1. Introduction
In this conceptual work we focus on the overall entropy/information content of electronic wavefunctions in the position representation of quantum mechanics (QM). Such quantum states are described by (complex) vectors in the molecular Hilbert space or by their statistical mixtures. Each state vector is characterized by its modulus (“length”) and phase (“orientation”) components in the complex plane. The square of the former determines the classical descriptor of the state probability distribution, while the gradient of the latter generates the density of electronic current and the associated velocity field reflecting the probability
convection. These physical descriptors summarize different aspects of the state electronic structure: the probability density represents the
static “structure of being”, while its flux characterizes the
dynamic “structure of becoming” [
1]. Indeed, in the underlying continuity equation for the (sourceless) probability distribution, the divergence of electronic flow, which shapes its time dependence, determines the local outflow of the probability density. The fundamental Schrödinger equation (SE) of QM ultimately determines the
time evolutions of the state wavefunction itself, its components, and expectation values of all physical observables.
As complementary descriptors of electronic structure and reactivity phenomena, both the modulus and phase parts of molecular states contribute to the overall (resultant) content of their entropy (uncertainty) and information (determinicity) descriptors [
2,
3,
4,
5,
6,
7,
8,
9]. The need for such generalized information-theoretic (IT) measures of the entropy/information content in electronic states has been emphasized elsewhere [
10,
11,
12,
13,
14]. Such descriptors combine the
classical terms due to wavefunction modulus (or probability density), and the
nonclassical contributions generated by the state phase (or its gradient determining the convection velocity). The overall gradient information, the quantum extension of Fisher’s intrinsic accuracy functional for locality events, then represents the dimensionless measure of the state electronic kinetic energy [
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15]. This proportionality relation between the state resultant information content and the average kinetic energy of electrons ultimately allows applications of the molecular virial theorem [
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29] in an information interpretation of the chemical bond and reactivity phenomena [
4,
6,
9,
30].
In principle, all these entropic contributions can be extracted by an experimental removal of the position and momentum uncertainties in the system quantum state [
31,
32]. For the parametrically specified particle location in the position representation of QM, both the probability distribution and its effective convectional velocity (current-per-particle) are uniquely specified by the system wavefunction. Therefore they both constitute bona fide sources of the information contained in electronic states, fully accessible in the separate position and momentum experiments.
In the stationary bound states, for the sharply specified energy, time-independent probability distribution and purely
time-dependent phase, the local phase component and probability convection identically vanish. It will be argued, however, that their (
phase-transformed)
equilibrium analogs exhibit latent electronic fluxes along probability contours, which do not affect the stationary probability density. These flows are related to the state local (“thermodynamic”) phase component, proportional to the negative logarithm of probability density, for which the
internal resultant IT descriptor of electronic state vanishes. Therefore, for this equilibrium criterion the average entropy measure in thermodynamic states becomes identical with von Neumann’s entropy [
33], the function of
external state probabilities defining the density operator of the ensemble mixed state.
In this analysis the quantum dynamics and continuity relations for the modulus (probability) and phase (current) degrees-of-freedom of electronic states are reexamined and their contributions to the resultant entropy/information descriptors are identified. The convection character of the net source of resultant gradient information is stressed, and equivalence of the energy and information criteria of chemical reactivity is emphasized. A distinction between classical (probability) and quantum (wavefunction) mappings is briefly discussed and the convection velocity of probability “fluid” is used to define fluxes of general physical and information properties. In such an approach, the system electrons thus act as carriers of the property densities. The latent electronic flows in the quantum stationary equilibrium, which do not affect the probability distribution, are also examined in some detail. Their quantum dynamics is examined and related to the “horizontal” phase component of “thermodynamic” equilibrium states. The local energy, probability acceleration, and force concepts are related to the state phase equalization and production. It is stressed that, contrary to the sourceless classical IT measures, the resultant descriptors exhibit finite local productions due to their nonclassical contributions.
2. Local Energy and Phase Equalization
Consider, for simplicity reasons, the quantum state |
ψ(
t)〉 of a single electron at time
t, and the associated (complex) wavefunction in position representation,
defined by its modulus
R(
r,
t) and phase
φ(
r,
t) ≥ 0 parts. The state logarithm then additively separates these two independent components:
where
p(
r,
t) =
R(
r,
t)
2 denotes the particle spatial probability density. Its real part determines the logarithm of the state classical (probability) component, while the imaginary part accounts for the nonclassical (phase) distribution:
The electron is moving in the external potential
v(
r), due to the fixed positions of the system constituent nuclei. In this Born–Oppenheimer (BO) approximation the (Hermitian) electronic Hamiltonian
determines the quantum dynamics of this molecular state, in accordance with the time-dependent SE.
This fundamental equation and its complex conjugate ultimately imply the associated dynamic equations for the wavefunction components or temporal evolutions of the associated physical distributions of the spatial probability and current densities (see the next section).
Consider the
stationary state corresponding to the sharply specified energy
Est.,
where
φst.(
r,
t) = −
ωst.t ≡
φst.(
t). In this state the probability distribution is
time-independent,
and the probability current exactly vanishes:
These eigenstates of electronic Hamiltonian,
correspond to the spatially equalized local energy
This equalization principle can be also interpreted as the related equalization rule for the state spatial phase. Indeed, introducing the local wave-number/phase concepts,
directly implies their spatial equalization in the stationary electronic state:
The
stationary equilibrium in QM is thus marked by the local phase equalization throughout the whole physical space. It should be realized that due to the complex nature of wavefunctions, the local energy of Equation (10) is also complex in character:
E(
r,
t) ≠
E(
r,
t)
*. This further implies the complex concepts of the local phase or wave-number,
which determines dynamic equations for the additive components of the state wavefunction of Equations (2) and (3). Rewriting SE in terms of complex wave-number components gives:
The real terms in this complex equation determine the
modulus dynamics,
while its imaginary terms determine the time evolution of the wavefunction phase:
For more SE identification of these wave-number components, the reader is referred to Equations (65) and (66) in
Section 5.
