
A Novel Hybrid Monte Carlo Algorithm for Sampling Path Space

Abstract
1. Introduction
2. Time Evolution of the Hamiltonian
2.1. Splitting: ABA
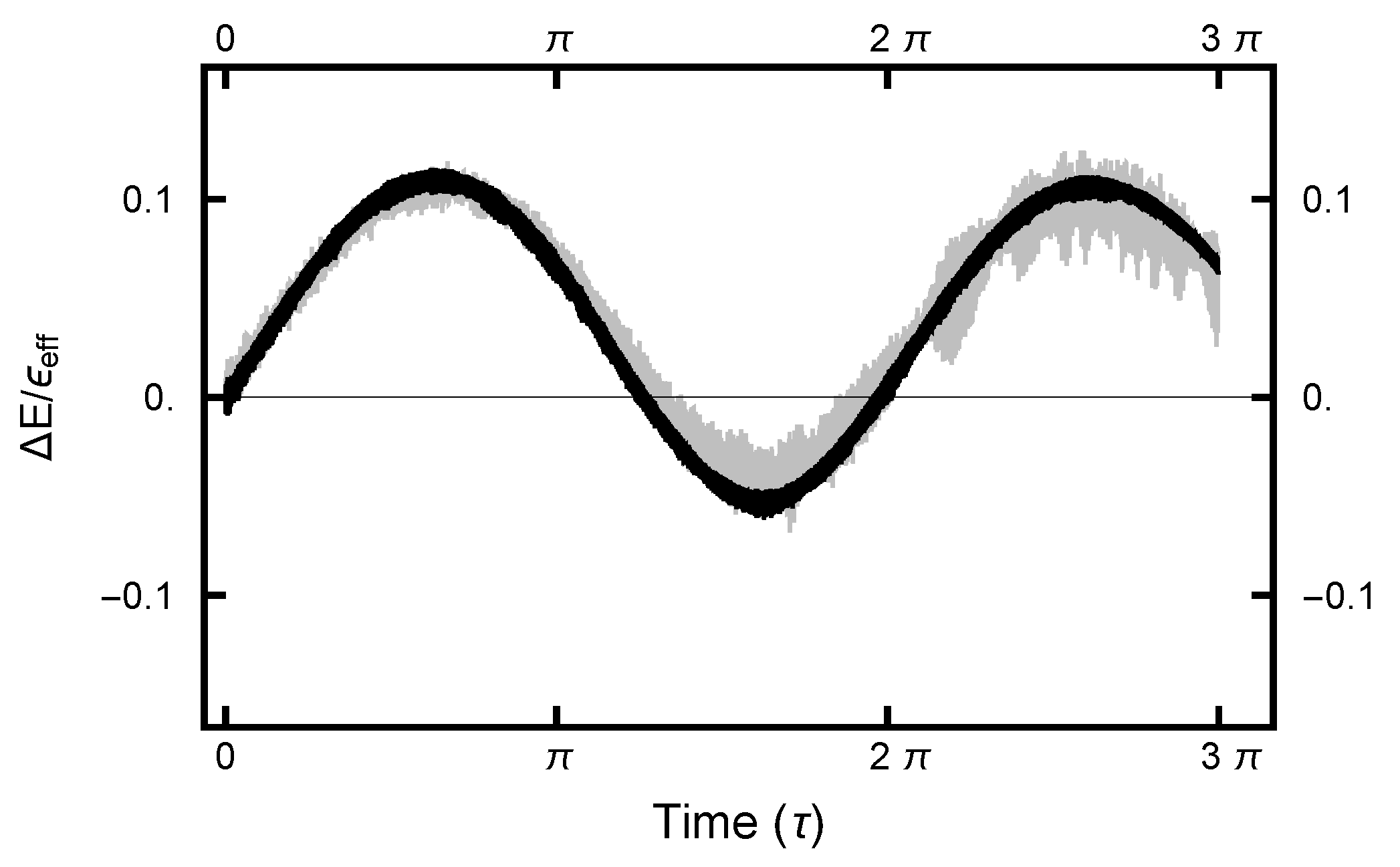
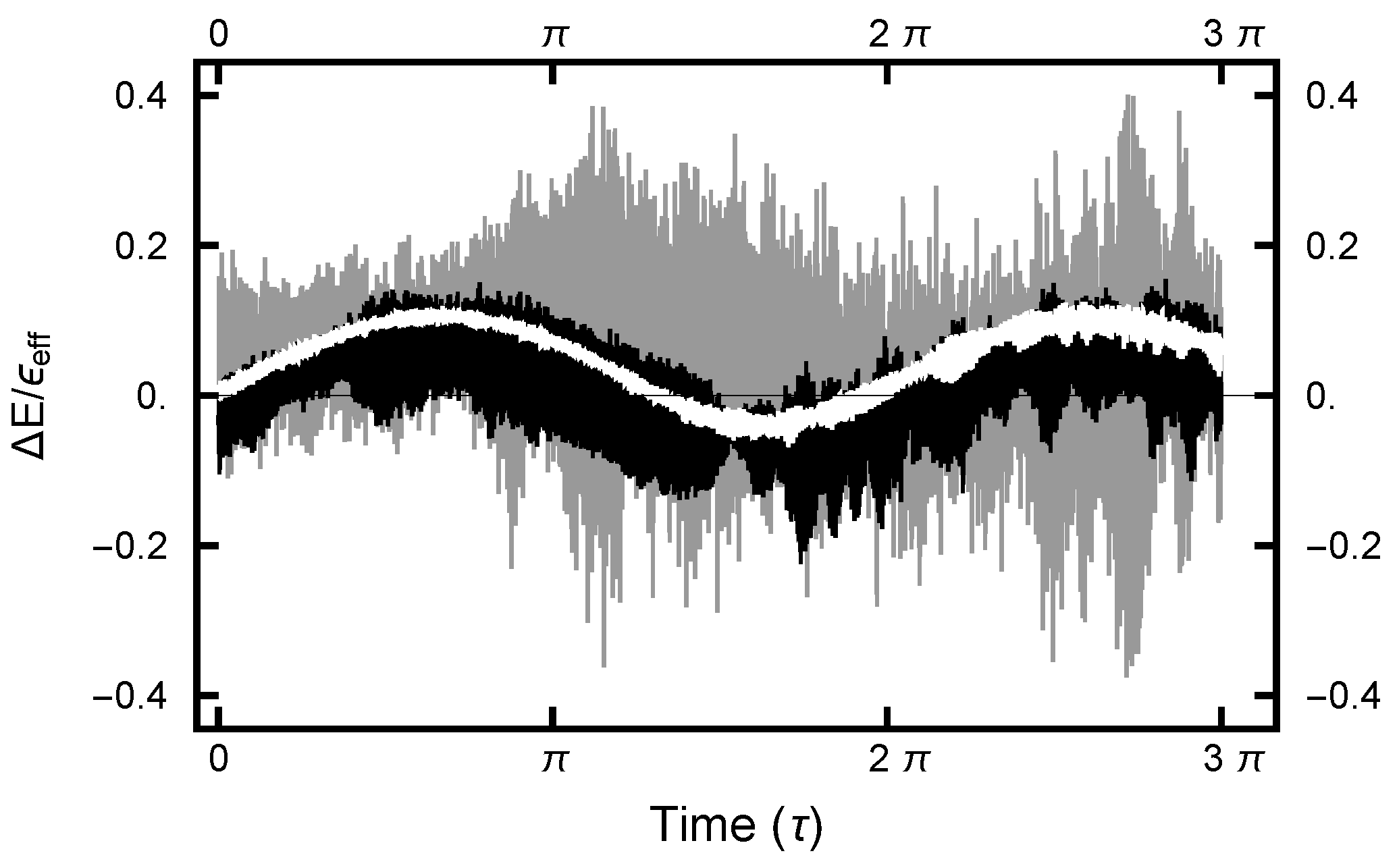
2.2. “Energy” Error
3. Numerical Experiments
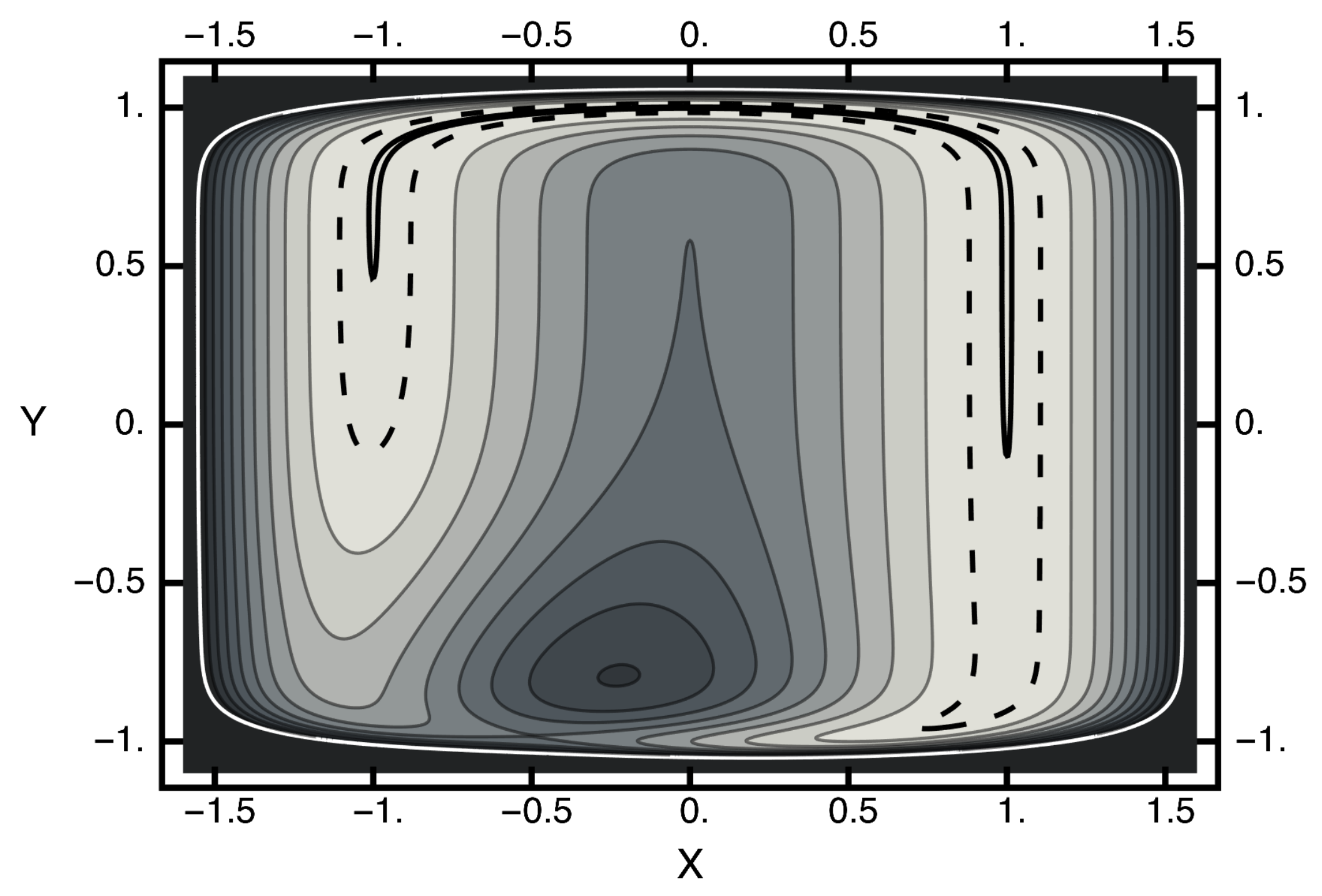
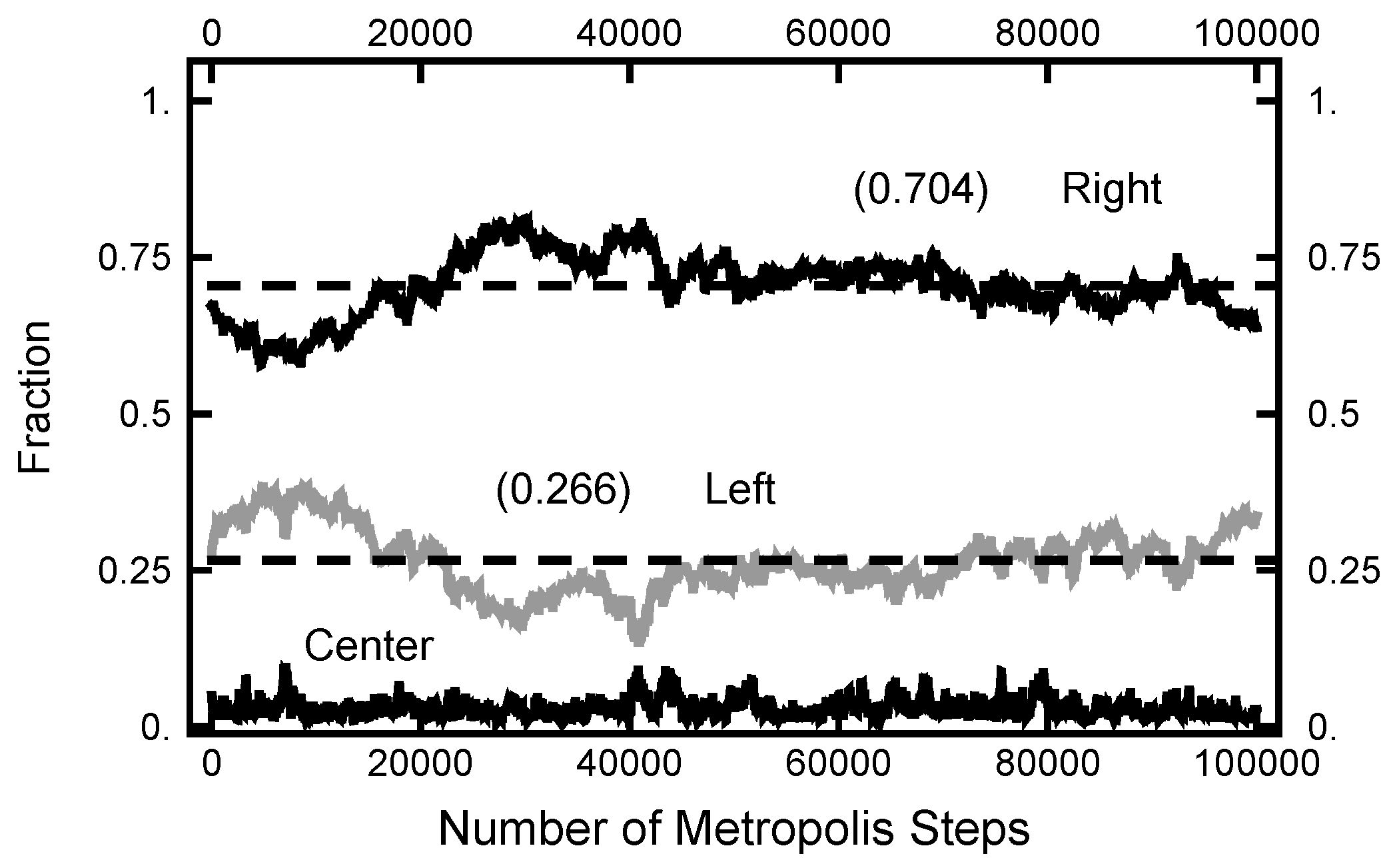
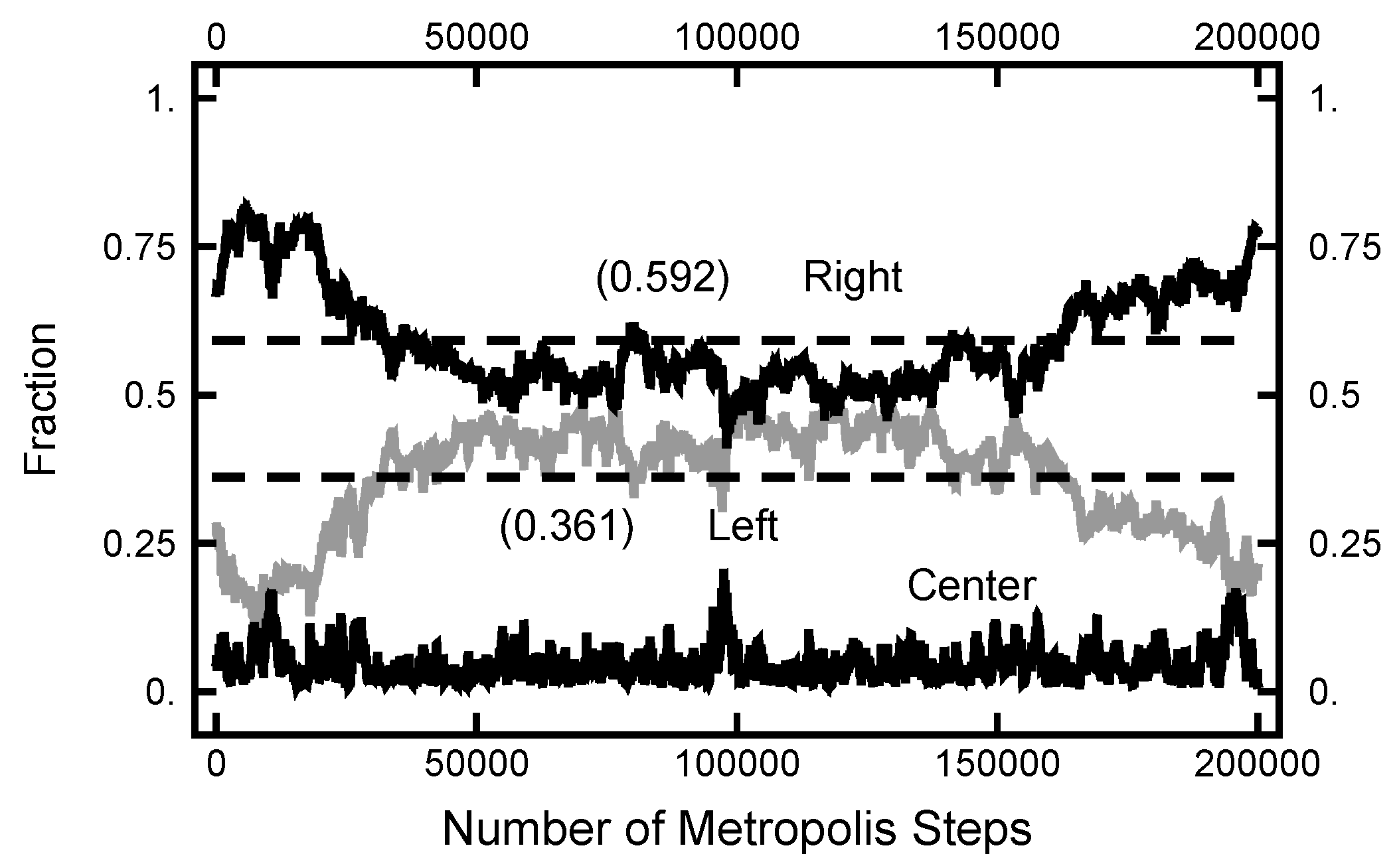
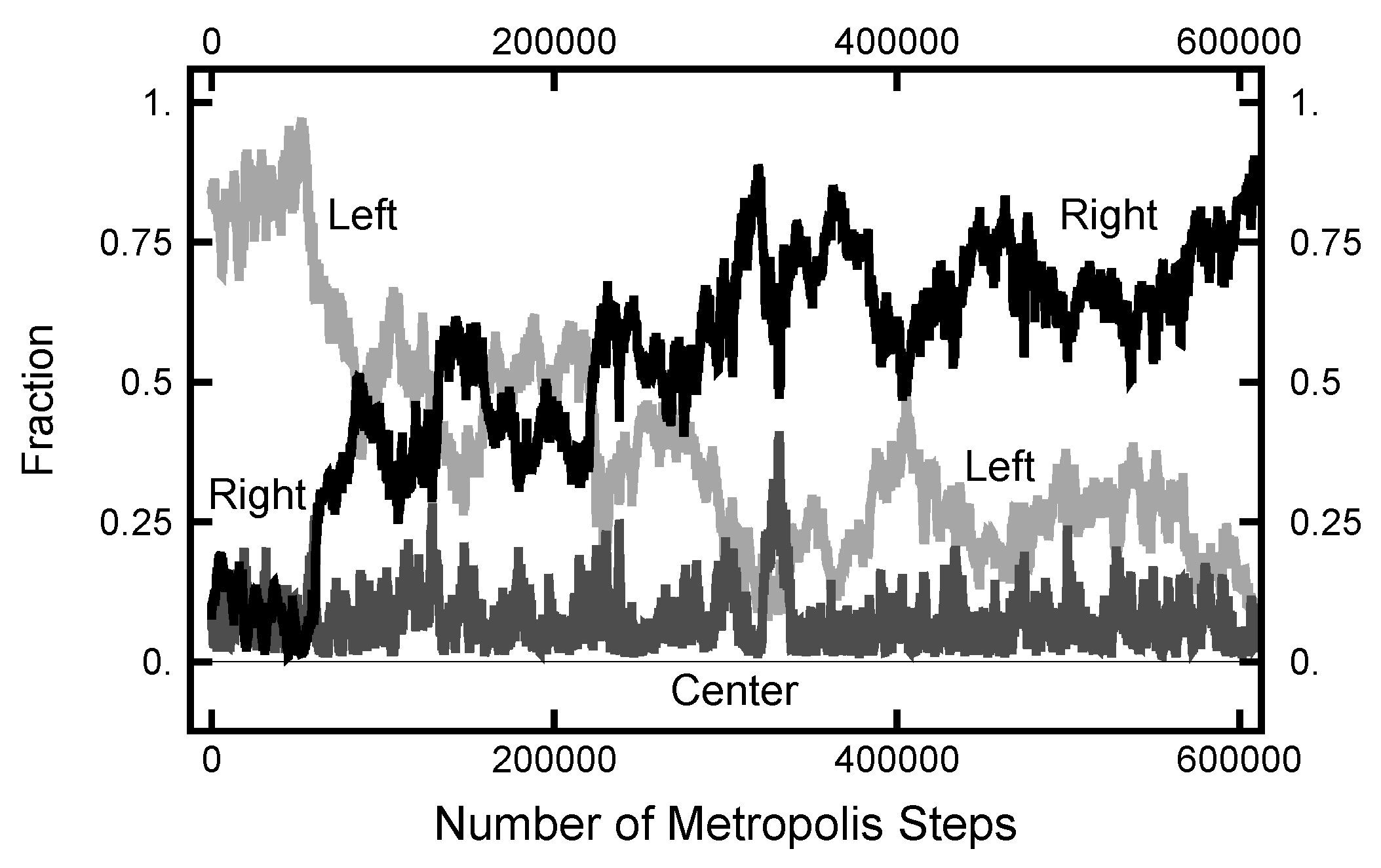
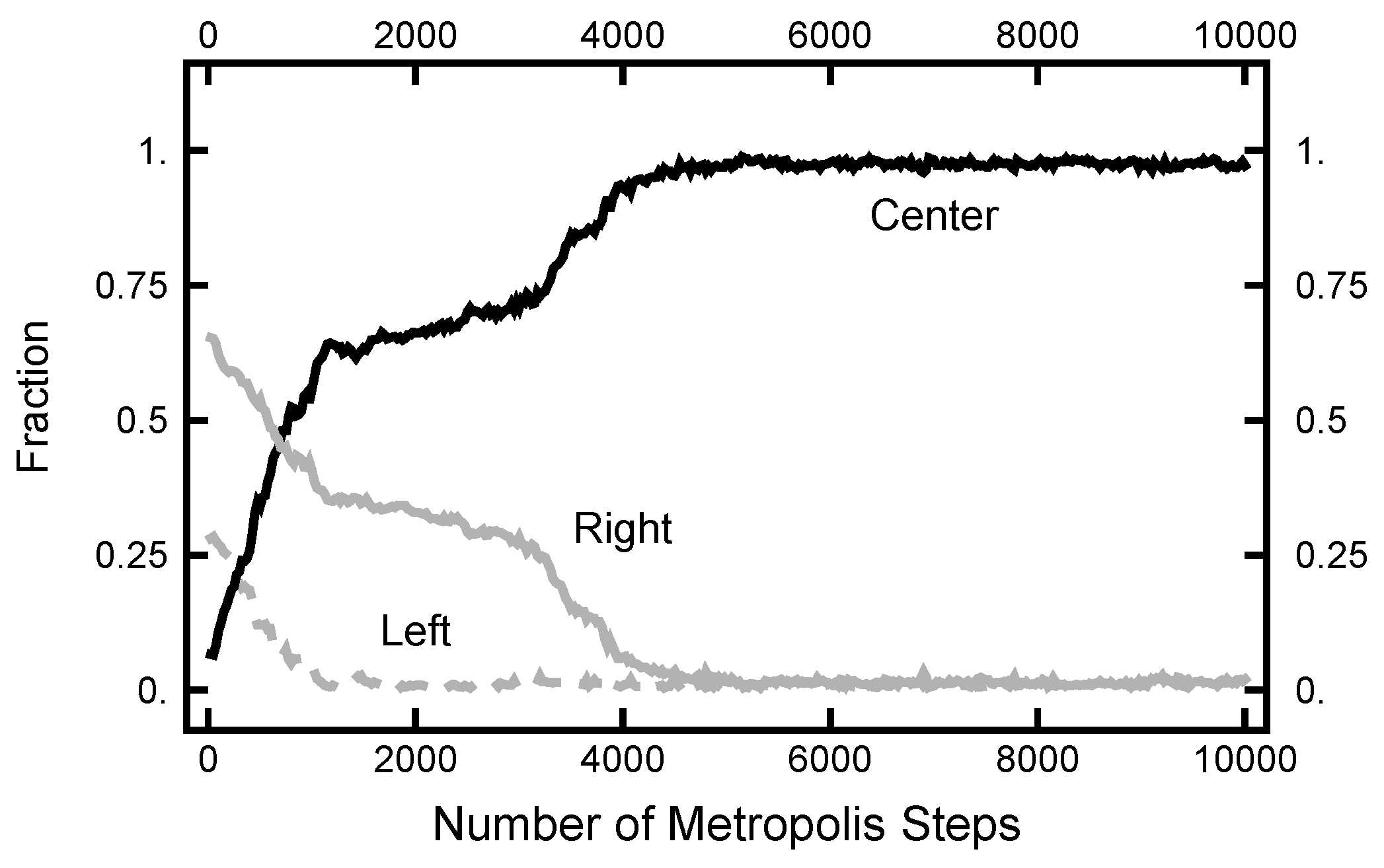
3.1. Equilibrium Distribution
3.2. Parameter Tuning
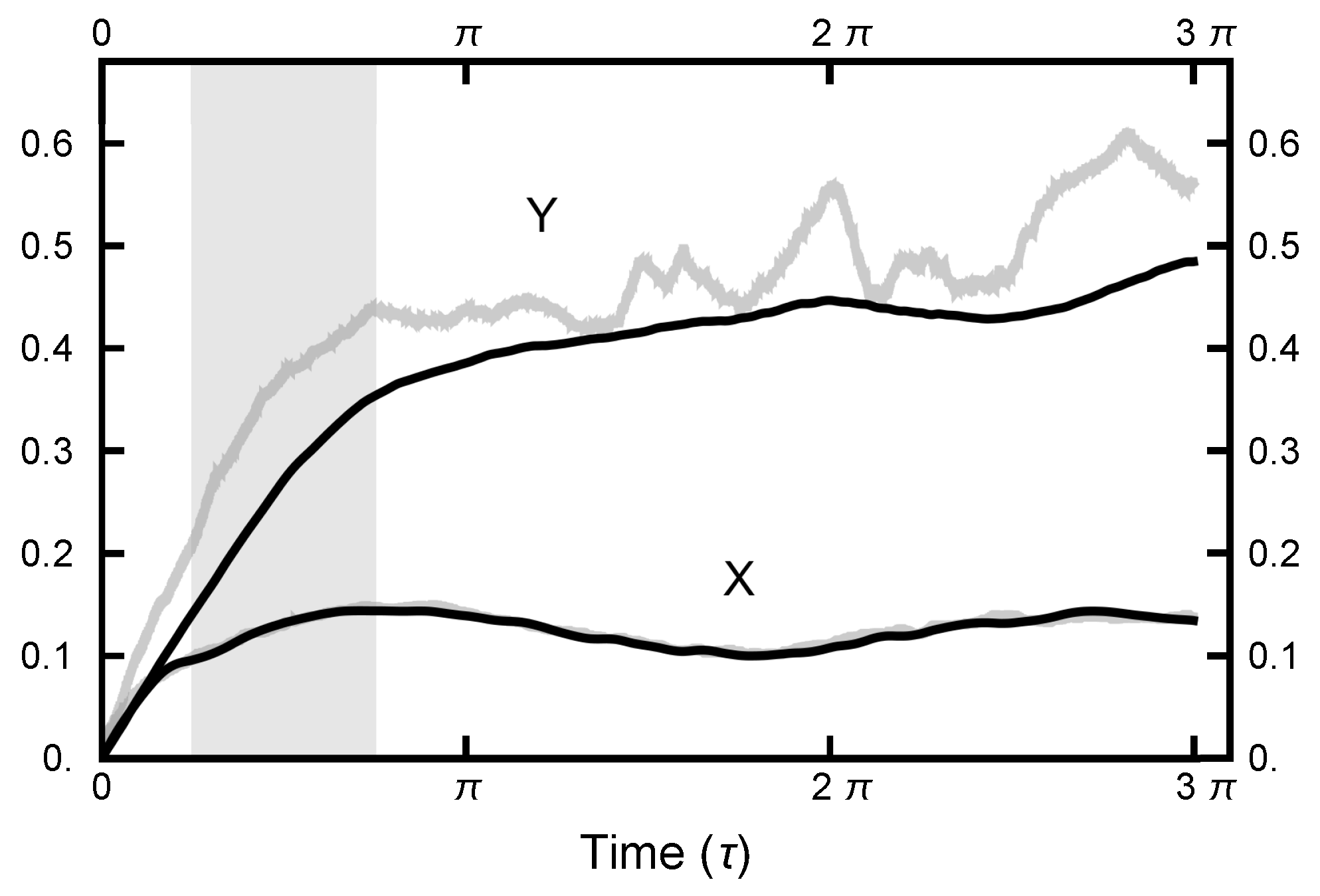
3.2.1. Path Length
3.2.2. Deterministic Integration Time
3.2.3. OU Bridge Parameter
