
The Dissipative Photochemical Origin of Life: UVC Abiogenesis of Adenine


Abstract
1. Introduction
2. Photochemistry
2.1. Quantum Selection Rules
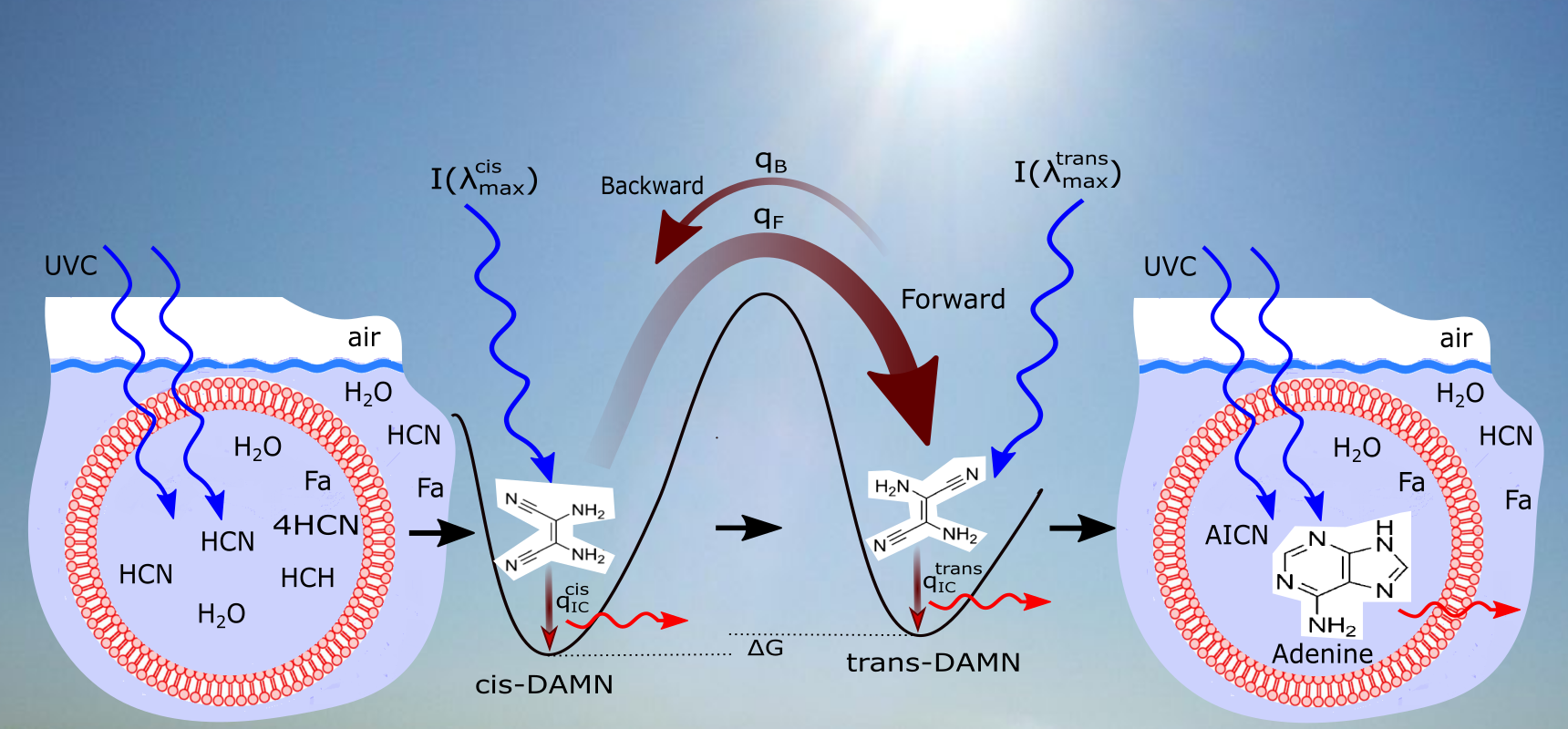
2.2. Conical Intersections
2.3. Excited State Reaction Mechanisms
3. The Thermodynamic Dissipation Theory of the Origin of Life
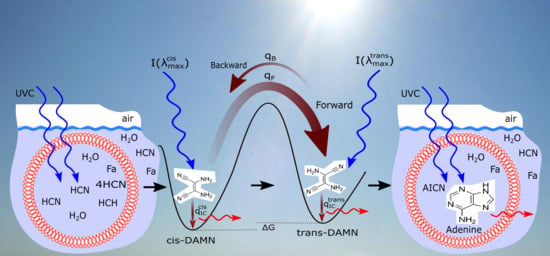
4. Example: The Dissipative Structuring of Adenine
4.1. The Model

4.2. The Kinetic Equations

| Name | Chemical Formula | Abbrev. in Text | Abbrev. in Kinetics | Figure 4 | nm | M cm | [D] | TPSA [Å] |
|---|---|---|---|---|---|---|---|---|
| hydrogen cyanide | HCN | HCN | H | 1 | 2.98 | 23.8 | ||
| formamide | HN-CHO | formamide | F | 220 | 60 [89,90] | 4.27 [91] | 43.1 | |
| formimidic acid | H(OH)C=NH | formimidic acid (trans) | Fa | 220 | 60 | 1.14 [91] | 43.1 * | |
| ammonium formate | NHHCO | ammonium formate | Af | +/−, 2.0 * | 41.1 | |||
| diaminomaleonitrile | CHN | cis-DAMN (DAMN) | C | 2 | 298 | 14,000 [92] | 6.80 [93] | 99.6 |
| diaminofumaronitrile | CHN | trans-DAMN (DAFN) | T | 3 | 313 | 8500 [92] | 1.49 [93] | 99.6 |
| 2-amino-3-iminoacrylimidoyl cyanide | CHN | AIAC | J | 4 | 275 | 9000 [68,71] | 1.49 | 99.6 * |
| 4-aminoimidazole-5-carbonitrile | CHN | AICN | I | 7 | 250 | 10,700 [92] | 3.67 | 78.5 |
| 4-aminoimidazole-5-carboxamide | CHNO | AICA | L | 266 [94] | 10,700 * | 3.67 * | 97.8 | |
| 5-(N’-formamidinyl)-1H-imidazole-4-carbonitrileamidine | CHN | amidine | Am | 250 | 10,700 [95] | 6.83 * | 80.5 * | |
| adenine | CHN | adenine | A | 8 | 260 | 15,040 [96] | 6.83 [97] | 80.5 |
| hypoxanthine | CHNO | hypoxanthine | Hy | 250 | 12,500 [98] | 3.16 | 70.1 |
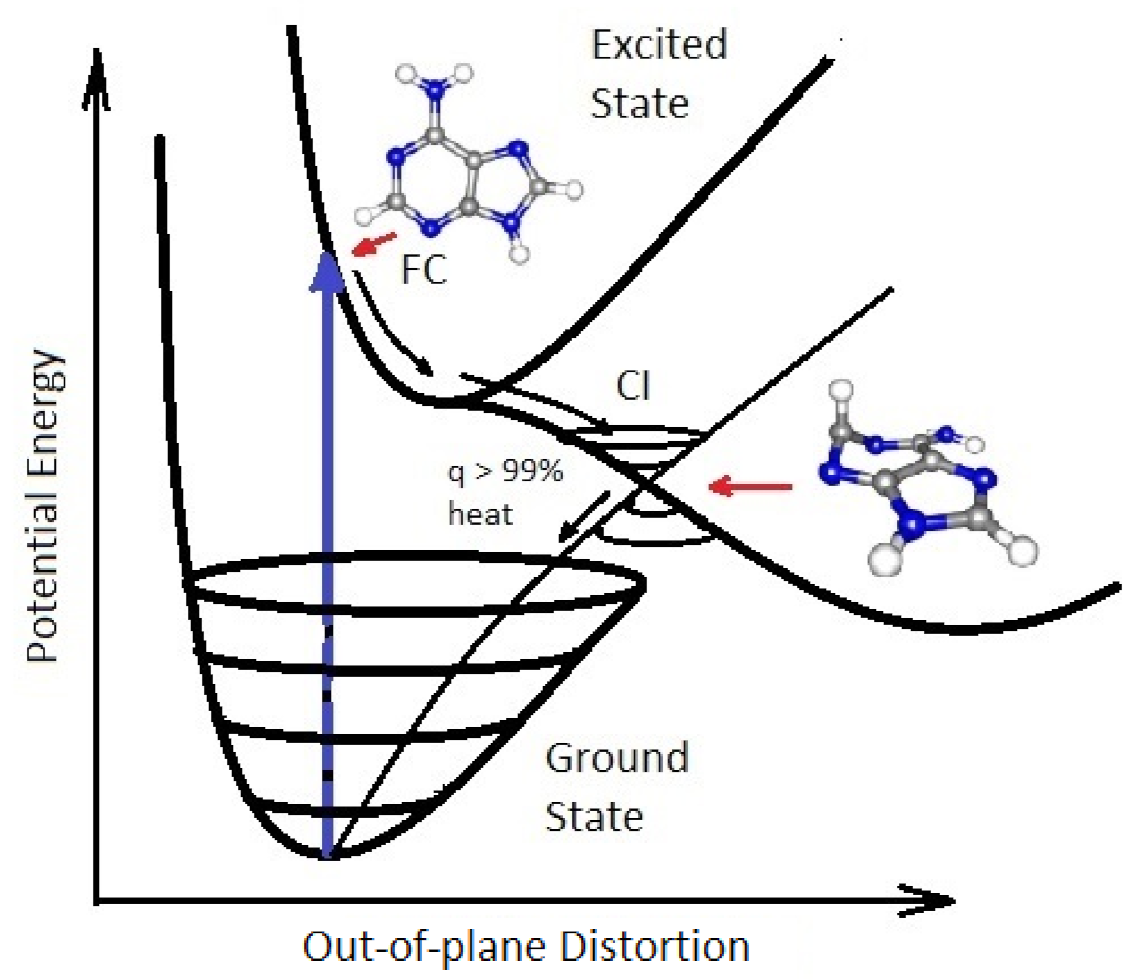
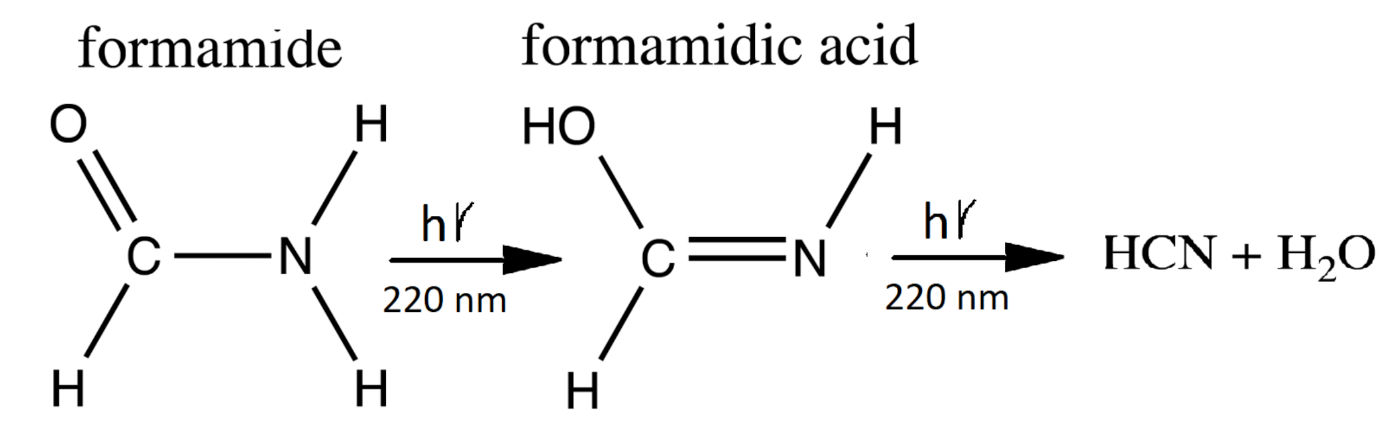