To summarize, the (complex) local energy generates a transparent description of the time evolution of wave-function components: its real contribution shapes the
phase dynamics, while the
modulus dynamics is governed by the imaginary components of
E(
r,
t) or
ω(
r,
t). In QM the spatial equalization of these wave-number or local-phase concepts marks the stationary state corresponding to the sharply specified energy, purely time-dependent phase, and time-independent probability distribution. We argue in
Section 7 and
Section 8 that these equilibrium states may still exhibit finite “hidden” flows of electrons, along probability contours, which can be associated with the local “horizontal” phase defining the
phase-transformed, “thermodynamic” states.
3. Origins of Information Content in Electronic States
The independent (real) parts of the complex electronic wavefunction of an electron in Equation (1) ultimately define the state physical descriptors of the spatial probability density
p(
r,
t) =
R(
r,
t)
2 and its current
The effective probability velocity introduced in the preceding equation measures a density of the current-per-particle,
and reflects the local convection momentum
P(
r,
t) ≡
ħ k(
r,
t), with
k(
r,
t) = ∇
φ(
r,
t) standing for its wave-vector factor.
The real and imaginary components of Equation (3), in the wavefunction logarithm of Equation (2), determine the independent probability and velocity densities, respectively. They account for the “static” and “dynamic” (convection) aspects of the state probability distribution, which we call the molecular structures of “being” and “becoming”. Both these organization levels ultimately contribute to the overall entropy or gradient-information contents in quantum electronic states and their thermodynamic mixtures [
2,
10,
11,
12,
13,
14].
The probability IT functionals
S[
p] and
I[
p], due to the logarithm of the state probability density of Equation (2), constitute the classical IT concepts of Shannon’s global entropy [
34,
35],
and Fisher’s information functional for locality events [
36,
37]:
In the associated resultant measures [
2,
10,
11,
12,
13,
14] these probability functionals are supplemented by the average nonclassical contributions
S[
φ] and
I[
φ] =
I[
j], due to the state phase or its gradient generating the probability velocity:
We also introduce the combined measure of the gradient-entropy,
The nonclassical entropy terms
S[
φ] and
M[
φ] ≡ −
I[
φ] = −
I[
j] are negative since the current pattern introduces an extra dynamic “order” into the system electronic “organization”, compared to the corresponding classical descriptors
S[
p] and
M[
p] =
I[
p], thus decreasing the state overall “uncertainty” content. These generalized descriptors of the resultant uncertainty (entropy) content
S[
ψ] in the quantum state
ψ, or of its overall (gradient) information
I[
ψ] [
2,
10,
11,
12,
13,
14], have been used to describe the phase equilibria in the substrate subsystems and to monitor electronic reconstructions in chemical reactions [
3,
4,
5,
13,
14,
38,
39,
40].
To summarize, in the resultant IT descriptors of the pure quantum state
ψ, the classical probability functionals, of Shannon’s global entropy or Fisher’s intrinsic accuracy for locality events, are supplemented by the corresponding nonclassical complements
S[
φ] or
I[
φ] =
I[
j], respectively, due to the wavefunction phase or the electronic current it generates. In the overall (“scalar”) entropy [
2,
10], the (positive) classical descriptor is combined with the (negative) average phase contribution,
while the complex (“vector”) entropy [
2,
12] represents the expectation value of the state (non-Hermitian) entropy operator
S = −2ln
ψ:
Therefore, the negative nonclassical entropy effectively lowers the state classical uncertainty measure
S[
p]. Indeed, the presence of finite currents implies more state spatial “order”, i.e., less electronic “disorder”. The resultant measure of the state average gradient information [
2,
10,
11,
12,
13,
14,
15],
then reflects the (dimensionless) kinetic energy of electrons:
T[
ψ] = 〈
ψ|T|
ψ〉 =
κ−1 I[
ψ].
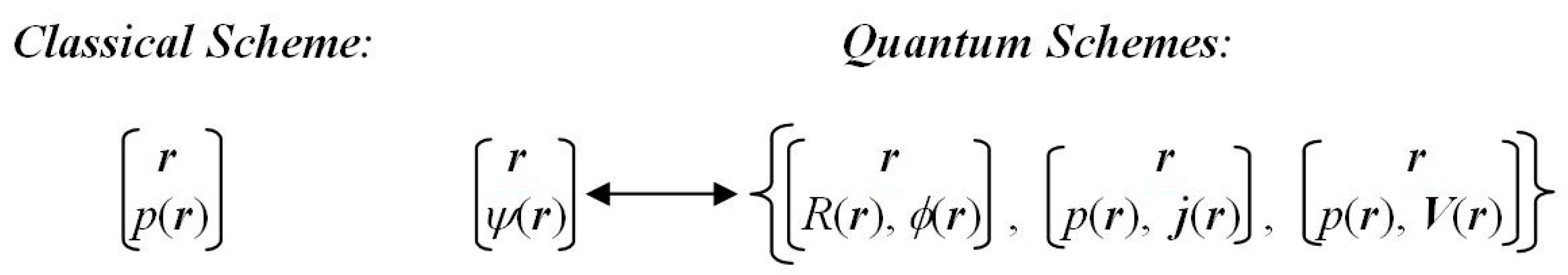
In both the classical IT and in position representation of QM the admissible locations {
r} of an electron exhaust the whole physical space and constitute the complete set of elementary particle-position events. The associated infinite and continuous
probability scheme of the classical mapping {
r→
p(
r)} in
Figure 1 thus describes a state of the position
indeterminacy (uncertainty). It is best reflected by Shannon’s global entropy
S[
p], measuring a “spread” (width) of the probability distribution, since we know only the probabilities
p(
r) = |
ψ(
r)|
2 of possible definite outcomes of the underlying localization experiment in the pure quantum state
ψ. Another suitable classical probe of the average information content in
p(
r) is provided by Fisher’s probability functional
I[
p]. This gradient measure of the position
determinacy reflects the “compactness” (height) of the probability distribution, thus complementing the Shannon global descriptor.