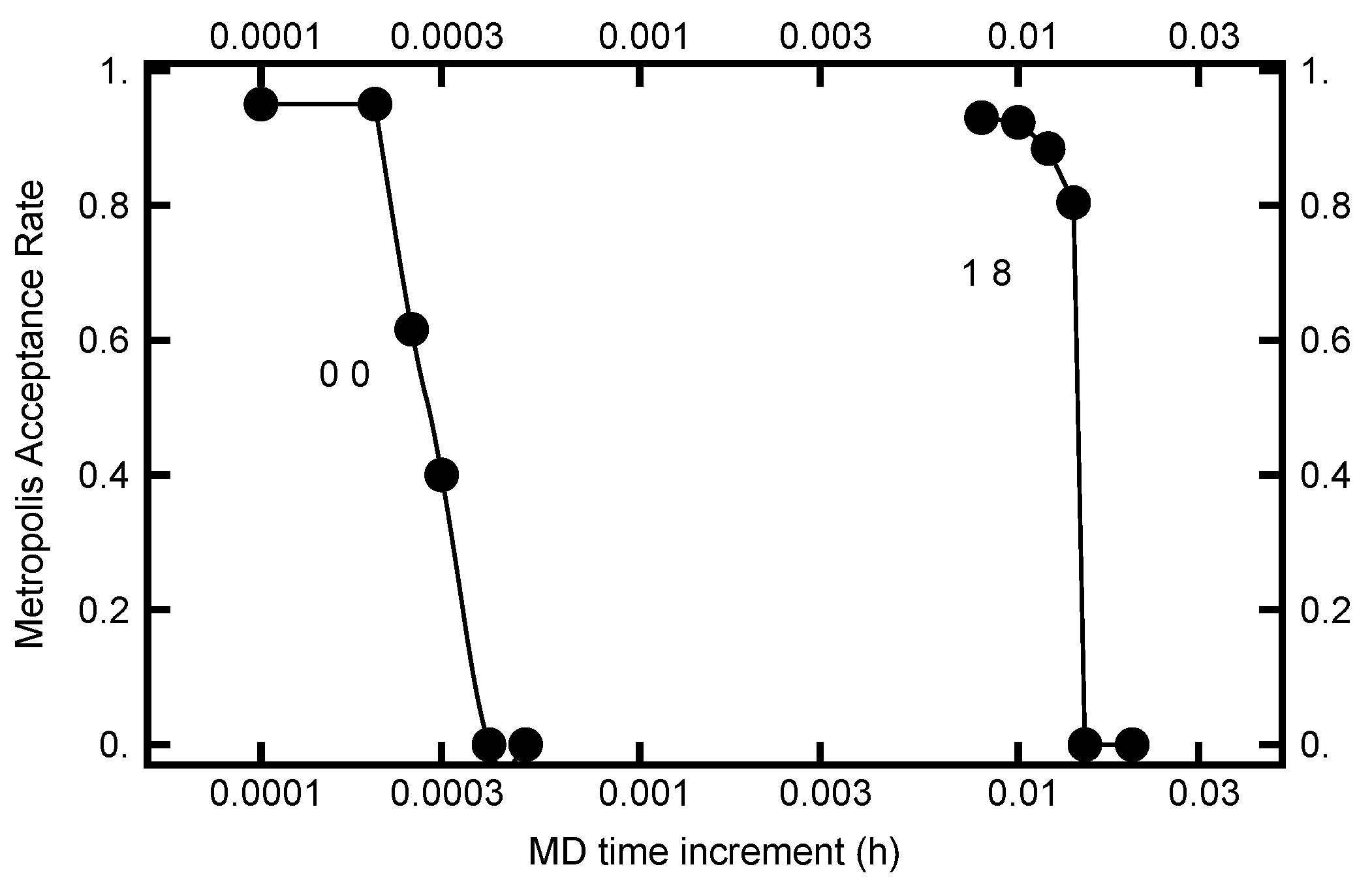
3.2.4. MD Time Step Size (h)
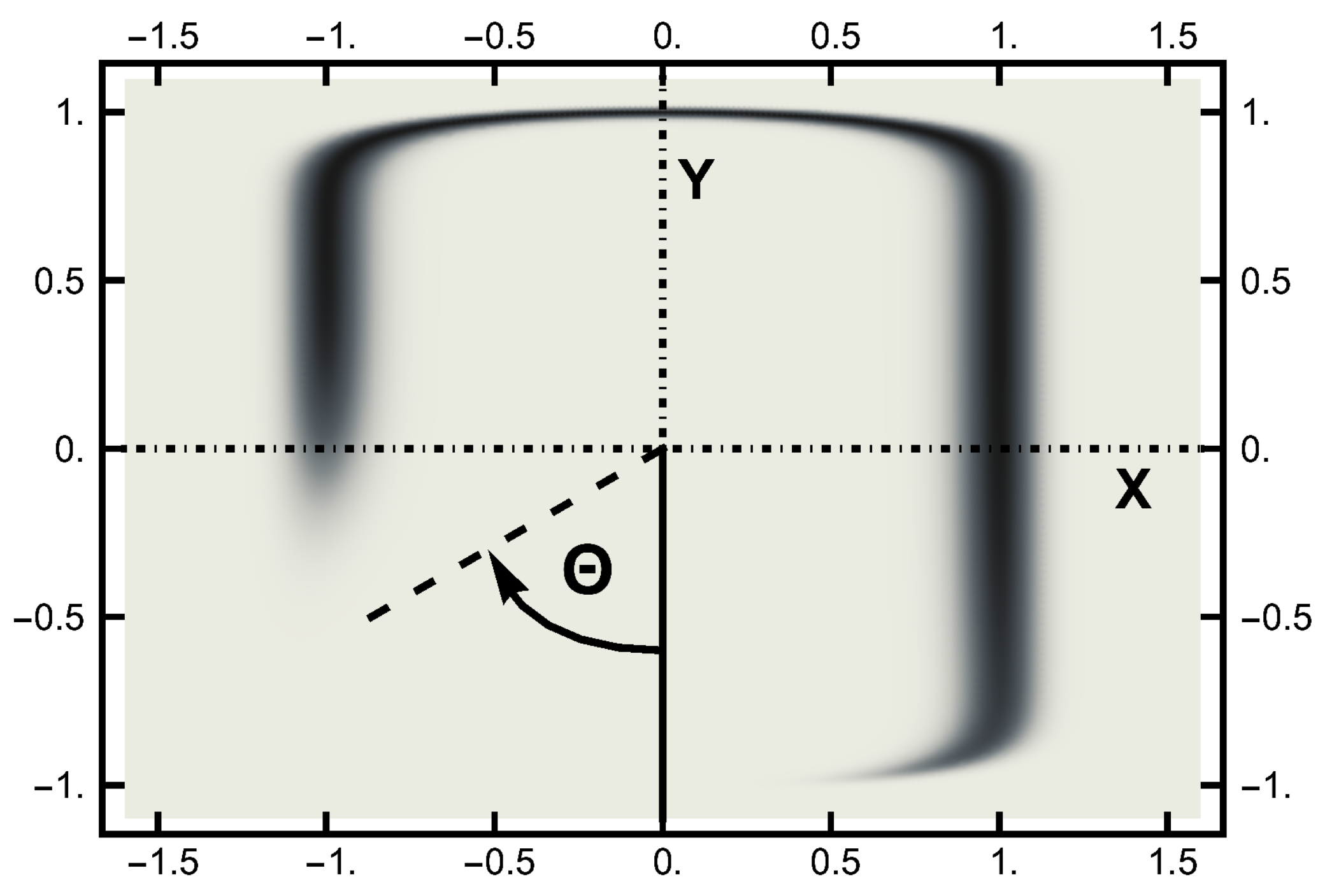
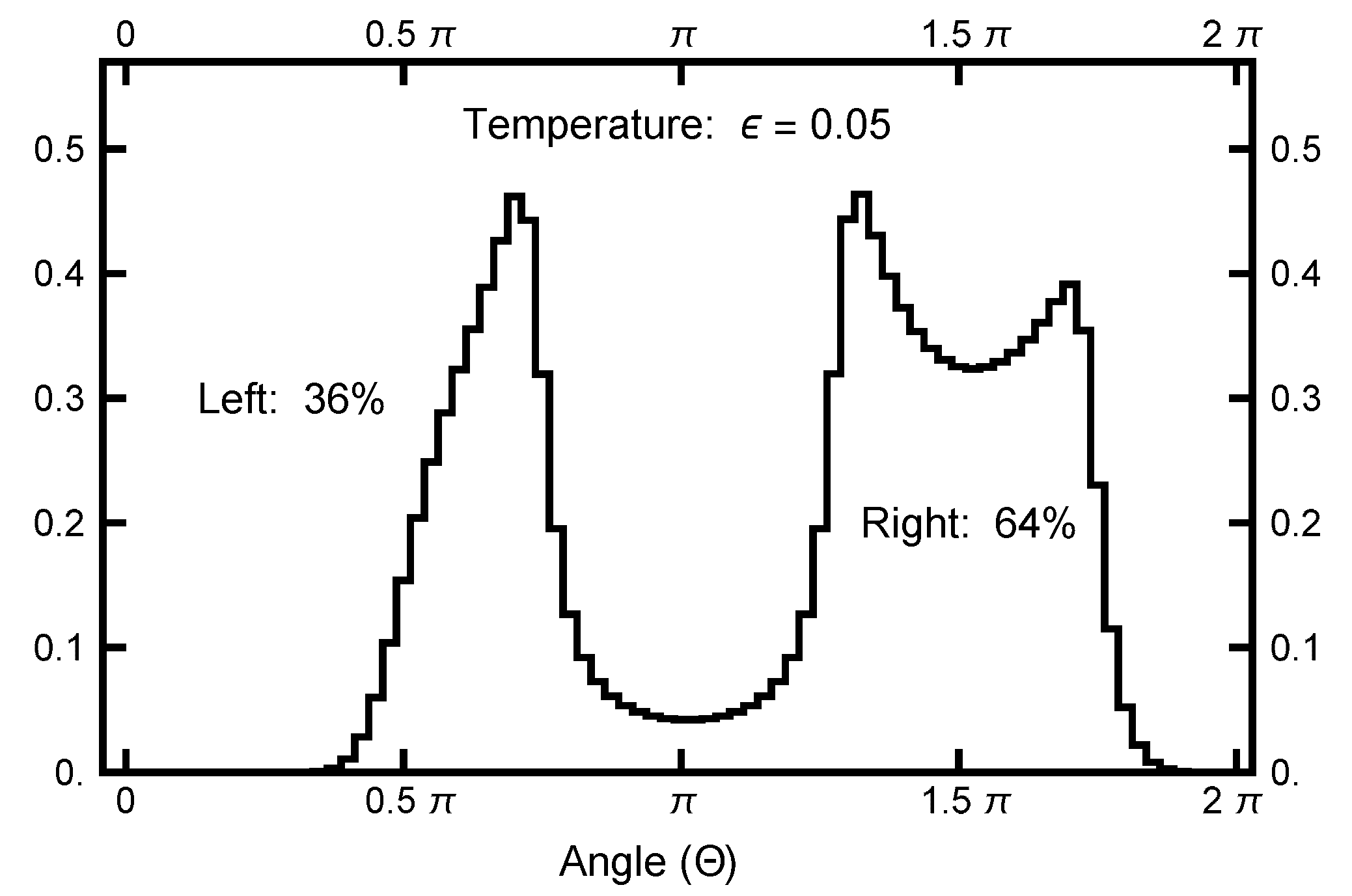
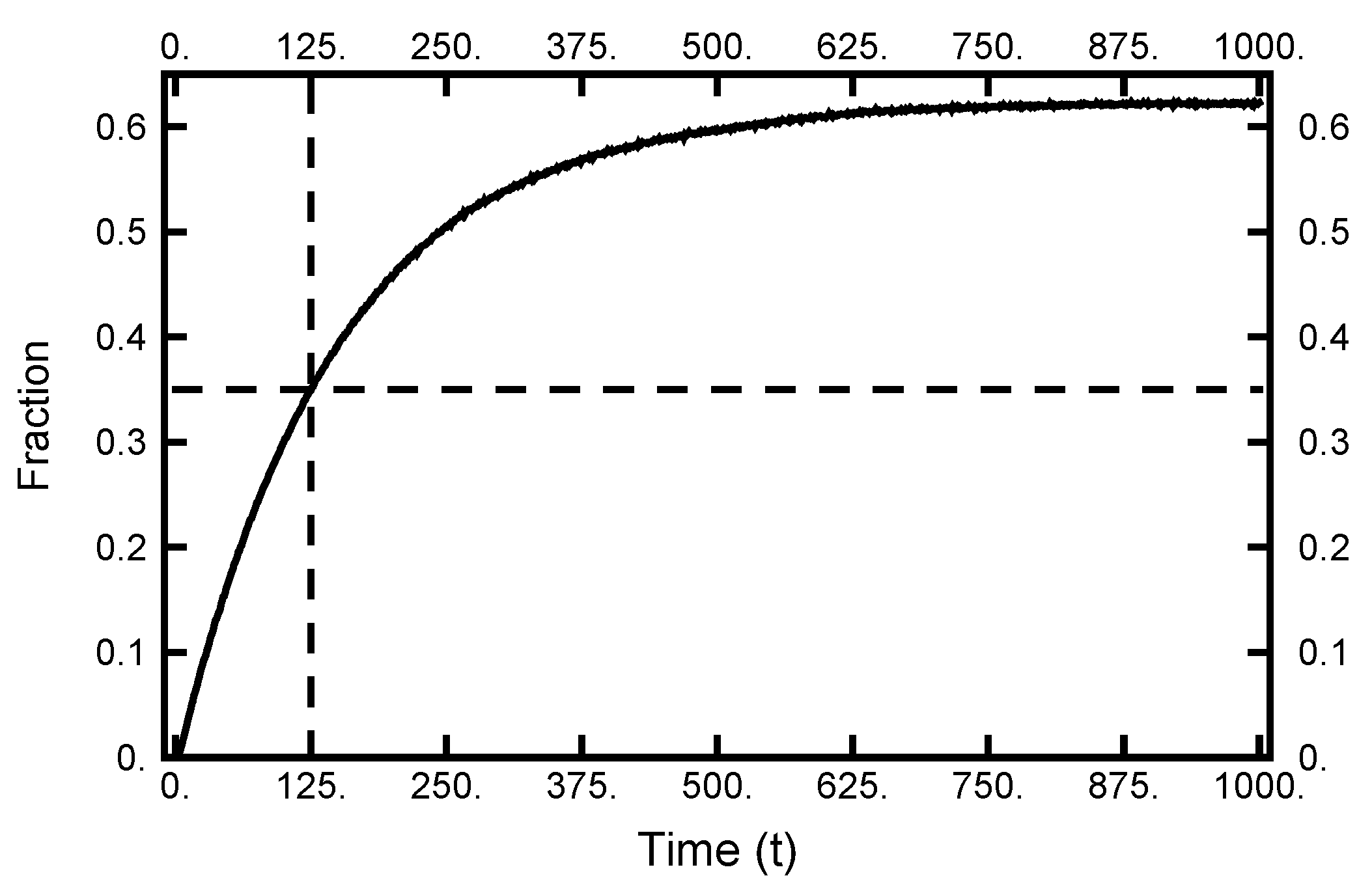
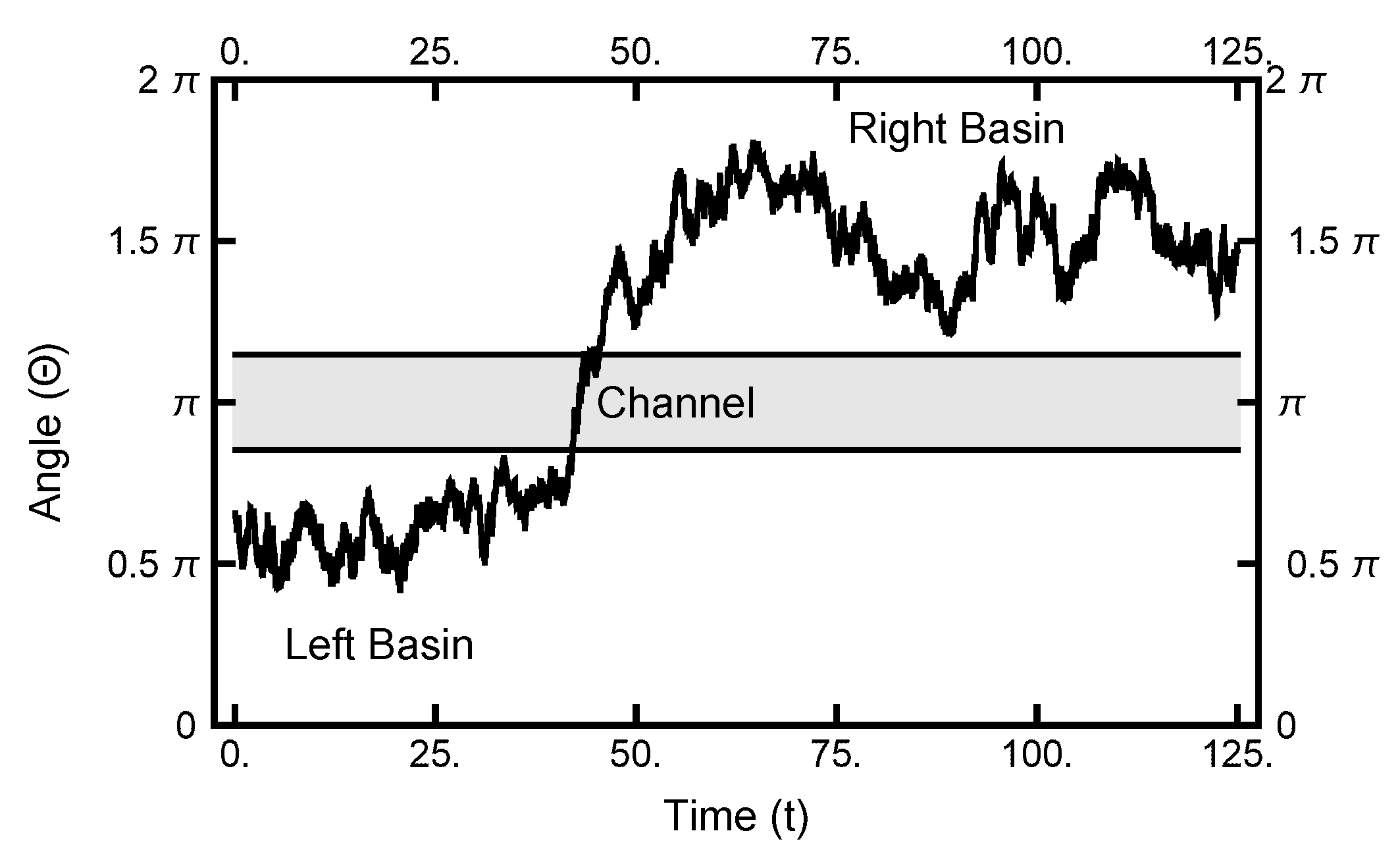
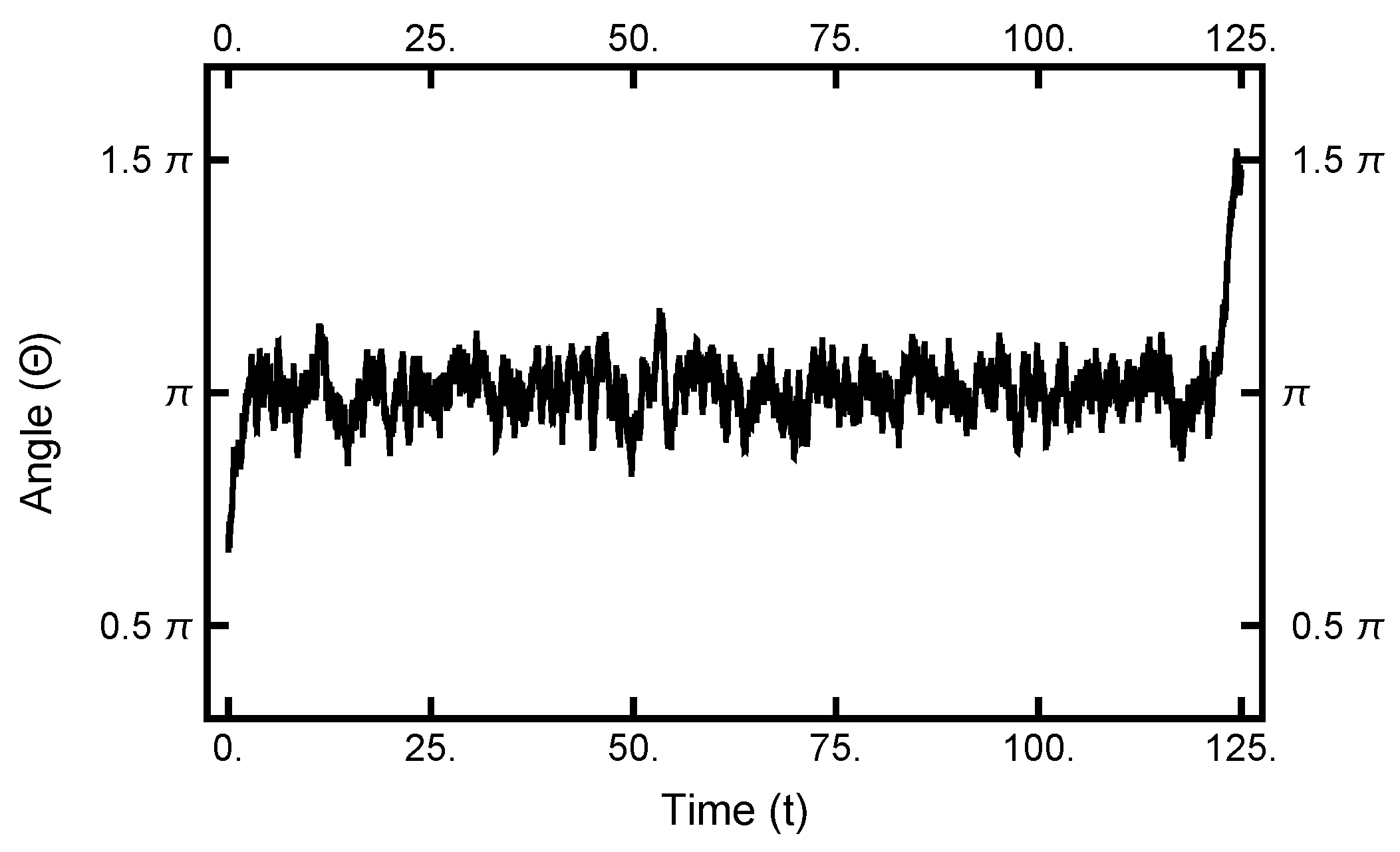
4. Path Sampling
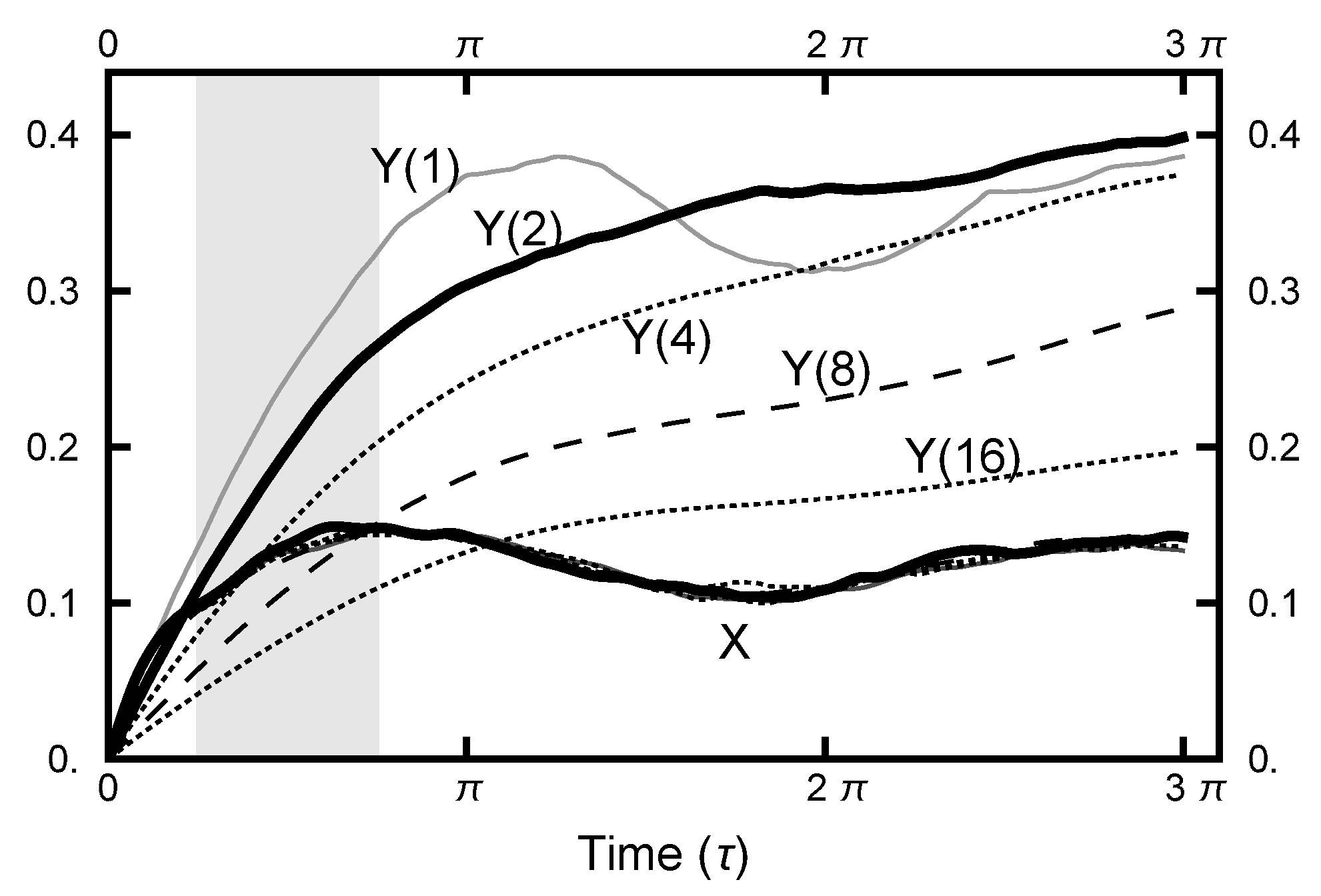
5. Continuous-Time Limit
6. Discussion
7. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Onsager–Machlup Functionals
Appendix B. Constructing Ornstein–Uhlenbeck Bridges
Appendix C. BAB Splitting: Numerical Integration
Appendix D. BAB Splitting: Energy Error
References