- A photon-induced tautomerization converts formamide (F) into formimidic acid (Fa). Basch et al. [90] have measured the electronic excitation spectrum of formamide (F) and find a peak in absorption at 55,000 cm (182 nm) with a molar extinction of 11,000 M cm. However, a shoulder exists on the main absorption peak which extends down to 40,000 cm (250 nm). Duvernay et al. [102] suggest that this shoulder arises from the resonant excitation of the forbidden transition located at 219 nm (130 kcal mol) and not from the main transition located at 182 nm. Maier and Endres [101] have determined that irradiation of formamide (F) at 248 nm rapidly converts it into basically two tautomeric isomers of formimidic acid (Fa), H(OH)C=NH, which are both about 3.6 kcal mol in energy above formamide and separated from it by a transition barrier of height of kcal mol (gas phase). Similarly, Duvernay et al. [102] have shown that under UVC light of 240 nm, formamide (F) tautomerizes into formimidic acid (Fa) and their calculation gives a similar transition state barrier height of 47.8 kcal mol. Wang et al. calculate a transition state barrier of 49.8 kcal mol [110] but show that this is reduced to kcal mol in the presence of only a single water molecule. The energy needed to overcome this barrier is in the infrared (1265 nm) but Cataldo et al. have shown that there is no evidence of thermal excitation until about 220 C [111]. Our model, therefore, assumes that the F → Fa tautomerization requires the absorption of a photon and we take the wavelength region for tautomerization due to the transition of 10 nm and assign an average molar extinction coefficient to that region of 60 M cm as measured by Basch et al. [90] and also by Petersen et al. [89].
- Duvernay et al. [102] have shown that formimidic acid (Fa) can, in turn, be photo-lysed into HCN (H) (or HNC) plus HO, (dehydration) with maximal efficiency at about 198 nm [104]. However, the absorption spectrum of formimidic acid also has a shoulder extending to about 250 nm due to the same excitation as in formamide. For example, Duvernay et al. observe a small amount of dehydration of formimidic acid at 240 nm. Given that our surface solar spectrum during the Archean (Figure 2) is extinguished below about 205 nm, here we likewise assume an absorption wavelength for photo-lysing of nm and a similar average molar extinction coefficient as for the tautomerization of fomamide (F) of 60 M cm which is in accordance with the findings of Gingell et al. [104]. Combining photo-reactions #2 and #3, we thus recuperate some of the HCN lost to thermal hydrolysis as described by reaction #1 (see Figure 6). Barks et al. [103] have shown that if neat formamide is heated (130 C), thereby exciting vibrational states, a photon-induced excitation at even longer wavelengths (254 nm) also leads to the disintegration of formamide into HCN and HO, and they believe that this is the route to the production of the purines, adenine, guanine, and hypoxanthine, that they detect. Their yields are increased when including the inorganic catalysts sodium pyrophosphate and calcium carbonate, indicating that heating and inorganic catalysts can improve the photochemical reaction steps #2 and #3. Formamide also disintegrates thermally into HCN and HO, without requiring the absorption of a photon, but only at temperatures greater than about 220 C [111] because of high barriers [100].Figure 6. The production of formamidic acid (Fa) from formamide (F) (photoreaction #2) and its subsequent decay into HCN (H) and water (photoreaction #3).Figure 6. The production of formamidic acid (Fa) from formamide (F) (photoreaction #2) and its subsequent decay into HCN (H) and water (photoreaction #3).

- The hydrolysis of formamide (F) gives the salt ammonium formate (Af) at a rate of 1% in 24 h at 100 C [103]. The temperature dependent rate equation given in Table 2 was determined by Kua and Thrush [100] at pH 7.0 from the experimental data of Miyakawa et al. [99]. The ammonium and formate parts of this salt become useful for the thermal reaction leading to the final addition of an HCN (H) to AICN (I) catalyzed by formamide (F) to give adenine (A) (reaction #13) [105,106].
