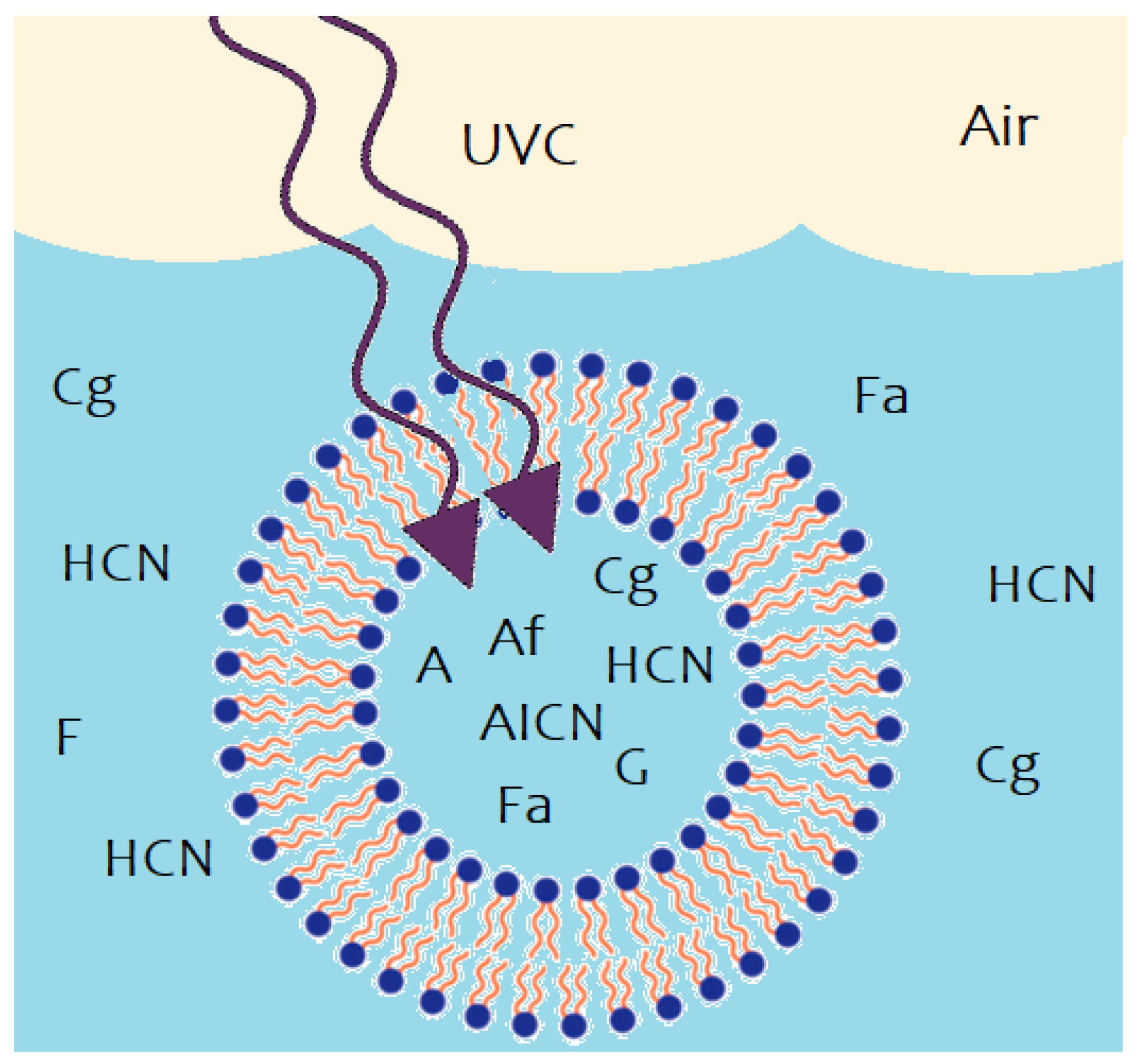
3.4. Vesicle Permeability and Internal Diffusion
3.5. Initial Conditions
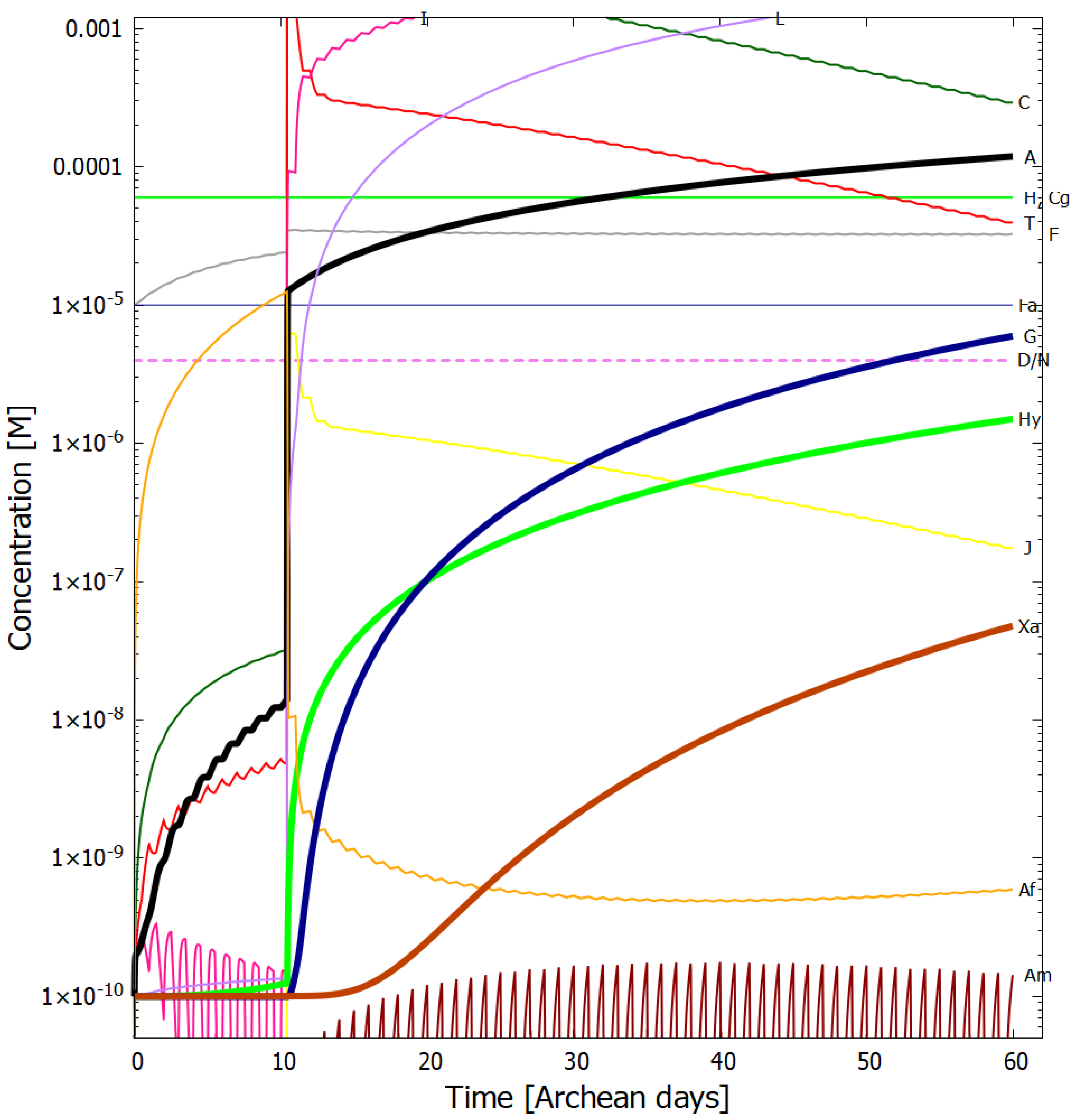
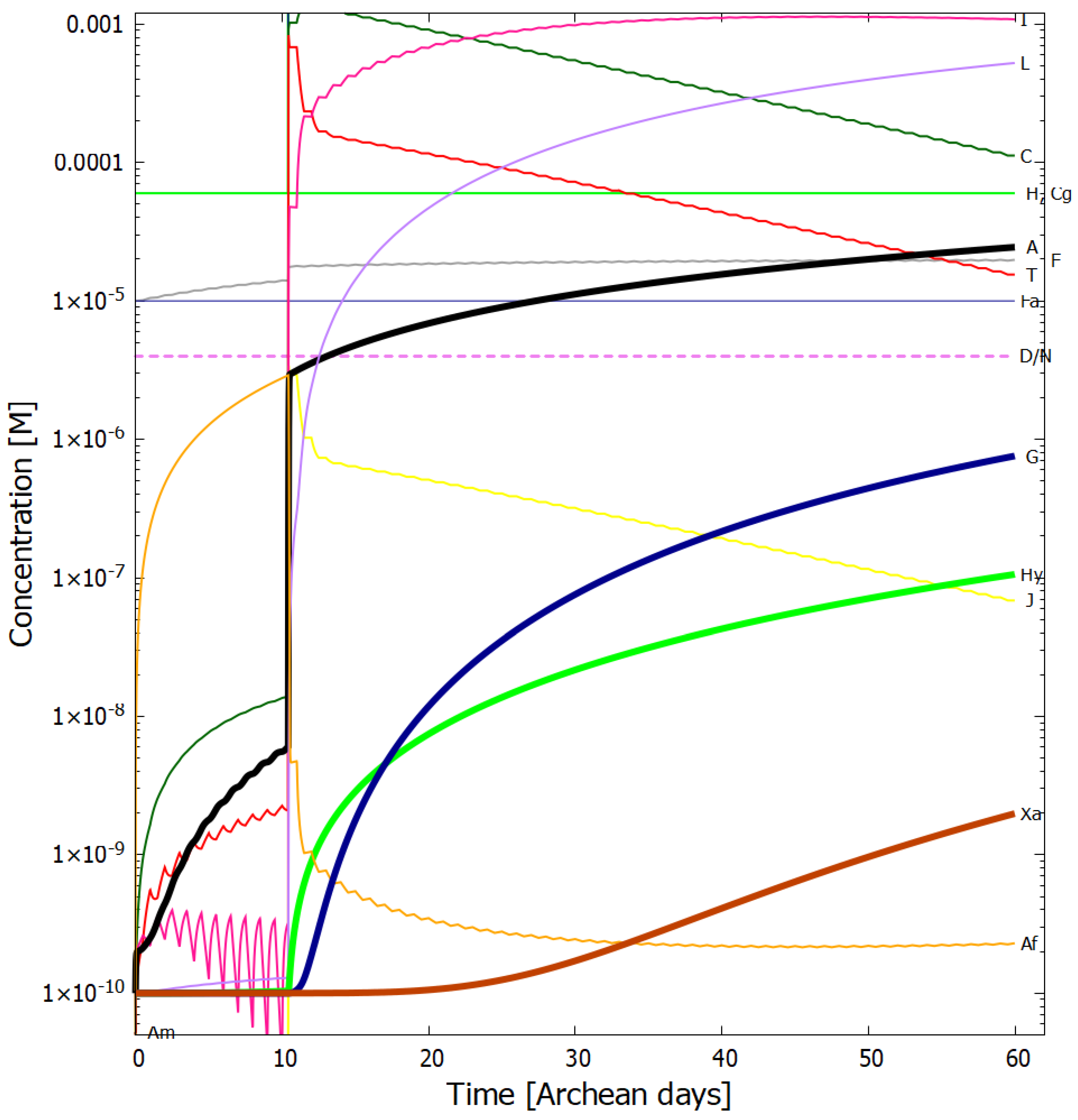
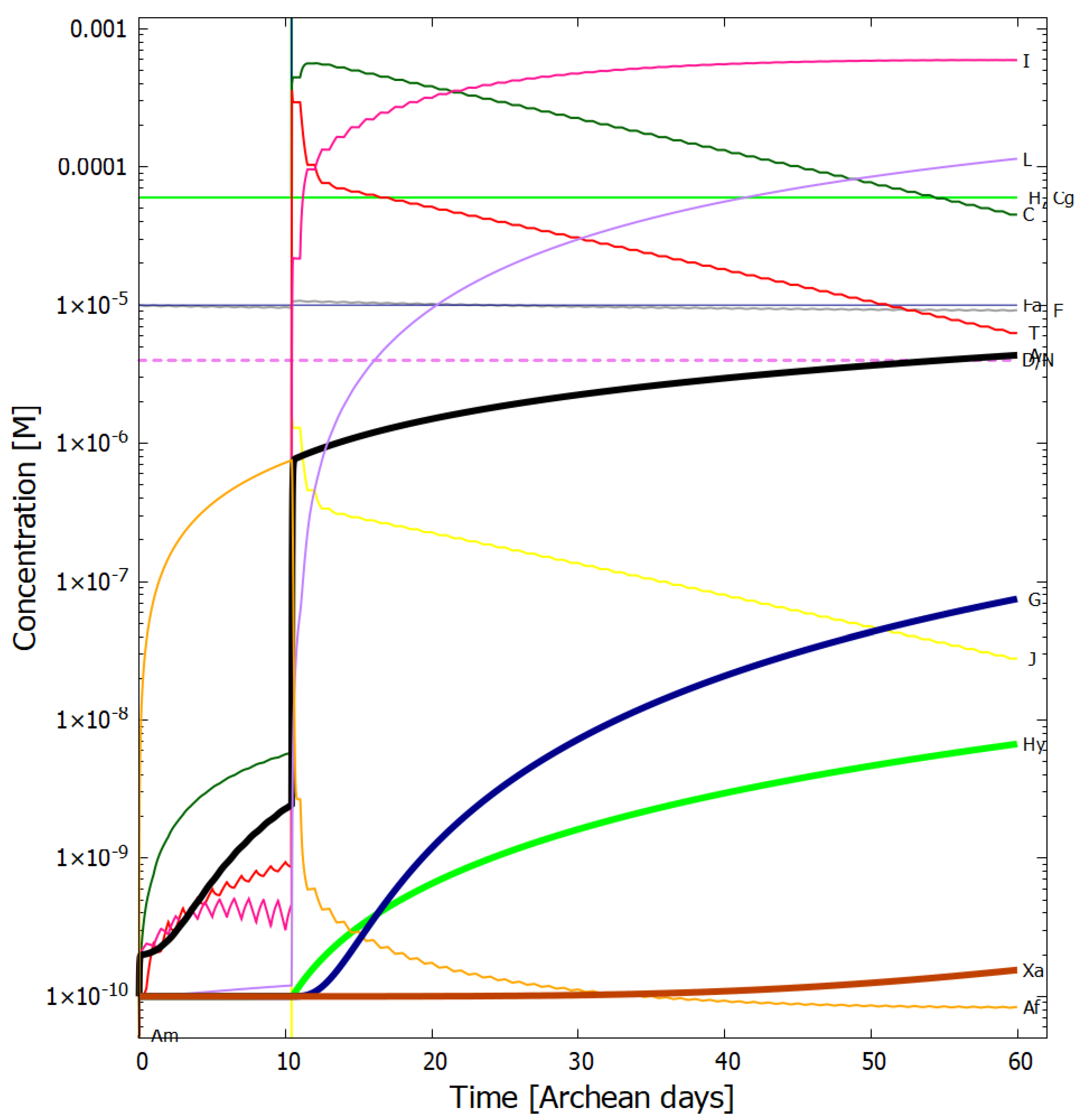
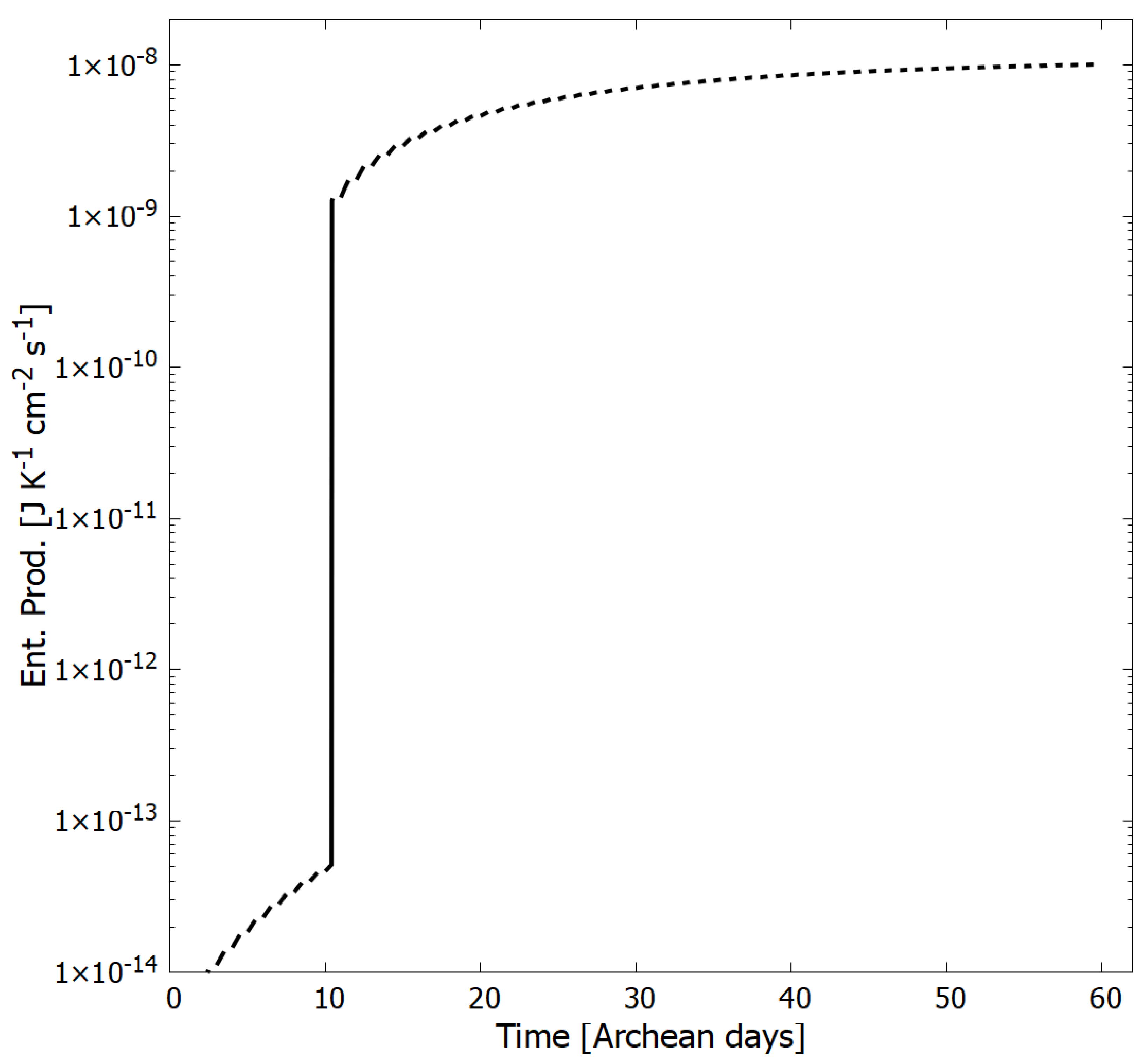
4. Results
Evolution of the Concentration Profile
5. Discussion
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIAC | 2-amino-3-iminoacrylimidoyl cyanide |
| AICA | 4-aminoimidazole-5-carboxamide |
| AICN | 4-aminoimidazole-5-carbonitrile |
| CIT | Classical Irreversible Thermodynamics |
| DAMN | diaminomaleonitrile |
| DAFN | diaminofumaronitrile |
| PAHs | Polyaromatic Hydrocarbons |
| UV-A | light in the region 360–400 nm |
| UV-B | light in the region 285–360 nm (only the region 310–360 nm is relevant here) |
| UV-C | light in the region 100–285 nm (only the region 210–285 nm is relevant here) |
| UVTAR | Ultraviolet and Temperature Assisted Replication |
References
- Miller, S.L.; Urey, H.C. Origin of Organic Compounds on the Primitive Earth and in Meteorites. Science 1959, 130, 245. [Google Scholar] [CrossRef]
- Oró, J. Synthesis of adenine from ammonium cyanide. Biochem. Biophys. Res. Commun. 1960, 2, 407–412. [Google Scholar] [CrossRef]
- Oró, J.; Kimball, A. Synthesis of purines under possible primitive earth conditions: I. Adenine from hydrogen cyanide. Arch. Biochem. Biophys. 1961, 94, 217–227. [Google Scholar] [CrossRef]
- Oró, J.; Kimball, A. Synthesis of purines under possible primitive earth conditions: II. Purine intermediates from hydrogen cyanide. Arch. Biochem. Biophys. 1962, 96, 293–313. [Google Scholar] [CrossRef]
- Sanchez, R.A.; Ferris, J.P.; Orgel, L.E. Studies in Prebiodc Synthesis II: Synthesis of Purine Precursors and Amino Acids from Aqueous Hydrogen Cyanide. J. Mol. Biol. 1967, 80, 223–253. [Google Scholar]
- Sanchez, R.A.; Ferris, J.P.; Orgel, L.E. Studies in Prebiodc Synthesis IV: Conversion of 4-Aminoimidazole-5-carbonitrile Derivatives to Purines. J. Mol. Biol. 1968, 38, 121–128. [Google Scholar] [CrossRef]
- Schwartz, A.; Goverde, M. Acceleration of HCN oligomerization by formaldehyde and related compounds: Implications for prebiotic syntheses. J. Mol. Evol. 1982, 18, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Voet, A.; Schwartz, A. Prebiotic adenine synthesis from HCN—Evidence for a newly discovered major pathway. Bioorg. Chem. 1983, 12, 8–17. [Google Scholar] [CrossRef]
- Levy, M.; Miller, S.L. The Prebiotic Synthesis of Modified Purines and Their Potential Role in the RNA World. J. Mol. Evol. 1999, 48, 631–637. [Google Scholar] [CrossRef]
- Levy, M.; Miller, S.L.; Oró, J.J. Production of Guanine from NH4CN Polymerizations. J. Mol. Evol. 1999, 49, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Saladino, R.; Crestini, C.; Costanzo, G.; Negri, R.; Di Mauro, E. A possible prebiotic synthesis of purine, adenine, cytosine, and 4(3H)-pyrimidinone from formamide: Implications for the origin of life. Bioorg. Med. Chem. 2001, 9, 1249–1253. [Google Scholar] [CrossRef]
- Hill, A.; Orgel, L. Synthesis of Adenine from HCN Tetramer and Ammonium Formate. Orig. Life Evol. Biosph. 2002, 32, 99–102. [Google Scholar] [CrossRef]