- Englander, S.W.; Mayne, L. The case for defined protein folding pathways. Proc. Natl. Acad. Sci. USA 2017, 114, 8253–8258. [Google Scholar] [CrossRef] [PubMed]
- Onsager, L.; Machlup, S. Fluctuations and irreversible processes. Phys. Rev. 1953, 91, 1505. [Google Scholar] [CrossRef]
- Beskos, A.; Pinski, F.; Sanz-Serna, J.; Stuart, A. Hybrid Monte Carlo on Hilbert spaces. Stoch. Process. Their Appl. 2011, 121, 2201–2230. [Google Scholar] [CrossRef]
- Duane, S.; Kennedy, A.; Pendleton, B.J.; Roweth, D. Hybrid Monte Carlo. Phys. Lett. B 1987, 195, 216–222. [Google Scholar] [CrossRef]
- Neal, R.M. Handbook of Markov Chain Monte Carlo; Brooks, S., Gelman, A., Jones, G., Meng, X., Eds.; CRC Press: Boca Raton, FL, USA, 2011; Chapter 5; pp. 113–162. [Google Scholar]
- Uhlenbeck, G.E.; Ornstein, L.S. On the Theory of the Brownian Motion. Phys. Rev. 1930, 36, 823–841. [Google Scholar] [CrossRef]
- Malsom, P.J.; Pinski, F.J. Role of Ito’s lemma in sampling pinned diffusion paths in the continuous-time limit. Phys. Rev. E 2016, 94, 042131. [Google Scholar] [CrossRef] [PubMed]
- Malsom, P. Rare Events and the Thermodynamic Action. Ph.D. Thesis, University of Cincinnati, Cincinnati, OH, USA, 2015. [Google Scholar]
- Trotter, H.F. On the product of semi-groups of operators. Proc. Am. Math. Soc. 1959, 10, 545–551. [Google Scholar] [CrossRef]
- Strang, G. On the construction and comparison of difference schemes. SIAM J. Numer. Anal. 1968, 5, 506–517. [Google Scholar] [CrossRef]