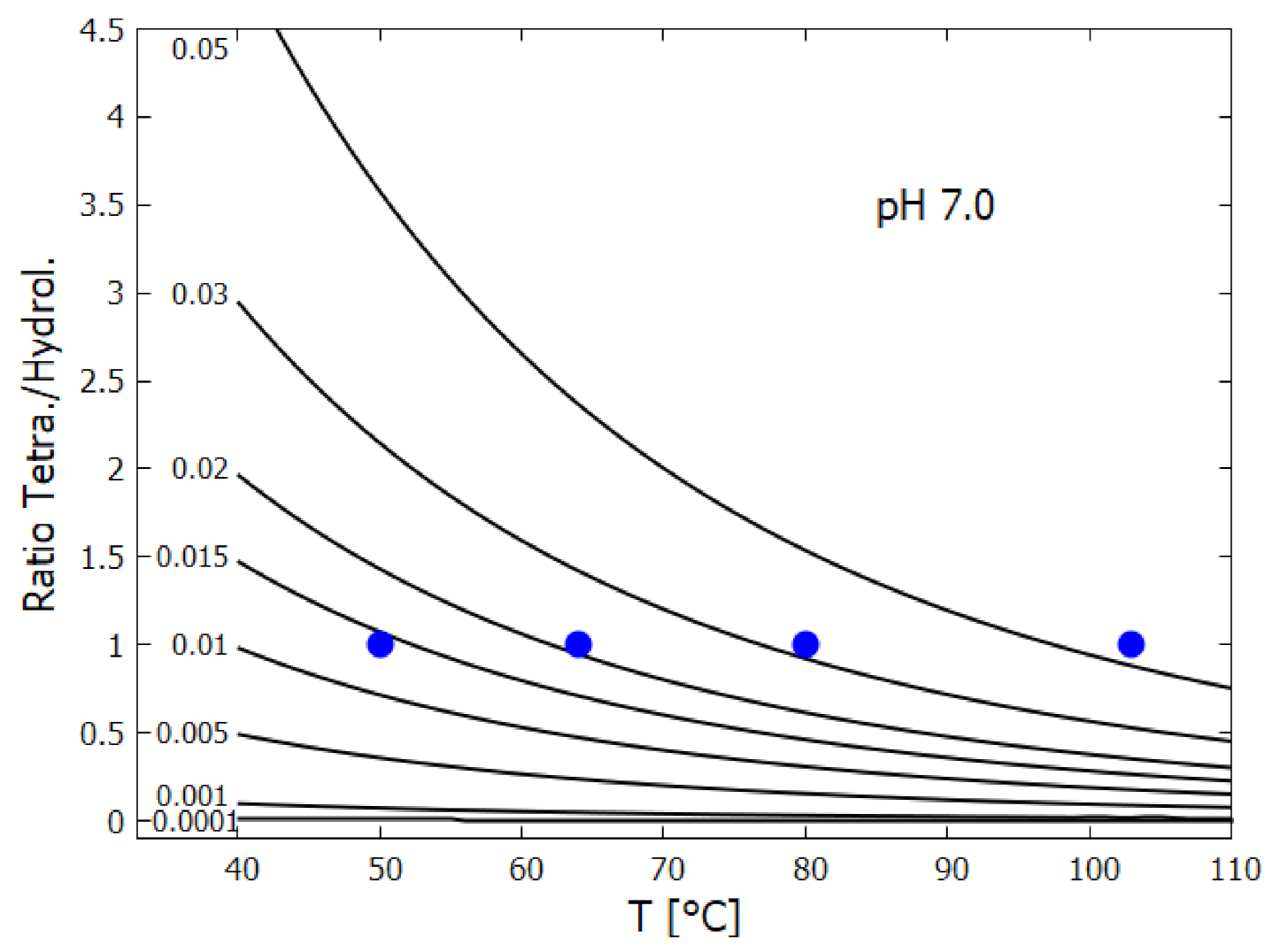
- HCN (H) thermally polymerizes into (HCN) with its most stable tetramer () known as cis-DAMN (C) being the preferred polymer from which more complex polymers can be synthesized [111]. The tetramization of 4HCN is an exothermic thermal reaction and occurs most rapidly at a solvent pH at its pKa value, which decreases with increasing temperature (pKa at 60 C and at 100 C) [68]. The tetramization of HCN into DAMN is not elementary but involves successive polymerization of HCN with H and CN ions [68] so is second order in the concentration of HCN. The temperature dependence of the rate of conversion of HCN to DAMN has been measured by Sanchez et al. [68]. We assume transition state theory and an Arrhenius equation of form,From the conversion rates for a 1 M solution of HCN with 0.01 M tetramer catalyst as given in Table 5 of Sanchez et al. [68], a straight line can be fitted to the graph of vs giving values of and , or kcal mol. However, this would be the rate equation for tetramerization of HCN at its pKa value which would be about 8.2 at 80 C [68]. To obtain the rate equation at the lower pH value assumed here of 7.0 we note that from Figure 14 of Sanchez et al. [68] the half-lives for tetramization of HCN are the same for pH 7.0 at 80 C as they are for pH 8.2 at 64 C (1 day). Setting the rate constants as the same for these two conditions leads to a value of for pH 7.0. Finally, since HCN can polymerize into either cis-DAMN (C) or trans-DAMN (T), and since trans-DAMN (T) has a free energy of [68] (0.56 [93]) kcal mol higher than cis-DAMN (C) a Boltzmann factor of is included for cis-DAMN and the same factor but with for trans-DAMN.The rates for hydrolysis and polymerization are similar for concentrations of HCN (H) between approximately 0.01 M and 0.1 M (equal rates at 0.03 M for pH 7, T = 80 C, Figure 15 of reference [68]). At lower concentrations, hydrolysis dominates while at higher concentrations polymerization dominates [68].