- Borquez, E.; Cleaves, H.; Lazcano, A.; Miller, S. An Investigation of Prebiotic Purine Synthesis from the Hydrolysis of HCN Polymers. Orig. Life Evol. Biosph. 2005, 35, 79–90. [Google Scholar] [CrossRef]
- Michaelian, K. Thermodynamic origin of life. arXiv 2009, arXiv:0907.0042. [Google Scholar]
- Michaelian, K. Thermodynamic dissipation theory for the origin of life. Earth Syst. Dyn. 2011, 224, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Michaelian, K. The biosphere: A thermodynamic imperative. In The Biosphere; INTECH: Location, UK, 2012. [Google Scholar]
- Michaelian, K. A non-linear irreversible thermodynamic perspective on organic pigment proliferation and biological evolution. J. Phys. 2013, 475, 012010. [Google Scholar] [CrossRef]
- Michaelian, K.; Simeonov, A. Fundamental molecules of life are pigments which arose and co-evolved as a response to the thermodynamic imperative of dissipating the prevailing solar spectrum. Biogeosciences 2015, 12, 4913–4937. [Google Scholar] [CrossRef] [Green Version]
- Michaelian, K. Thermodynamic Dissipation Theory of the Origina and Evolution of Life: Salient Characteristics of RNA and DNA and Other Fundamental Molecules Suggest an Origin of Life Driven by UV-C Light; Printed by CreateSpace; Self-Published: Mexico City, Mexico, 2016; ISBN 9781541317482. [Google Scholar]
- Michaelian, K.; Simeonov, A. Thermodynamic explanation of the cosmic ubiquity of organic pigments. Astrobiol. Outreach 2017, 5, 156. [Google Scholar] [CrossRef]
- Michaelian, K. Microscopic Dissipative Structuring and Proliferation at the Origin of Life. Heliyon 2017, 3, e00424. [Google Scholar] [CrossRef]
- Michaelian, K. Homochirality through Photon-Induced Denaturing of RNA/DNA at the Origin of Life. Life 2018, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Michaelian, K.; Rodriguez, O. Prebiotic fatty acid vesicles through photochemical dissipative structuring. Rev. Cuba. QuíMica 2019, 31, 354–370. [Google Scholar]
- Michaelian, K.; Santillan, N. UVC photon-induced denaturing of DNA: A possible dissipative route to Archean enzyme-less replication. Heliyon 2019, 5, e01902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejía Morales, J.; Michaelian, K. Photon Dissipation as the Origin of Information Encoding in RNA and DNA. Entropy 2020, 22, 940. [Google Scholar] [CrossRef]
- Michaelian, K. The Dissipative Photochemical Origin of Life: UVC Abiogenesis of Adenine. Entropy 2021, 23, 217. [Google Scholar] [CrossRef]
- Michaelian, K.; Mateo, R.E.C. A Photon Force and Flow for Dissipative Structuring: Application to Pigments, Plants and Ecosystems. Entropy 2022, 24, 76. [Google Scholar] [CrossRef]
- Michaelian, K. Non-Equilibrium Thermodynamic Foundations of the Origin of Life. Foundations 2022, 2, 308–337. [Google Scholar] [CrossRef]
- Sagan, C. Ultraviolet Selection Pressure on the Earliest Organisms. J. Theor. Biol. 1973, 39, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Sagan, C.; Chyba, C. The Early Faint Sun Paradox: Organic Shielding of Ultraviolet-Labile Greenhouse Gases. Science 1997, 276, 1217–1221. [Google Scholar] [CrossRef]