The information given us by carrying out the given experiment consists of removing the uncertainty existing before the experiment [
32]. If we carry out the particle-localization probe we obtain some information, since its outcome means that we then know exactly, which position has actually been detected. This implies that, after repeated trials performed for the specified quantum state, the initial uncertainty contained in the position
probability scheme has been completely eliminated. The average information gained by such tests thus amounts to the removed position uncertainty. The larger the uncertainty in
p(
r), the larger the amount of information obtained when we eventually find out which electron position has actually been detected after the experiment. In other words, the amount of information given us by the realization of the classical, probability scheme alone equals the global entropy in the classical probability scheme of
Figure 1 [
31,
32].
In QM, however, one deals with the
wavefunction scheme {
r →
ψ(
r)} of
Figure 1, in which the classical probability map {
r →
p(
r)} constitutes only a part of the overall (complex) mapping. In fact, the wavefunction mapping implies a simultaneous ascription to the parametrically specified electron position of the local modulus (static) and phase/current (dynamic) arguments of the state wavefunction, or the related local probability and probability velocity descriptors. This two-level scheme in QM ultimately calls for the
resultant measures of the entropy/information content in quantum states, combining classical (probability) and nonclassical (phase/current/velocity) contributions. The difference between the resultant and classical information contents can be best compared to that between the (phase-dependent) hologram and (phase-independent) ordinary photograph.
The resultant IT measures are in principle experimentally accessible, since the local probability velocity in physical space, defined by the velocity of probability current, is uniquely specified in QM. In other words, all static and dynamic arguments of the resultant IT descriptors are all sharply specified by the corresponding expectation values of the associated observables. However, the localization experiment alone cannot remove all the uncertainty contained in a general electronic state, which exhibits a nonvanishing local phase component
φ(
r,
t) and hence gives rise to a finite current density
j(
r,
t). This probability flux vanishes only in the stationary state of Equation (6), for the purely time-dependent stationary phase
φst.(
t):
jst.(
r,
t) =
Vst.(
r,
t) =
0. For such states an experimental determination of electronic position removes completely all the uncertainty contained in the spatial wavefunction
Rst.(
r) and the probability distribution
pst.(
r) =
Rst.(
r)
2. Indeed, the quantum scheme of
Figure 1 then reduces to the classical mapping alone.
Since the current operator
j(
r) includes the momentum operator of an electron,
P(
r) = −i
ħ∇,
which does not commute with the position operator
r(
r) =
r,
the incompatible observables
r and
j(
r) do not have common eigenstates. In other words, these quantities cannot be
simultaneously defined sharply, in accordance with Heisenberg’s uncertainty principle of QM. Therefore, the position dispersion
σr cannot be simultaneously eliminated with the current dispersion
σj in a single type of experiment, e.g., that of the particle localization. Indeed, a removal of
σj ultimately calls for an additional momentum experimental setup, which is incompatible with that required for determining the electronic position. Only the repeated,
separate localization and momentum experiments, performed on molecular systems in the same quantum state, can fully eliminate the position and current uncertainties contained in a general electronic state. Neertheless, both the particle position
r and the local convection velocity
V(
r) of the probability distribution are precisely defined as expectation values of the associated Hermitian operators. Therefore, their resultant IT functionals are all uniquely specified, with their densities exhibiting vanishing spatial dispersions.
The nonclassical uncertainty S[φ], proportional to the state average phase 〈φ〉ψ, effectively lowers the information received from the localization-only experiment. The removable uncertainty in ψ(r) is then less than its classical content S[ρ] or M[ρ] = I[ρ]. In other words, the nonvanishing current pattern introduces an extra (dynamic) determinacy in the system electronic structure, which diminishes its resultant uncertainty (indeterminacy) descriptors.
The
phase equilibria corresponding to
phase-transformed quantum states,
have been explored elsewhere [
2,
10,
11,
12,
13,
14]. The optimum local (“thermodynamic”) phase component
φeq.[
p,
r] ≡
φeq.(
r) for the specified probability density
p(
r)
= pst.(
r) in the stationary state
ψ =
ψst. of Equation (6) marks the exact cancellation of the state classical (
S[
p]) and nonclassical (
S[
φeq.]) entropy contributions:
We argue in the next section that this exact reduction of the “internal” (resultant) entropy content in the equilibrium “thermodynamic” state is essential for the consistency between the von Neumann thermodynamic entropy [
33] and the overall IT entropy in the
grand ensemble.
The above condition determines the equilibrium (“thermodynamic”, horizontal) local phase for the conserved (stationary) probability distribution,
proportional to the negative logarithm of probability density:
The same prediction follows from the condition of the vanishing gradient measure of the resultant entropy content in
ψeq.:
Indeed, solving this equation for
φeq. ≥ 0 (phase convention) gives:
We can also observe that writing the average functionals for resultant entropy measures as expectations of the corresponding (multiplicative) operators,
and
makes it possible to formally interpret the equilibrium phase of Equations (32) and (34) as the optimum solution defined by the extrema of these
wavefunction functionals:
4. Equilibrium States and Thermodynamic Entropy
Consider now the mixed quantum state in the
grand ensemble, the statistical mixture of molecular stationary states {|Ψ
ji〉 ≡ |Ψ
j(
Ni)〉} for different numbers of electrons {
Ni}, defined by the corresponding density operator,
where, O
ji = |Ψ
ji〉〈Ψ
ji| stands for the state projector. The average entropy or information—say, the resultant IT quantity
G represented by the associated operator G, possibly state-dependent, G = G[Ψ
ji] ≡ G
ji, is given by the weighted average of the property state-expectations {
Gji = 〈Ψ
ji|G|Ψ
ji〉}:
For example, the ensemble entropy of von Neumann [
33],
corresponds to the state entropy operator S
ji =
Sji O
ji and the expectation value of entropy in state Ψ
ji
This average value depends solely on the state
external probability
Pji in the mixture, shaped by thermodynamic conditions, and is devoid of any local (
internal) content of the constituent wavefunction distributions.