- Rowlands, G. A numerical algorithm for Hamiltonian systems. J. Comp. Phys. 1991, 97, 235–239. [Google Scholar] [CrossRef]
- Toxvaerd, S. Hamiltonians for discrete dynamics. Phys. Rev. E 1994, 50, 2271. [Google Scholar] [CrossRef] [PubMed]
- Øksendal, B. Stochastic Differential Equations; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Stuart, A.; Voss, J.; Wiberg, P. Conditional Path Sampling of SDEs And The Langevin MCMC Method. Comm. Math. Sci. 2004, 2, 685. [Google Scholar] [CrossRef]
- Korol, R.; Rosa-Raíces, J.L.; Bou-Rabee, N.; Miller, T.F., III. Dimension-free path-integral molecular dynamics without preconditioning. J. Chem. Phys. 2020, 152, 104102. [Google Scholar] [CrossRef] [PubMed]
- Pinski, F.J.; Stuart, A.M. Transition paths in molecules at finite temperature. J. Chem. Phys. 2010, 132, 184104. [Google Scholar] [CrossRef]
- Leimkuhler, B.; Matthews, C. Efficient molecular dynamics using geodesic integration and solvent–solute splitting. Proc. R. Soc. A Math. Phys. Eng. Sci. 2016, 472, 20160138. [Google Scholar] [CrossRef] [PubMed]
- Ottobre, M.; Pillai, N.S.; Pinski, F.J.; Stuart, A.M. A function space HMC algorithm with second order Langevin diffusion limit. Bernoulli 2016, 22, 60–106. [Google Scholar] [CrossRef]