- Berkner, L.; Marshall, L. Origin and Evolution of the Oceans and Atmosphere; John Wiley and Sons: Hoboken, NJ, USA, 1964; pp. 102–126. [Google Scholar]
- Cnossen, I.; Sanz-Forcada, J.; Favata, F.; Witasse, O.; Zegers, T.; Arnold, N.F. The habitat of early life: Solar X-ray and UV radiation at Earth’s surface 4–3.5 billion years ago. J. Geophys. Res. 2007, 112, E02008. [Google Scholar] [CrossRef]
- Meixnerová, J.; Blum, J.D.; Johnson, M.W.; Stüeken, E.E.; Kipp, M.A.; Anbar, A.D.; Buick, R. Mercury abundance and isotopic composition indicate subaerial volcanism prior to the end-Archean “whiff” of oxygen. Proc. Natl. Acad. Sci. USA 2021, 118, e2107511118. [Google Scholar] [CrossRef]
- Schuurman, M.S.; Stolow, A. Dynamics at Conical Intersections. Annu. Rev. Phys. Chem. 2018, 69, 427–450. [Google Scholar] [CrossRef] [PubMed]
- Ferris, J.P.; Orgel, L.E. An Unusual Photochemical Rearrangement in the Synthesis of Adenine from Hydrogen Cyanide. J. Am. Chem. Soc. 1966, 88, 1074. [Google Scholar] [CrossRef]
- Sagan, C.; Khare, B.N. Long-Wavelength Ultraviolet Photoproduction of Amino Acids on the Primitive Earth. Science 1971, 173, 417–420. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Bermejo, M.; Zorzano, M.P.; Osuna-Esteban, S. Simple Organics and Biomonomers Identified in HCN Polymers: An Overview. Life 2013, 3, 421–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pflüger, E. Beitragë zur Lehre von der Respiration. I. Ueber die physiologische Verbrennung in den lebendigen organismen. Arch. Ges. Physiol. 1875, 10, 641–644. [Google Scholar] [CrossRef]
- Ferris, J.; Joshi, P.; Edelson, E.; Lawless, J. HCN: A plausible source of purines, pyrimidines and amino acids on the primitive Earth. J. Mol. Evol. 1978, 11, 293–311. [Google Scholar] [CrossRef]
- Yuasa, S.; Flory, D.; Basile, B.; Oro, J. On the abiotic formation of amino acids. I. HCN as a precursor of amino acids detected in extracts of lunar samples. II. Formation of HCN and amino acids from simulated mixtures of gases released from lunar samples. J. Mol. Evol. 1984, 20, 52–58. [Google Scholar] [CrossRef]
- Ritson, D.; Sutherland, J. Prebiotic synthesis of simple sugars by photoredox systems chemistry. Nat. Chem. 2012, 4, 895–899. [Google Scholar] [CrossRef] [Green Version]
- Das, T.; Ghule, S.; Vanka, K. Insights Into the Origin of Life: Did It Begin from HCN and H2O? ACS Cent. Sci. 2019, 5, 1532–1540. [Google Scholar] [CrossRef] [Green Version]
- Matthews, C. Dark matter in the solar system: Hydrogen cyanide polymers. Orig. Life Evol. Biosph. 1991, 21, 421–434. [Google Scholar] [CrossRef]
- Matthews, C.; Minard, R. Hydrogen cyanide polymers, comets and the origin of life. Faraday Discuss. 2006, 133, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Rolffs, R.; Schilke, P.; Zhang, Q.; Zapata, L. Structure of the hot molecular core G10.47+0.03. Astron. Astrophys. 2011, 536, A33. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.; House, C.; Arevalo, R.D.; Dworkin, J.P.; Callahan, M.P. Organometallic compounds as carriers of extraterrestrial cyanide in primitive meteorites. Nat. Commun. 2019, 10, 2777. [Google Scholar] [CrossRef] [PubMed]