One would expect a similar feature in the overall IT description of molecular ensembles. In the pure quantum state |Ψ
ji〉 the probability of finding an electron at the specified location
r is given by the state internal distribution,
the shape factor of the associated electron density
ρji(
r). In thermodynamic ensemble it is given by the weighted average over such internal state densities {
pji(
r)}, with the state (external) probability weights {
Pji}:
The probability product
P(Ψ
ji,
r) represents the normalized
joint probability of finding in state Ψ
ji an electron at
r, with both its factors thus acquiring the status of
conditional probabilities:
The Shannon entropy in the ensemble joint distribution then separates into the “external” entropy
S[{
Pji}] of von Neumann and the weighted average of “internal” state contributions
For a consistent IT description of the equilibrium
mixed states of the open reactive complexes and their substrate subsystems, it would be desirable that in each
phase-transformed
pure state,
defined by its local (horizontal) phase
φeq.[
pji,
r] ≡
φ(h)(
r) (see also
Section 7 and
Section 8), equilibrium for the specified state probability density
pji(
r), the second (internal) contribution of Equation (45), exactly vanishes. This is indeed the case when the internal entropy of each equilibrium state is exactly zero:
In statistical mixtures of the equilibrium stationary states the only source of uncertainty is then generated by von Neumann’s ensemble entropy, determined by the “external” probabilities alone. This consistency requirement thus identifies the state equilibrium phase of Equation (32) [
2,
10,
11,
12,
13,
14]:
In such “horizontally”
phase-transformed states the thermodynamic and resultant equilibrium entropies are thus consistent with one another:
To summarize, the equilibrium “thermodynamic” (horizontal) phase is proportional to the local probability logarithm. This is very much in spirit of density-functional theory (DFT) [
41,
42,
43,
44,
45,
46]: the equilibrium stationary state is the unique functional of the system electron distribution
ρji(
r) =
Ni pji(
r), Ψ
ji,eq. = Ψ
eq.[
ρji], since both Ψ
ji = Ψ
ji[
ρji], by the first Hohenberg–Kohn (HK) [
41] theorem, and the equilibrium “thermodynamic” phase
φeq. =
φeq.[
pji].
Therefore, when the state “thermodynamic” phase satisfies the “equilibrium” criterion of Equation (30), the introduction of the
phase-transformed states for conserved (stationary) probability distribution generates the mutual consistency between the external (ensemble) and internal (resultant) entropy descriptors. It implies that for the single stationary state the resultant global and gradient uncertainty descriptors of the specified wavefunction vanish in equilibrium, as indeed does von Neumann’s [
33] entropy of the pure quantum state. In such states, the internal nonclassical (phase/current) contribution exactly cancels out the classical (probability) term. The equilibrium-phase condition of the state vanishing “internal” (resultant) IT descriptor then consistently predicts the equilibrium (horizontal) phase being related to the negative logarithm of the stationary probability distribution [
2,
10,
11,
12,
13,
14]:
5. Continuity Relations
It is of crucial importance for continuity laws of QM to distinguish between the reference frame moving with the particle (Lagrangian frame) and the reference frame fixed to the prescribed coordinate system (Eulerian frame). The total derivative
d/
dt is the time change appearing to an observer who moves with the probability flux, while the partial derivative ∂/∂
t is the local time rate of change observed from a fixed point in the Eulerian reference. These derivatives are related to each other by the chain-rule transformation,
where the velocity-dependent part
V(
r,
t) ⋅∇ generates the probability “convection” term.
In Schrödinger’s dynamical picture the state vector |
ψ(
t)〉 introduces an
explicit time dependence of the system wavefunction, while the dynamics of the basis vector |
r(
t)〉 of the position representation is the source of an additional,
implicit time dependence of the electronic wavefunction
ψ(
r,
t) =
ψ[
r(
t),
t], due to the moving reference (monitoring) point. This separation applies to wavefunctions, their components, and expectation values of physical observables. In
Table 1 we summarize the dynamic equations for the wavefunction modulus and phase components together with the continuity relations for the state probability, current, and information densities, which directly follow from the wavefunction dynamics of SE.
It directly follows from the SE that the probability field is sourceless:
Indeed, separating the explicit and implicit time dependencies in probability density
p(
r,
t) =
p[
r(
t),
t] gives:
Above, the total
time derivative
dp(
r,
t)/
dt determines the vanishing local probability “source”:
σp(
r,
t) = 0. It measures the time rate of change in an infinitesimal volume element of probability fluid
moving with probability velocity
V(
r,
t) =
dr(
t)/
dt, while the partial derivative ∂
p[
r(t),
t]/∂
t refers to a volume element around the
fixed point in space. The divergence of probability flux in the preceding equation,
thus implies the vanishing divergence of the velocity field
V(
r,
t), related to the phase Laplacian ∇
2φ(
r,
t) = Δ
φ(
r,
t):
As in fluid dynamics, in these transport equations the operators (
V⋅∇) and ∇
2 = Δ represent the “convection” and “diffusion”, respectively. Thus, in Equation (52), the local evolution of the particle probability is governed by the density “convection”, while the preceding equation implies the vanishing “diffusion” of the phase distribution.
In
Table 1 we summarize local continuity equations for the wavefunction components, the state physical descriptors, and information densities. For example, it follows from the table that the resultant gradient information exhibits a nonvanishing net production
σI(
t) due to a finite phase source
σφ(
r,
t). The classical contribution to
σI(
t) identically vanishes due to the probability continuity of Equation (52). These relations directly follow from the molecular SE and identify the relevant local sources of the distributions of interest.
As an example, consider continuities of the wavefunction components. When expressed in terms of the state modulus and phase parts the SE reads:
where we have used Equation (55). Dividing both sides by
ħR(
r,
t) and multiplying by exp[−i
φ(
r,
t)] gives the following (complex) dynamic relation linking the wavefunction components:
Comparing its
imaginary parts generates the time evolution of the modulus part of electronic state,
which can be directly transformed into the probability continuity equation
Equating the
real parts of Equation (57) similarly determines the phase dynamics
The preceding equation ultimately determines the production term
σφ(
r,
t) =
dφ(
r,
t)/
dt in the
phase-continuity relation
since the effective velocity
V(
r,
t) of the
probability current
j(
r,
t) =
p(
r,
t)
V(
r,
t) also determines the
phase flux and its divergence, the convection term in the continuity Equation (61):
This complementary flow descriptor ultimately identifies the finite phase production
Finally, using Equation (60) gives the following expression for the
phase source:
This production of the local phase is seen to group the probability-diffusion and phase-convection terms supplemented by the external potential contribution.