- HCN (H) can also thermally polymerize into trans-DAMN (T) which has a free energy of 0.61 kcal mol [68] higher than cis-DAMN (C). We therefore assume that the rate constant for the polymerization into trans-DAMN is the same as that for cis-DAMN multiplied by a temperature dependent Boltzmann factor .
- Trans-diaminomaleonitrile, trans-DAMN (T), produced through the thermal reaction #6, or through the UV photon-induced transformation of cis-DAMN (C) into trans-DAMN (see reaction #9), is a good catalyst because it has electronic donor parts (–NH groups) and acceptor parts (–CN group) linked by a double bond. As such, it can act as a catalyst for the tetramization of 4HCN into cis-DAMN [69]. Cis-DAMN is also a catalyst for the same thermal reactions, but has significantly less activity than trans-DAMN [68] and therefore its catalytic activity is neglected in our analysis. As can be surmised from the discussion of Table 7 of reference [68], including 0.01 M of the tetramer trans-DAMN increases the rate of tetramization by a factor of 12 at 20 C which would correspond to a reduction in the activation energy of 1.45 kcal mol. This change in the barrier height is therefore included in the rate constant for this catalyzed reaction.
- Trans-DAMN also acts as an auto-catalyst for its own thermal production from 4HCN [69] and we assume a similar reduction in barrier height as for its catalysis of the production of cis-DAMN from 4HCN (reaction #7).
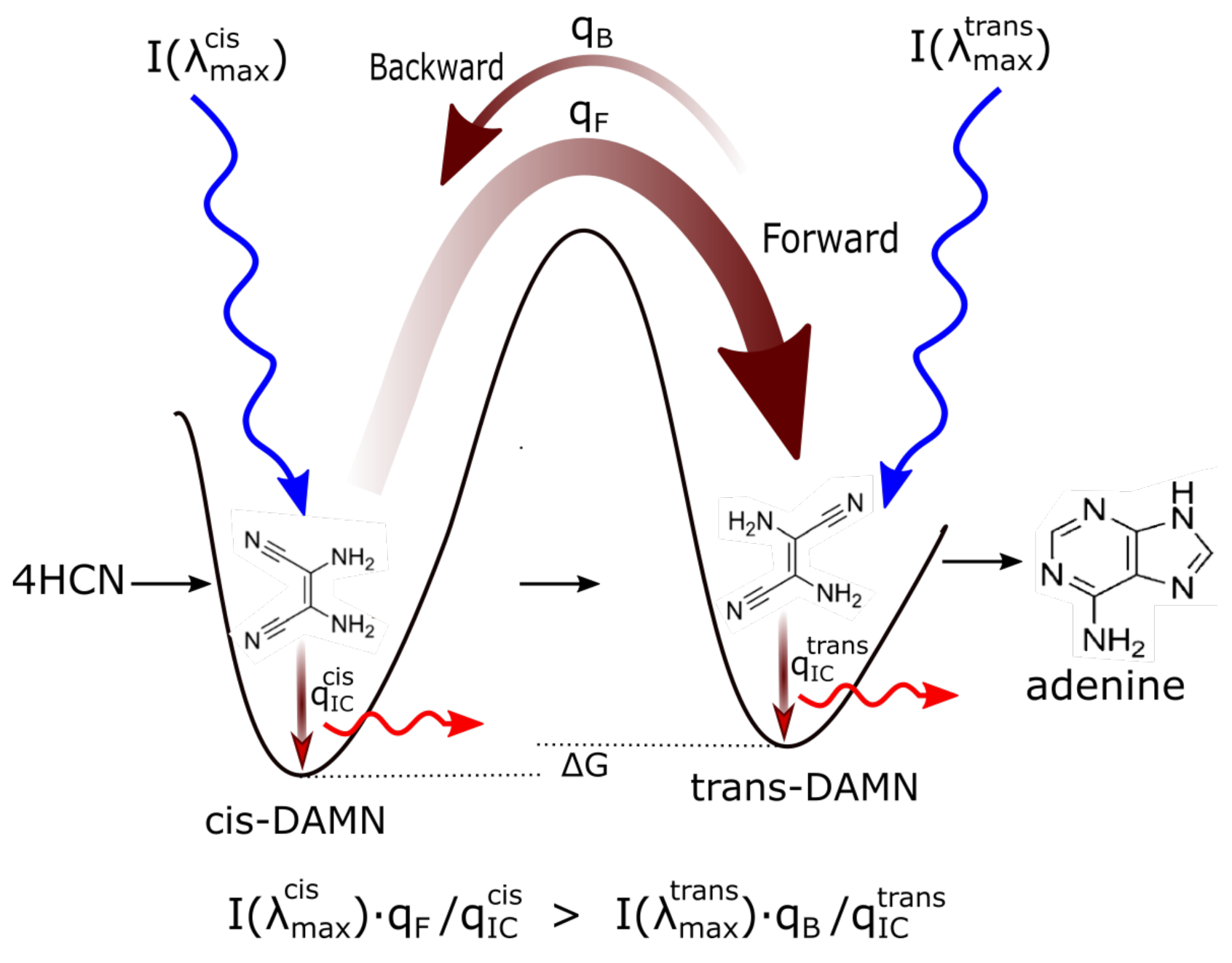
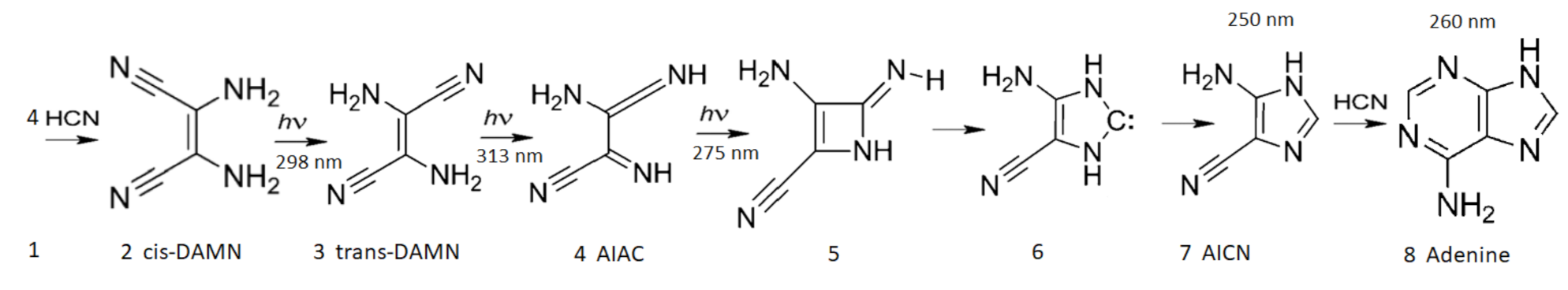
- (a) cis-DAMN can transform into trans-DAMN (3), step (2) of Figure 4 through a rotation around the double covalent carbon-carbon bond by absorbing a high energy photon (298 nm) to overcome the large energy barrier for rotation, calculated to be 58.03 kcal mol [93] (>4 eV [71]). The quantum yield has been measured by Koch and Rodehorst to be [92].