- Trainer, M.G.; Jimenez, J.L.; Yung, Y.L.; Toon, O.B.; Tolbert, M.A. Nitrogen Incorporation in CH4-N2 Photochemical Aerosol Produced by Far UV Irradiation. Astrobiology 2012, 12, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Pearce, B.K.D.; Molaverdikhani, K.; Pudritz, R.E.; Henning, T.; Hébrard, E. HCN Production in Titan’s Atmosphere: Coupling Quantum Chemistry and Disequilibrium Atmospheric Modeling. Astrophys. J. 2020, 901, 110. [Google Scholar] [CrossRef]
- Airey, P.L.; Dainton, F.S. The photochemistry of aqueous solutions of Fe(II) II. Processes in acidified solutions of potassium ferrocyanide at 25 °C. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1966, 291, 478–486. [Google Scholar] [CrossRef]
- Fox, S.W.; Harada, K. Synthesis of Uracil under Conditions of a Thermal Model of Prebiological Chemistry. Science 1961, 133, 1923–1924. [Google Scholar] [CrossRef] [PubMed]
- Zahnle, K.J. Photochemistry of methane and the formation of hydrocyanic acid (HCN) in the Earth’s early atmosphere. J. Geophys. Res. Atmos. 1986, 91, 2819–2834. [Google Scholar] [CrossRef]
- Miller, S.L.; Lazcano, A. The Origin of Life—Did It Occur at High Temperatures? Mol. Evol. 1995, 41, 689–692. [Google Scholar] [CrossRef]
- Bada, J.L.; Lazcano, A. Some Like It Hot, But Not the First Biomolecules. Science 2002, 296, 1982–1983. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, S.; Cleaves, H.J.; Miller, S.L. The cold origin of life: B. Implications based on pyrimidines and purines produced from frozen ammonium cyanide solutions. Orig. Life Evol. Biosph. 2002, 32, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.; Dahl, N.H.; Jensen, S.K.; Poulsen, J.A.; Thøgersen, J.; Keiding, S.R. Femtosecond Photolysis of Aqueous Formamide. J. Phys. Chem. A 2008, 112, 3339–3344. [Google Scholar] [CrossRef] [PubMed]
- Basch, H.; Robin, M.B.; Kuebler, N.A. Electronic Spectra of Isoelectronic Amides, Acids, and Acyl Fluorides. J. Chem. Phys. 1968, 49, 5007–5018. [Google Scholar] [CrossRef]
- Lelj, F.; Adamo, C. Solvent effects on isomerization equilibria: An energetic analysis in the framework of density functional theory. Theor. Chim. Acta 1995, 91, 199–214. [Google Scholar] [CrossRef]
- Koch, T.; Rodehorst, R. Quantitative investigation of the photochemical conversion of diaminomaleonitrile to diaminofumaronitrile and 4-amino-5-cyanoimidazole. J. Am. Chem. Soc. 1974, 96, 6707–6710. [Google Scholar] [CrossRef]
- Gupta, V.; Tandon, P. Conformational and vibrational studies of isomeric hydrogen cyanide tetramers by quantum chemical methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 89, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, E.; Anoop, A.; Nachtigallova, D.; Thiel, W.; Barbatti, M. Photochemical Steps in the Prebiotic Synthesis of Purine Precursors from HCN. Angew. Chem. Int. 2013, 52, 8000–8003. [Google Scholar] [CrossRef] [Green Version]
- Glaser, R.; Hodgen, B.; Farrelly, D.; McKee, E. Adenine synthesis in interstellar space: Mechanisms of prebiotic pyrimidine-ring formation of monocyclic HCN-pentamers. Astrobiology 2007, 7, 455–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavaluzzi, M.J.; Borer, P.N. Revised UV extinction coefficients for nucleoside-5’- monophosphates and unpaired DNA and RNA. Nucleic Acids Res. 2004, 32, e13. [Google Scholar] [CrossRef] [Green Version]