- Lavenda, B. Thermodynamic criteria governing the stability of fluctuating paths in the limit of small thermal fluctuations: Critical paths in the limit of small thermal fluctuations: Critical paths and temporal bifurcations. J. Phys. A Math. Gen. 1984, 17, 3353. [Google Scholar] [CrossRef]
- Stratonovich, R. On the probability functional of diffusion processes. Selected Trans. Math. Stat. Prob. 1971, 10, 273–286. [Google Scholar]
- Maruyama, G. Continuous Markov processes and stochastic equations. Rend. Circ. Mat. Palermo 1955, 4, 48–90. [Google Scholar] [CrossRef]
- Barczy, M.; Kern, P. Sample path deviations of the Wiener and the Ornstein–Uhlenbeck process from its bridges. Braz. J. Probab. Stat. 2013, 27, 437–466. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinski, F.J. A Novel Hybrid Monte Carlo Algorithm for Sampling Path Space. Entropy 2021, 23, 499. https://doi.org/10.3390/e23050499
Pinski FJ. A Novel Hybrid Monte Carlo Algorithm for Sampling Path Space. Entropy. 2021; 23(5):499. https://doi.org/10.3390/e23050499
Chicago/Turabian StylePinski, Francis J. 2021. "A Novel Hybrid Monte Carlo Algorithm for Sampling Path Space" Entropy 23, no. 5: 499. https://doi.org/10.3390/e23050499
APA StylePinski, F. J. (2021). A Novel Hybrid Monte Carlo Algorithm for Sampling Path Space. Entropy, 23(5), 499. https://doi.org/10.3390/e23050499