The component SE (57) also allows one to identify the wave-number distributions introduced in Equations (14)–(17):
and
To summarize, the effective velocity of the probability current also determines the phase flux in molecular states. The source (net production) of the classical
probability variable of electronic states identically vanishes, while that of their nonclassical
phase part remains finite. In overall descriptors of the state information or entropy contents they ultimately generate finite production terms. For example, the nonclassical information
I[
φ] generates the nonvanishing (integral) source of the average resultant gradient information
I[
ψ]:
Its density-per-electron
σI(
r,
t) is determined by a product of the local probability “flux”
j(
r,
t) and “affinity“ factor proportional to the gradient of the phase source. It also follows from this local information source in
Table 1, that it is determined by the “convection” of the phase source
σφ(
r,
t):
6. Principle of Stationary Resultant Information and Charge-Transfer Descriptors of Open Systems
The equilibrium subsystems in the specified (pure) state of the molecular system as a whole require the
mixed-state description in terms of
ensemble-average physical quantities [
31,
47,
48,
49,
50]. The same applies to the (externally) open microscopic systems in the applied thermodynamic conditions. In reactivity problems the specified temperature
T of the “heat bath”
𝕭(
T) and electronic chemical potential
μ (or electronegativity
χ = −
μ) of the macroscopic “electron reservoir” 𝓡(
μ) call for the
grand-ensemble approach [
44,
51,
52]. The equilibrium quantum state is then represented by the statistical mixture of the system
pure (stationary) states, defined by the externally imposed (equilibrium) state probabilities. Indeed, only the
ensemble-average value of the overall number of electrons 𝓝 ≡ 〈
N〉
ens. exhibits a continuous (fractional) spectrum of values justifying the
populational derivatives defining the reactivity criteria [
44,
51,
52]. The externally open molecule M(
v), identified by its external potential
v(
r) due to the system fixed nuclei, then constitutes a part of the
composed system 𝓜 = [M(
v)¦𝓡(
μ)] consisting of the mutually
open (microscopic) molecular fragment M(
v) and an external (macroscopic) electron reservoir 𝓡(
μ). In the theory of chemical reactivity one adopts such populational derivatives of the system ensemble-average energy and its underlying Taylor expansion in predicting reactivity behavior of molecules (
single-reactant criteria) or bimolecular reactive systems (
two-reactant criteria in situ) [
44,
53,
54,
55,
56,
57,
58,
59].
Such 𝓝-derivatives of electronic energy are indeed involved in definitions of several reactivity criteria, e.g., the chemical potential/electronegativity [
44,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62] or hardness/softness [
46,
56,
57,
58,
59,
63] and Fukui function (FF) [
44,
56,
57,
58,
59,
64] descriptors of the reaction complex. In IT treatments one introduces analogous concepts of the populational derivatives of the ensemble average (resultant) gradient information. Since reactivity phenomena involve electron flows between the
mutually open (polarized) substrates, only in such a generalized, ensemble framework can one precisely define the relevant reactivity criteria, determine the hypothetical states of the promoted subsystems, and eventually predict effects of their chemical coordination. It has been demonstrated that, in such an ensemble approach, the energetic and information principles are exactly equivalent, giving rise to identical predictions of thermodynamic equilibria, charge relaxation, and average descriptors of molecular systems and their fragments [
9,
10,
30,
65,
66].
The populational derivatives of the average energy and resultant information in reactive systems thus invoke the composite representation 〈M(
v)〉
ens. of the equilibrium state of the molecular system M(
v) in the grand ensemble. Thermodynamic conditions in the (microscopic) molecular system are thus imposed by the hypothetical (macroscopic) heat bath
𝕭(
T) and external electron reservoir 𝓡(
μ). The mixed state then corresponds to the equilibrium probabilities
P(
μ,
T;
v) ≡ {
Pji(
μ,
T;
v)} of the pure (stationary) states {|Ψ
ji〉 ≡ |Ψ
j(
Ni)〉}, with |Ψ
ji〉 denoting the
j-th state for
Ni (integer) number of electrons, which define the equilibrium density operator of Equation (37):
This statistical mixture of molecular states gives rise to the ensemble average values of the system electronic energy and its resultant gradient information. The former is defined by the quantum expectations of electronic Hamiltonians {H
i = H(
Ni,
v)},
while the latter corresponds to the quantum expectation of (Hermitian) operator for the resultant gradient information of
Ni electrons, {I
i ≡ I(
Ni) ≡ ∑
k I(
k)}, related to the corresponding kinetic-energy operators {T
i ≡ T(
Ni) = ∑
k T(
k)},
k = 1, 2, …,
Ni,
Thus the average gradient information
𝓘(D) reflects the (dimensionless) average kinetic energy
The equilibrium probabilities
P(
μ,
T;
v) result from the minimum principle of the
grand potential
Ω(D):
Here, the average number of electrons
and the thermodynamic entropy of the ensemble
with
kB denoting the Boltzmann constant.
The entropy-constrained
energy principle of Equation (74) can be also interpreted as an equivalent (potential-energy constrained)
information rule [
5,
6,
9,
67,
68,
69], for the minimum of the ensemble resultant gradient-information
𝓘(D):
It contains the additional constraint of the fixed overall potential energy, 〈
W〉
ens. = 𝓦(D), multiplied by the Lagrange multiplier
λ = −
κ, and includes the “scaled”
information intensities associated with the remaining constraints:
potential ζ = κ μ, enforcing the prescribed electron population 𝓝(D) = N;
temperature τ ≡ κ T, for the subsidiary entropy condition, Տ(D) = S.
The extrema of the ensemble principles of Equations (74) and (77) determine the same equilibrium probabilities P(μ, T; v) of electronic states. The physical equivalence of the energy and information principles indicates that energetic and information reactivity concepts are mutually related, being both capable of describing charge-transfer (CT) phenomena in acid(A)–base(B) systems.