- Franz, J.; Gianturco, F. Low-energy positron scattering from DNA nucleobases: The effects from permanent dipoles. Eur. Phys. J. D 2014, 68, 279. [Google Scholar] [CrossRef] [Green Version]
- Stimson, M.M.; Reuter, M.A. Ultraviolet Absorption Spectra of Nitrogenous Heterocycles. VII. The Effect of Hydroxy Substitutions on the Ultraviolet Absorption of the Series: Hypoxanthine, Xanthine and Uric Acid1. J. Am. Chem. Soc. 1943, 65, 153–155. [Google Scholar] [CrossRef]
- Yam, S.C.; Zain, S.M.; Sanghiran Lee, V.; Chew, K.H. Correlation between polar surface area and bioferroelectricity in DNA and RNA nucleobases. Eur. Phys. J. E Soft. Matter. 2018, 41, 86. [Google Scholar] [CrossRef]
- Oró, J. Chemical synthesis of lipids and the origin of life. J. Biol. Phys. 1995, 20, 135–147. [Google Scholar] [CrossRef]
- Walde, P.; Wick, R.; Fresta, M.; Mangone, A.; Luisi, P.L. Autopoietic Self-Reproduction of Fatty Acid Vesicles. J. Am. Chem. Soc. 1994, 116, 11649–11654. [Google Scholar] [CrossRef]
- Deamer, D.; Georgiou, C. Hydrothermal Conditions and the Origin of Cellular Life. Astrobiology 2015, 15, 1091–1095. [Google Scholar] [CrossRef]
- Deamer, D. The Role of Lipid Membranes in Life’s Origin. Life 2017, 7, 5. [Google Scholar] [CrossRef]
- Karhu, J.; Epstein, S. The Implication of the Oxygen isotope Records in Coexisting Cherts and Phosphates. Geochim. Cosmochim. Acta 1986, 50, 1745–1756. [Google Scholar] [CrossRef]
- Knauth, L.P. Isotopic Signatures and Sedimentary Records. In Lecture Notes in Earth Sciences #43; Springer: Berlin, Germany, 1992; pp. 123–152. [Google Scholar]
- Knauth, L.P.; Lowe, D.R. High Archean climatic temperature inferred from oxygen isotope geochemistry of cherts in the 3.5 Ga Swaziland group, South Africa. Geol. Soc. Am. Bull. 2003, 115, 566–580. [Google Scholar] [CrossRef]
- Lowe, D.R.; Tice, M.M. Geologic evidence for Archean atmospheric and climatic evolution: Fluctuating levels of CO2, CH4, and O2 with an overriding tectonic control. Geology 2004, 32, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Knauth, L.P. Temperature and salinity history of the Precambrian ocean: Implications for the course of microbial evolution. Paleogeogr. Paleoclimatol. Paleoecol. 2005, 219, 53–69. [Google Scholar] [CrossRef]
- Schwartzman, D.; Lineweaver, C.H. The hyperthermophilic origin of life revisited. Biochem. Soc. Transact. 2004, 32, 168–171. [Google Scholar] [CrossRef]
- Fan, Y.; Fang, Y.; Ma, L. The self-crosslinked ufasome of conjugated linoleic acid: Investigation of morphology, bilayer membrane and stability. Colloids Surf. B Biointerfaces 2014, 123, 8–14. [Google Scholar] [CrossRef]
- Han, J.; Calvin, M. Occurrence of fatty acids and aliphatic hydrocarbons in a 3.4 billion-year-old sediment. Nature 1969, 224, 576–577. [Google Scholar] [CrossRef]
- Van Hoeven, W.; Maxwell, J.; Calvin, M. Fatty acids and hydrocarbons as evidence of life processes in ancient sediments and crude oils. Geochim. Cosmochim. Acta 1969, 33, 877–881. [Google Scholar] [CrossRef]