(b) Because of the high energy barrier 58.03 kcal mol between trans-DAMN and cis-DAMN, thermal energy, even at our high temperatures, is insufficient to significantly reverse the rotation about the double covalent bond. However, trans-DAMN (T) can absorb a photon at 313 nm which would provide it with sufficient energy to isomerize back into cis-DAMN. The quantum efficiency for this reversal was determined to be by fitting to the experimental data given in Figure 1 of Koch and Rodehorst [92] as explained in the description of Figure 8.
- The absorption of a photon at 313 nm excites trans-DAMN which then transforms into AIAC through proton transfer from one of the amino groups [71]. Although there does not appear to exist a quantum yield for this photochemical reaction in the literature, the quantum yield for trans-DAMN to AICN (T→ I) has been measured by Koch and Rodehorst [92] to be 0.0034. By fitting our model concentration results to the experimental data of Koch and Rodehorst (see discussion of Figure 8) it is found that a best value for the quantum yield of trans-DAMN to AIAC (T→J), , is 0.006 and therefore the quantum yield for AIAC to AICN (J→ I) would be .
- AIAC (J) on absorbing a photon at 275 nm then transforms through photon-induced cyclicization (ring closure) into an azetene intermediate (5 of Figure 4) in an excited state, which then transforms to the N-heterocyclic carbene (6 of Figure 4) and finally this tautomerizes to give the imidazole AICN (I) [71]. As noted above, the quantum yield for this process (J→I) is taken to be 0.5833 to give the overall quantum yield for trans-DAMN to AICN (T→ I) to be 0.0034 [92]. AICN absorbs maximally at wavelength 250 nm.
- The imidazole, 4-aminoimidazole-5-carbonitrle, AICN (I) created in the previous photochemical reaction (reaction #11) gives the hydrolysis product 4-aminoimidazole-5-carboxamide, AICA (L). The rate equation for this first-order reaction was determined from the data of Sanchez et al. [69] at different temperatures (their Table 1). The barrier to hydrolysis was determined to be 19.93 kcal mol and the frequency factor to be .