The ensemble interpretation applies to all populational, 𝓝-derivatives of the average energy or information functionals. For example, in energy representation the global chemical hardness [
44,
63] reflects the 𝓝-derivative of the chemical potential,
while the information hardness measures the 𝓝-derivative of the information potential:
The positive signs of these “diagonal” (hardness) derivatives assure the external stability of 〈M(
v)〉
ens., with respect to charge flows between the molecular system M(
v) and its electron reservoir, in accordance with the Le Châtelier and Le Châtelier–Braun principles of thermodynamics [
70].
The global FF [
44,
56,
57,
58,
59,
64] is defined by the “mixed” second derivative of the ensemble average energy:
where we have applied the Maxwell cross-differentiation identity. It can be thus interpreted as either the density response per unit populational displacement, or as the response in the global chemical potential to unit displacement in the local external potential. The analogous derivative of the average gradient information similarly reads:
The in situ CT derivatives of the average resultant gradient information in the reactive system R = A–B include the CT potential quantity, related to
μCT,
and the CT hardness descriptor, related to
ηCT =
SCT−1,
which is the inverse of the CT softness
θCT = ∂
NCT/∂
ζ. In terms of these CT descriptors, the optimum amount of the B→A electron transfer in the donor–acceptor reactive system,
thus reads:
Above, {
NX0} and {𝓝
X} denote electron populations of the mutually
closed and
open reactants in M
+ = (A
+|B
+) and M
* = (A
*¦B
*) = M, respectively.
Therefore, the in situ derivatives {ζCT, ωCT = θCT−1} of the average content of the resultant gradient information provide alternative reactivity descriptors, equivalent to the chemical potential and hardness or softness indices {μCT, ηCT = SCT−1} of the classical, energy-centered theory of chemical reactivity. This again demonstrates the physical equivalence of the energy and information principles in describing the CT phenomena in molecular systems. One thus concludes that the resultant gradient information, the quantum generalization of the classical Fisher measure, constitutes a reliable basis for an “entropic” description of reactivity phenomena.
7. Latent Probability Flows in Stationary Equilibrium
Consider again the stationary state
ψst.(
r,
t) of an electron (Equation (6)) corresponding to the sharply specified energy
Est.. The wavefunction phase is then purely time dependent,
φst.(
r,
t) = −
ωst.t ≡
φst.(
t), with the state local aspect being described solely by its modulus part
Rst.(
r), the eigenfunction (see Equation (9)) of the electronic Hamiltonian of Equation (4). This stationary “equilibrium” thus generates the vanishing probability current
jst.(
r) =
pst.(
r)
Vst.(
r), where the time-independent probability distribution
pst.(
r) =
Rst.(
r)
2 and the vanishing flux-velocity
Vst.(
r) = (
ħ/
m) ∇
φst.(
t) =
0. As indicated in
Section 2, the eigenstates of the electronic Hamiltonian correspond to the equalized local energy,
Est.(
r,
t) =
Est., marking the equalized local phase:
φst.(
r,
t) =
φst.(
t).
Clearly, the stationary probability distribution and its vanishing current/velocity in such states do not imply that the particle is then at rest. The electrons are incessantly moving around the fixed nuclei, with the experimentally (sharply) unobserved instantaneous particle velocity W(r, t) = dr(t)/dt = P(r, t)/m reflecting its momentum P(r, t) = ħ k(r, t). Indeed, the system stability requires that centrifugal forces of these fast movements compensate for the nuclear attraction as, e.g., in Bohr’s historic, “planetary” model of the hydrogen atom. The tightly bound inner (“core”) electrons have to move faster than less confined outer (“valence”) electrons. The natural question then arises: how to describe the presence of these unceasing (latent) instantaneous motions in the dynamics of the probability “fluid”?
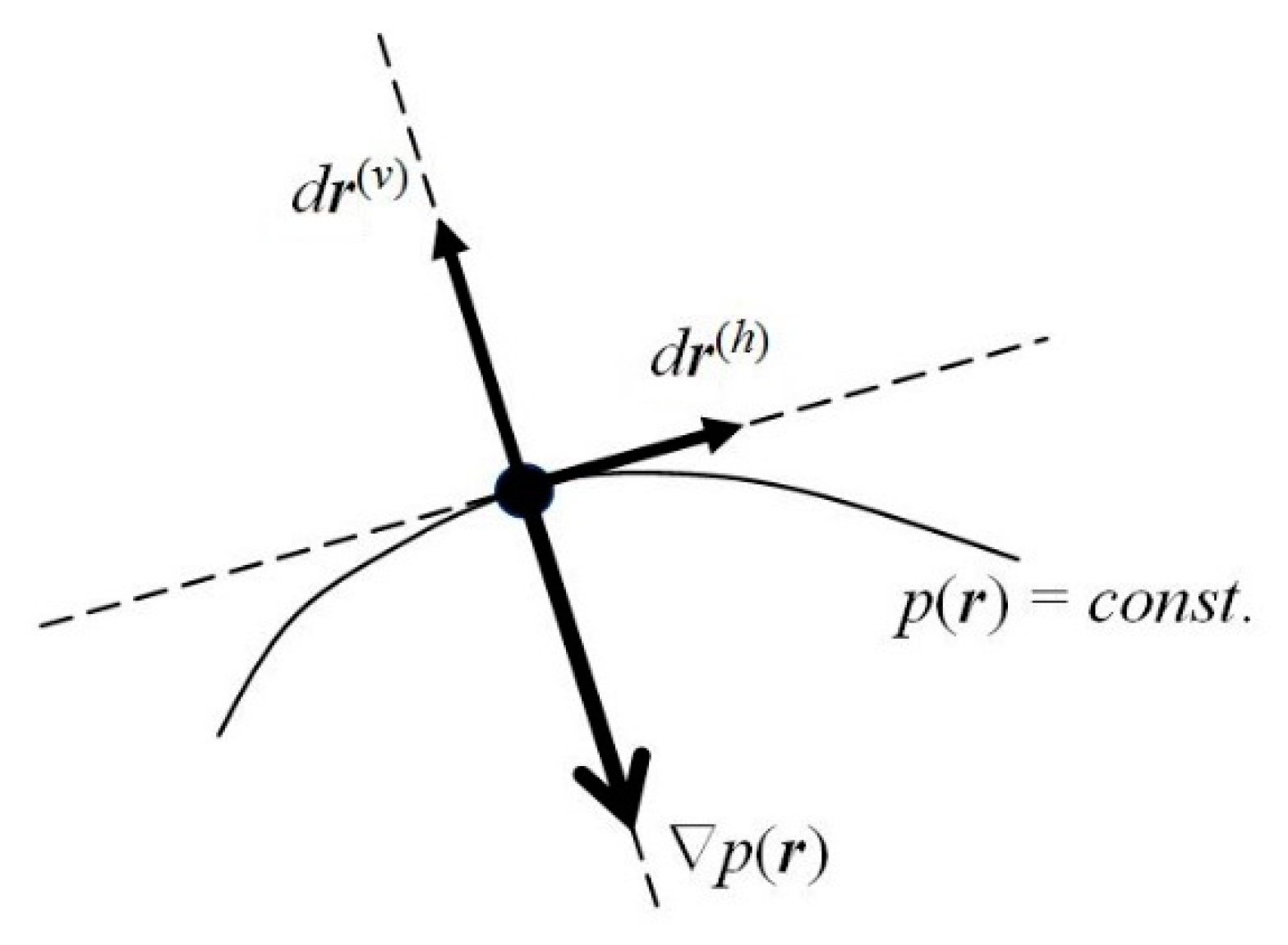