- Carballeira, N.; Sostre, A.; Stefanov, K.; Popov, S.; Kujumgiev, A.; Dimitrova-Konaklieva, S.; Tosteson, C.; Tosteson, T. The fatty acid composition of a Vibrio alginolyticus associated with the alga Cladophora coelothrix. Identification of the novel 9-methyl-10-hexadecenoic acid. Lipids 1997, 32, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Hippchen, B.; Röll, A.; Poralla, K. Occurrence in soil of thermo-acidophilic bacilli possessing ω-cyclohexane fatty acids and hopanoids. Arch. Microbiol. 1981, 129, 53–55. [Google Scholar] [CrossRef]
- Ponnamperuma, C.; Mariner, R. Formation of Ribose and Deoxyribose by Ultraviolet Irradiation of Formaldehyde in Water. Rad. Res. 1963, 19, 183. [Google Scholar]
- Ponnamperuma, C.; Mariner, R.; Sagan, C. Formation of Adenosine by Ultraviolet Irradiation of a Solution of Adenine and Ribose. Nature 1963, 198, 1199–1200. [Google Scholar] [CrossRef]
- Schoffstall, A.M. Prebiotic phosphorylation of nucleosides in formamide. Orig. Life Evol. Biosph. 1976, 7, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, G.; Saladino, R.; Crestini, C.; Ciciriello, F.; Di Mauro, E. Nucleoside phosphorylation by phosphate minerals. J. Biol. Chem. 2007, 282, 16729–16735. [Google Scholar] [CrossRef] [Green Version]
- Ponnamperuma, C.; Sagan, C.; Mariner, R. Synthesis of adenosine triphosphate under possible primitive Earth conditions. Nature 1963, 199, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Pasek, M.A.; Harnmeijer, J.P.; Buick, R.; Gull, M.; Atlas, Z. Evidence for reactive reduced phosphorus species in the early Archean ocean. Proc. Natl. Acad. Sci. USA 2013, 110, 10089–10094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turing, A.M. The Chemical Basis of Morphogenesis. Philos. Trans. R. Soc. Lond. Ser. B 1952, 237, 37–72. [Google Scholar]
- Glansdorff, P.; Prigogine, I. Thermodynamic Theory of Structure, Stability and Fluctuations; Wiley: Hoboken, NJ, USA, 1971. [Google Scholar]
- Levy, M.; Miller, S.L. The stability of the RNA bases: Implications for the origin of life. Proc. Natl. Acad. Sci. USA 1998, 95, 7933–7938. [Google Scholar] [CrossRef] [Green Version]
- Maier, G.; Endres, J. Isomerization of Matrix-Isolated Formamide: IR-Spectroscopic Detection of Formimidic Acid. Eur. J. Org. Chem. 2000, 2000, 1061–1063. [Google Scholar] [CrossRef]
- Miyakawa, S.; Cleaves, H.J.; Miller, S.L. The Cold Origin of Life: A. Implications Based on the Hydrolytic Stabilities of Hydrogen Cyanide and Formamide. Orig. Life Evol. Biosph. 2002, 32, 195–208. [Google Scholar] [CrossRef]
- Duvernay, F.; Trivella, A.; Borget, F.; Coussan, S.; Aycard, J.P.; Chiavassa, T. Matrix Isolation Fourier Transform Infrared Study of Photodecomposition of Formimidic Acid. J. Phys. Chem. A 2005, 109, 11155–11162. [Google Scholar] [CrossRef]
- Barks, H.L.; Buckley, R.; Grieves, G.A.; Di Mauro, E.; Hud, N.V.; Orlando, T.M. Guanine, Adenine, and Hypoxanthine Production in UV-Irradiated Formamide Solutions: Relaxation of the Requirements for Prebiotic Purine Nucleobase Formation. ChemBioChem 2010, 11, 1240–1243. [Google Scholar] [CrossRef]