- The final coupling of a fourth HCN to AICN (I) and its cyclization to form adenine (A) is a very exothermal overall, kcal mol, but there are numerous large energy barriers on the path to its completion [70]. The first step is the coupling of an HCN molecule to AICN, and this appears to be rate limiting since it has the highest energy barrier, calculated in the gas phase, of 39.7 kcal mol [70]. However, it is catalyzed by both bulk solvent and specific water molecules which reduce the barrier to 29.6 kcal mol, or by ammonium molecules with bulk water solvent which reduce the barrier further to 27.6 kcal mol [70]. A number of experimental works [103,105,106,112] have revealed that ammonium formate (Af) could provide a route with an even lower barrier, but the rate is still too slow to allow significant adenine production from AICN and ammonium formate, unless a strong concentration mechanism existed, for example, dehydration [103], or perhaps the buildup of concentration inside the vesicle, or the reaction-diffusion self-organizing occurring within the vesicle, as will be considered below.A solution to this rate problem may exist, however, without requiring high concentrations. As early as 1974 Yonemitsu et al. [105] showed that including formamide, the hydrolysis product of HCN (reaction #1), in aqueous solution, or by itself (neat solution), along with ammonium formate could dramatically speed up the reaction as long as the temperature was above approximately 80 C, leading to a successful industrial patent for the production of adenine from cis-DAMN (C) or trans-DAMN (T) and formamide with ammonium formate. From examples 1 and 12 of the experiments of Yonemitsu et al. carried out at 150 and 100 C (using 135 g of formamide, 30 g of ammonium formate, and 2.01 g of DAMN) giving rise to 43.5% and 30.0% product of adenine after 5 and 10 h at those temperatures respectively, it is possible to calculate an activation barrier for the overall reaction of kcal mol. Since ammonium formate is a salt, the probable pathway from AICN to adenine would be that proposed by Zubay and Mui [106] where the ammonium ion NH attacks the triple NC bond of AICN and the formate ion HCOO attacks the amine NH group of AICN (Figure 8 of reference [106]) both catalyzed by the proton transfer process involving formamide (see below), leading to this very low barrier. We therefore assume the reaction to be of second order and determined by the Arrhenius equation of form,where kcal mol and the pre-exponential frequency factor A was estimated from the reduced mass dependence of the Langevin model [113], for a charged ion - neutral molecule system where e is the ion electronic charge, is the polarizability of the neutral reactant, and is the reduced mass of the reactants [114]. Considering all factors being equal except the reduced mass, and then normalizing to the frequency factor of reaction #12 for the hydrolysis of AICN (I) by the inverse square root of the reduced mass for the reacting species, gives a value of .
- There exists a second possible route to adenine from AICN and HCN, without involving ammonium formate but considering the catalytic effect of formamide. Wang et al. [107] have studied, through ab initio DFT, the synthesis of adenine starting from pure formamide and propose what they call a “formamide self-catalytic mechanism”. This mechanism consists of; (1) a proton transfer from N to O of formamide to form the imidic acid tautomer, formimidic acid (Fa), potentially obtained in our case through a photon-induced proton transfer reaction #2; (2) a proton exchange between one imidic tautomer and one amide tautomer, resulting in two formimidic acids; and; (3) an interaction between these two imidic acids yielding formimidic acid, a water molecule, and HCN. This formamide self-catalytic mechanism has relevance to the entire adenine synthesis process starting from pure formamide since it reduces many of the barriers on route to adenine [107].Of importance to us here of Wang et al.’s results is the step of the attachment of HCN to the amine group (NH) of AICN. They show for their particular case of formiminylation of 5-aminoimidazole (Figure 13 of reference [107]) that this reaction can be formamide-catalyzed (as described above) and find the activation energy barrier for this to be 19.9 kcal mol (significantly lower than 46.1 kcal mol in the noncatalyzed process and 34.0 kcal mol in the water-assisted process) and that the subsequent dehydration process to give the amidine (Am) (our case) is calculated to be 14.0 kcal mol (34.3 kcal mol in the noncatalyzed reaction).Therefore, we assume that the attachment of HCN (H) to AICN (I) to form 5-(N’-formamidinyl)-1H-imidazole-4-carbonitrileamidine (Am) to be a formamide-catalyzed thermal reaction involving formimidic acid and formamide and we assume the rate of this reaction to be determined by the Arrhenius equation of formwhere kcal mol and the pre-exponential frequency factor A is again estimated from the reduced mass dependence of the Langevin model [113], considering again all factors equal except the reduced mass, and then normalizing to the reaction #12 for the hydrolysis of AICN (I) by the inverse square root of the reduced mass for the reacting species, giving a value of .Note that AICN (I) has a conical intersection for a charge transfer from the molecule in the excited state to a neighboring cluster of water molecules [115]. With AICN left in the charged state, this would significantly increase the rate of attachment, through charge-dipole interaction, to formamide, which has a dipole moment significantly larger than that of water (Table 1), effectively changing the reaction from third order to second order, thereby significantly increasing the overall rate of this last attachment of HCN to AICN through this formamide-catalyzed reaction.The possibility of a hot ground state reaction occurring to aid in overcoming the barrier to producing adenine (A) from AICN (I) and HCN (H) could also be considered during daylight periods. These occur within a narrow time window after photon excitation, calculated by Boulanger et al. for a molecule (trans-DAMN) which has a similar conical intersection as AICN, to be about 0.2 ps, which corresponds to the time at which the excess energy on the molecule has been reduced to about 1/3 of its initial value, allowing reactions to proceed with a maximum barrier height of about 30 kcal mol [71]. This possibility, however, is not included in the model. It would have the overall effect of increasing the rate of the production of adenine.
- After the attachment of a fifth HCN (H) to AICN (I) to form the amidine (Am), reaction #14, a subsequent tautomerization is required (calculated to have a high barrier of about 50 kcal mol) which, once overcome, allows the system to proceed through a subsequent barrier-less cyclicization to form adenine [95]. Such a high barrier to the final cyclicization means that, at the temperatures considered here, it cannot be a thermal reaction. Indeed, the fact that adenine has been found in space and in meteorites where temperatures are expected to be very low, indicated to Glaser et al. [95] that a photochemical route must be available. They suggested a photon-induced tautomerization of amidine, which absorbs strongly at 250 nm. Although oscillator strengths for the tautomerization have been calculated by Glaser et al., different ab initio approaches give significantly different values, so experiment will be required for its reliable determination. Therefore, until such data becomes available, we assume a similar molar extinction coefficient as for AICN and, being conservative, a quantum efficiency of but measure the effect on adenine production for a % variation of this parameter value (see Table 4). In fact, the results of Table 4 indicate that due to the large activation energy required, and the fact that the reactions are of second order, reactions #14 and #15 only come into play at very high temperature.