One observes that, for the probability density
pst.(
r) to remain conserved in time, its latent flows must follow the probability contours
pst.(
r) =
p(0) =
const. (see
Figure 2). Any motion in the direction perpendicular to the probability line passing a given location in space would imply a change in time of the probability value at this point, and hence the nonstationary character of the whole distribution. In other words, the latent flows of the stationary position-probability distribution must be “horizontal”, directed along the constant-probability lines. Such probability fluxes in
ψst., along the probability contours for the vanishing “vertical” velocity component, preserve in time the stationary character of the spatial probability distribution, which determines the vanishing probability flux. Therefore, the stationary character of the molecular electronic state does not preclude the latent local flows of electronic probability in horizontal directions generating the atomic vortices of
Figure 3.
The instantaneous
resultant (
r) velocity
V(r)(
r,
t) of probability “fluid” thus involves two independent components (see
Figure 2): the “
vertical” (current) velocity along the phase gradient,
perpendicular to the local direction of probability contour at time
t,
V ⊥ (
p =
const.), and hence parallel to ∇
p(
r,
t),
V||(∇
φ, ∇
p); and the “
horizontal” velocity
V(h)(
r,
t), along the probability contour,
The horizontal velocity
V(h)(
r,
t) of probability motions along the constant-probability lines,
V(h)||(
p =
const.), can also remain finite in the stationary electronic states of atomic or molecular systems, since it does not affect the conserved probability distribution. The vertical component
V of the probability current then reflects a common direction of gradients ∇
φ and ∇
p, of the distributions’ fastest increase, with a horizontal supplement perpendicular to both these gradients:
V(h)⊥(∇
φ, ∇
p).
These components of probability velocity imply the associated combination rules for the resultant probability and phase currents:
The above directional properties of the vertical and horizontal components then confirm the validity of the (vertical) continuity relations for the probability and phase distributions:
where we have recognized Equation (55) and observed that horizontal currents
j(h) and
J(h) generate vanishing divergences,
since
V(h) is perpendicular with respect to both ∇
p and ∇
φ.
Therefore, the phase and probability gradients are both perpendicular to the probability contour and, hence, ∇
φ(
r,
t) ∝ ∇
p(
r,
t). This directional character of the current velocity
V(
r,
t) suggests that the local aspect of the phase function itself should be related to the probability density:
Such a directional feature indeed characterizes the IT equilibrium (“thermodynamic”) phase of Equation (32) (see also
Section 4), resulting from extrema of the phase entropy/information functionals,
for which
The velocity of the latent, “horizontal” flows along the probability contours can be then attributed to the additional (local)
horizontal phase
φ(h)(
r) component, a “thermodynamic” addition to the purely time-dependent stationary phase
φst.(
t) in the resultant phase of the transformed state:
In order to study the time-dependent flows in liquids, the separate concepts of “streamline” and “pathline” are introduced [
71]. At the specified time, the former are tangential to the directional field of velocity “arrows”. Since the particles move in the direction of the streamlines, there is no motion perpendicular to the streamlines and the property flux per unit time between two streamlines remains constant. Patterns of streamlines describe the instantaneous state of a flow, indicating the direction of motion of all particles at a given time. For the time-dependent flows, the velocity field changes in time, with pathlines no longer coinciding with streamlines. Only for the time-independent flows do the particles move along streamlines, so that pathlines and streamlines coincide.