- Kua, J.; Thrush, K.L. HCN, Formamidic Acid, and Formamide in Aqueous Solution: A Free-Energy Map. J. Phys. Chem. B 2016, 120, 8175–8185. [Google Scholar] [CrossRef]
- Wang, S.; Hu, A. Comparative study of spontaneous deamination of adenine and cytosine in unbuffered aqueous solution at room temperature. Chem. Phys. Lett. 2016, 653, 207–211. [Google Scholar] [CrossRef]
- Zheng, H.; Meng, F. Theoretical study of water-assisted hydrolytic deamination mechanism of adenine. Struct. Chem. 2009, 20, 943–949. [Google Scholar] [CrossRef]
- Gingell, J.; Mason, N.; Zhao, H.; Walker, I.; Siggel, M. VUV optical-absorption and electron-energy-loss spectroscopy of formamide. Chem. Phys. 1997, 220, 191–205. [Google Scholar] [CrossRef]
- Yonemitsu, E.; Isshiki, T.; Kijima, Y. Process for Preparing Adenine. U.S. Patent 4,059,582, 31 July 1974. [Google Scholar]
- Zubay, G.; Mui, T. Prebiotic Synthesis of Nucleotides. Orig. Life Evol. Biosph. 2001, 31, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, J.; Nguyen, M.T.; Springsteen, G.; Leszczynski, J. From Formamide to Purine: A Self-Catalyzed Reaction Pathway Provides a Feasible Mechanism for the Entire Process. J. Phys. Chem. B 2013, 117, 9333–9342. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.J.; Hinner, M.J. Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. Methods Mol. Biol. 2015, 1266, 29–53. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Clancy, C.; Harvey, R. Mechanisms Restricting Diffusion of Intracellular cAMP. Sci. Rep. 2008, 6, 19577. [Google Scholar] [CrossRef]
- Bowen, W.J.; Martin, H.L. The diffusion of adenosine triphosphate through aqueous solutions. Arch. Biochem. Biophys. 1964, 107, 30–36. [Google Scholar] [CrossRef]
- Samanta, T.; Matyushov, D.V. Dielectric friction, violation of the Stokes-Einstein-Debye relation, and non-Gaussian transport dynamics of dipolar solutes in water. Phys. Rev. Res. 2021, 3, 023025. [Google Scholar] [CrossRef]
- Chyba, C.F.; Sagan, C. Endogenous production, exogenous delivery and impact-shock synthesis of organic molecules: An inventory for the origins of life. Nature 1992, 355, 125–132. [Google Scholar] [CrossRef]
- Stribling, R.; Miller, S.L. Energy Yields for Hydrogen Cyanide and Formaldehyde Syntheses: The HCN and Amino Acid Concentrations in the Primitive Ocean. Orig. Life 1986, 17, 261–273. [Google Scholar] [CrossRef]
- Hardy, J.T. The sea-surface Microlayer (1982) Biology, Chemistry and Anthropogenic Enrichment. Prog. Oceanogr. 1982, 11, 307–328. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, C.; Liu, L.; Yu, L.; Wang, Z. Study on Dissolved Trace Metals in Sea Surface Microlayer in Daya Bay. Chin. J. Oceanol. Limnol. 2004, 22, 54–63. [Google Scholar]
- Fábián, B.; Szőri, M.; Jedlovszky, P. Floating Patches of HCN at the Surface of Their Aqueous Solutions—Can They Make “HCN World” Plausible? J. Phys. Chem. C 2014, 118, 21469–21482. [Google Scholar] [CrossRef] [Green Version]
- Grammatika, M.; Zimmerman, W.B. Microhydrodynamics of flotation processes in the sea-surface layer. Dyn. Atmos. Ocean. 2001, 34, 327–348. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Chemical Formula | Abbrev. in Text | Abbrev. in Kinetics | Figure 3 | nm | M cm | [D] | TPSA [Å] |