In the stationary quantum mechanics the contours of molecular probability “fluid” at time
t =
t0,
p(
r,
t0) =
pst.(
r) similarly determine the streamlines of the latent (horizontal) flows of electronic probability, which preserve the “static” probability distribution
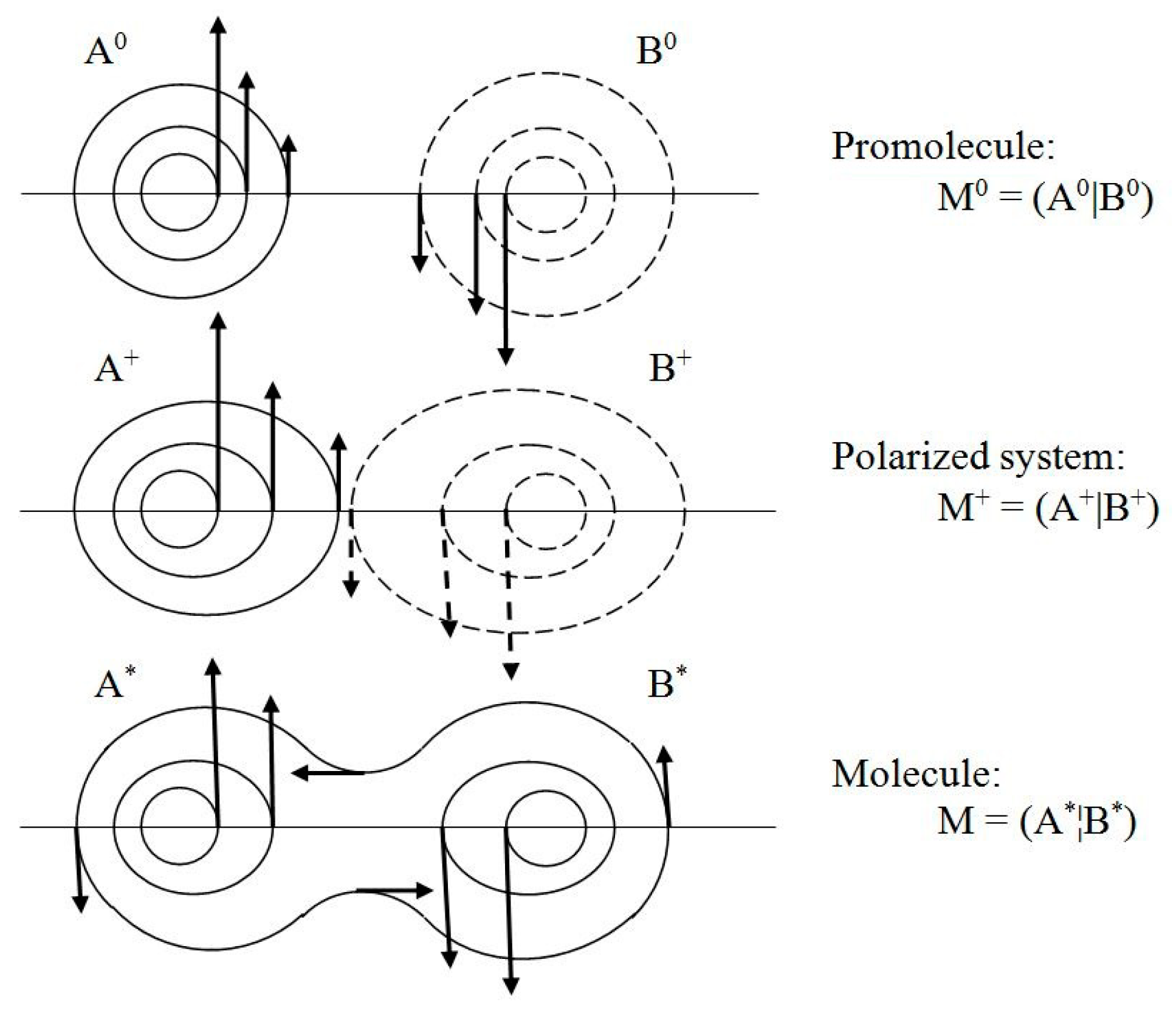
pst.(
r) of the stationary quantum state. They generate “vortices” of the latent “horizontal” velocity in spherical probability distributions of free atoms of the promolecule M
0, the deformed AIM distributions in the polarized system M
+, and in the equilibrium density of the molecule M = M* (see
Figure 3).
8. Component Dynamics in Equilibrium Stationary States
Consider again a general (complex) state of an electron (Equation (1)) and its quantum dynamics in Equation (5), determined by the Hamiltonian of Equation (4). Let us separate the local “vertical”
r(v) and “horizontal”
r(h) components of a general displacement in electronic position (see
Figure 2),
dr =
dr(v) +
dr(h), in directions perpendicular and parallel to the probability contour
p(
r) =
const., respectively. The former is consistent with the probability gradient ∇
p(
r), which reflects the direction of the distribution fastest increase.
The stationary (ground) state of an electron
ψ0, for the sharply specified energy,
corresponds to the time-independent modulus function
R0(
r) and time-dependent phase component
φ0(
t) = −(
E0/
ħ)
t ≡
−ω0 t. The associated equilibrium state then corresponds to the locally (horizontally) modified resultant phase,
in the
phase-transformed wavefunction,
which conserves the stationary probability distribution:
However, the expectation value of the energy,
differs from
E0 of Equation (97) by the “horizontal” kinetic energy,
related to the (horizontal) nonclassical information,
The normalization-constrained minimum principle for this average energy gives the following stationary SE, including the horizontal kinetic-energy contribution:
This
horizontally-generalized stationary SE thus includes the additional wave-number contribution
ω(h) =
T[
φ(h)]/
ħ. The DFT minimum principle of
Ev[
p], equivalent to the ordinary (stationary) SE,
determines the optimum probability distribution,
popt. =
p0 =
R02, and energy
Eopt. =
Ev[
p0] =
E0, while the equilibrium horizontal (“thermodynamic”) phase is determined by a supplementary IT rule (see
Section 3).
In the stationary equilibrium,
and the horizontal velocity of probability flux reflects the gradient of
φ(h):
The resultant probability velocity is then exclusively of a horizontal origin,
and both components of Φ contribute to the resultant
phase source in the associated continuity equation:
Therefore, in the stationary equilibrium, the
vertical source of the wavefunction phase remains constant,
σ0 =
dφ0/
dt =
−ω0, while the local horizontal-phase source assumes a purely convectional character:
The SE for components of Ψ
0 reads:
Its imaginary part confirms that
V(h) = (
ħ/
m)∇
φ(h), and hence also the associated probability current,
j(h) =
p0⋅
V(h), are indeed perpendicular to the probability gradient ∇
p0 = 2
R0∇
R0,
The real part of Equation (110) generates the associated phase dynamics,
where the horizontal phase current
J(h) =
φ(h)V(h), ∇⋅
J(h) = ∇
φ(h)⋅
V(h) and the resultant phase source is defined in Equation (108).






{kind=link}
{kind=link}
{kind=link}