|---|---|---|---|---|---|---|---|---|
| hydrogen cyanide | HCN | HCN | H | 1 | 2.98 | 23.8 | ||
| cyanogen | NCCN | NCCN | Cg | 10 | 0.00 | 47.6 | ||
| formamide | HN-CHO | formamide | F | 220 | 60 [55,56] | 4.27 [57] | 43.1 | |
| formimidic acid | H(OH)C=NH | formimidic acid (trans) | Fa | 220 | 60 | 1.14 [57] | 43.1 * | |
| ammonium formate | NHHCO | ammonium formate | Af | +/−, 2.0 * | 41.1 | |||
| diaminomaleonitrile | CHN | cis-DAMN (DAMN) | C | 2 | 298 | 14,000 [58] | 6.80 [59] | 99.6 |
| diaminofumaronitrile | CHN | trans-DAMN (DAFN) | T | 3 | 313 | 8500 [58] | 1.49 [59] | 99.6 |
| 2-amino-3-iminoacrylimidoyl cyanide | CHN | AIAC | J | 4 | 275 | 9000 [5,60] | 1.49 | 99.6 * |
| 4-aminoimidazole-5-carbonitrile | CHN | AICN | I | 7 | 250 | 10,700 [58] | 3.67 | 78.5 |
| 4-aminoimidazole-5-carboxamide | CHNO | AICA | L | 10 | 266 [39] | 10,700 * | 3.67 * | 97.8 |
| 5-(N’-formamidinyl)-1H-imidazole-4-carbonitrileamidine | CHN | amidine | Am | 250 | 10,700 [61] | 6.83 * | 80.5 * | |
| adenine | CHN | adenine | A | 8 | 260 | 15,040 [62] | 6.83 [63] | 80.5 |
| hypoxanthine | CHNO | hypoxanthine | Hy | 9 | 250 | 12,500 [64] | 3.16 | 70.1 |
| xanthine | CHNO | xanthine | Xa | 12 | 268 [4] | 9300 [2] | 4.46 [6] | 86.9 [11] |
| guanine | CHNO | guanine | G | 11 | 252 [62] | 14,090 [62] | 5.45 [65] | 96.2 [65] |
| # | Reaction | Reaction Constants |
|---|---|---|
| 1 | H F | ; s; hydrolysis of HCN [5,91,94] |
| 2 | F → Fa | [55,56,90,92,93] |
| 3 | Fa → H + HO | [92,93,97] |
| 4 | F Af | ; s; hydrolysis of formamide [93,94] |
| 5 | 4H C | ; M s; kcal mol [5] |
| 6 | 4H T | ; M s; tetramization [5] |
| 7 | 4H + T + Cg C + T + Cg | ; M s [5] |
| 8 | 4H + T + Cg 2T + Cg | ; M s [5] |
| 9a | + C → T | [58] |
| 9b | + T → C | [5,58,60] |
| 10 | + T → J | [5,58,60] |
| 11 | + J → I | ; T ; [5,60] |
| 12 | I L | ; s; kcal mol; hydrolysis of AICN [6] |
| 13 | I:F + Af A + F | ; M s; kcal mol [98,99] |
| 14 | I:F + Fa Am + Fa +HO | ; M s; kcal mol [100] |
| 15 | + Am → A | [61] |
| 16 | A Hy | ; s; valid for pH within 5 to 8; hydrolysis of adenine [89,95] |
| 17 | L + Cg G + H | ; Ms; AICA + Cyanogen, kcal mol [6] |
| 18 | G Xa | ; s; valid for pH within 5 to 8; hydrolysis of guanine [89,95] |
| 19 | + C → C | |
| 20 | + T → T | |
| 21 | + J → J | |
| 22 | + Am → Am | |
| 23 | + I → I | |
| 24 | + L → L | |
| 25 | + A → A | |
| 26 | + Hy → Hy | |
| 27 | + G → G | |
| 28 | + Xa → Xa |
| 3.84 | 2.26 | 4.52 | 3.64 | 1.11 | 2.81 | 2.81 | 1.86 | 1.66 | 1.18 | 1.18 | 2.16 | 6.76 | 1.28 | 1.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández, C.; Michaelian, K. Dissipative Photochemical Abiogenesis of the Purines. Entropy 2022, 24, 1027. https://doi.org/10.3390/e24081027
Hernández C, Michaelian K. Dissipative Photochemical Abiogenesis of the Purines. Entropy. 2022; 24(8):1027. https://doi.org/10.3390/e24081027
Chicago/Turabian StyleHernández, Claudeth, and Karo Michaelian. 2022. "Dissipative Photochemical Abiogenesis of the Purines" Entropy 24, no. 8: 1027. https://doi.org/10.3390/e24081027
APA StyleHernández, C., & Michaelian, K. (2022). Dissipative Photochemical Abiogenesis of the Purines. Entropy, 24(8), 1027. https://doi.org/10.3390/e24081027