- The temperature dependent rate equation for the destruction of adenine (A) through hydrolysis to give hypoxathine (Hy) which could then lead to guanine, or through deamination to some amino acids [116], was determined in careful experiments by Levy and Miller [108] (and by Wang and Hu [109]). Zheng and Meng calculated a transition state barrier for hydrolysis of 23.4 kcal mol [117].
- to 24. These reactions represent the absorption of a photon, in a 20 nm region centered on the wavelength of peak absorption, on the molecule which then decays through internal conversion at a conical intersection to the ground state on sub-picosecond time scales. All molecules listed in this set of photo-reactions are basically photo-stable because of a peaked conical intersection connecting the excited state with the ground state. These reactions, with large quantum efficiencies, represent the bulk of the flow of energy from the incident UVC spectrum to the emitted outgoing ocean surface spectrum in the infrared and therefore contribute most to photon dissipation, or entropy production.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4.3. Vesicle Permeability and Internal Diffusion
4.4. Initial Conditions
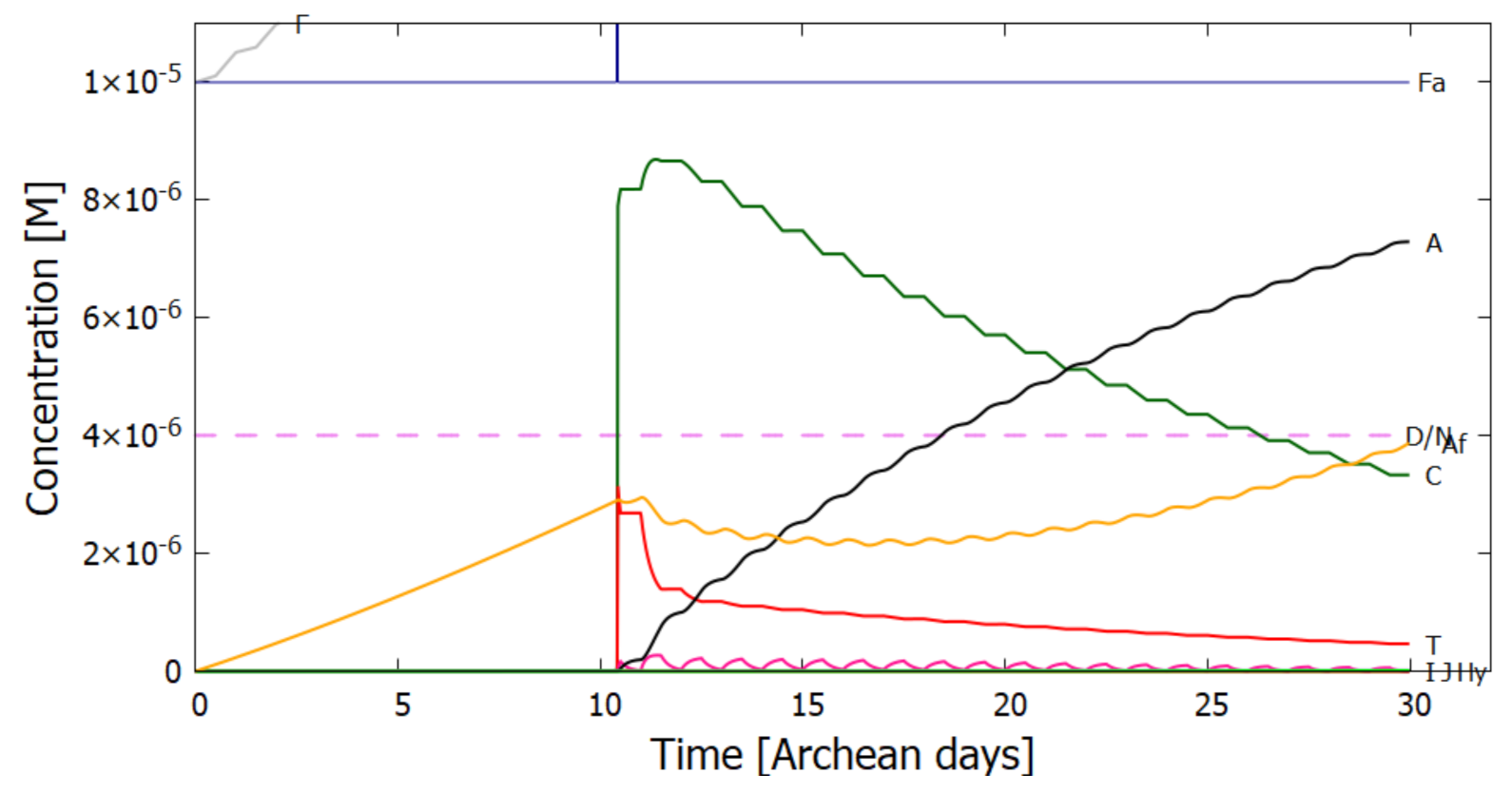
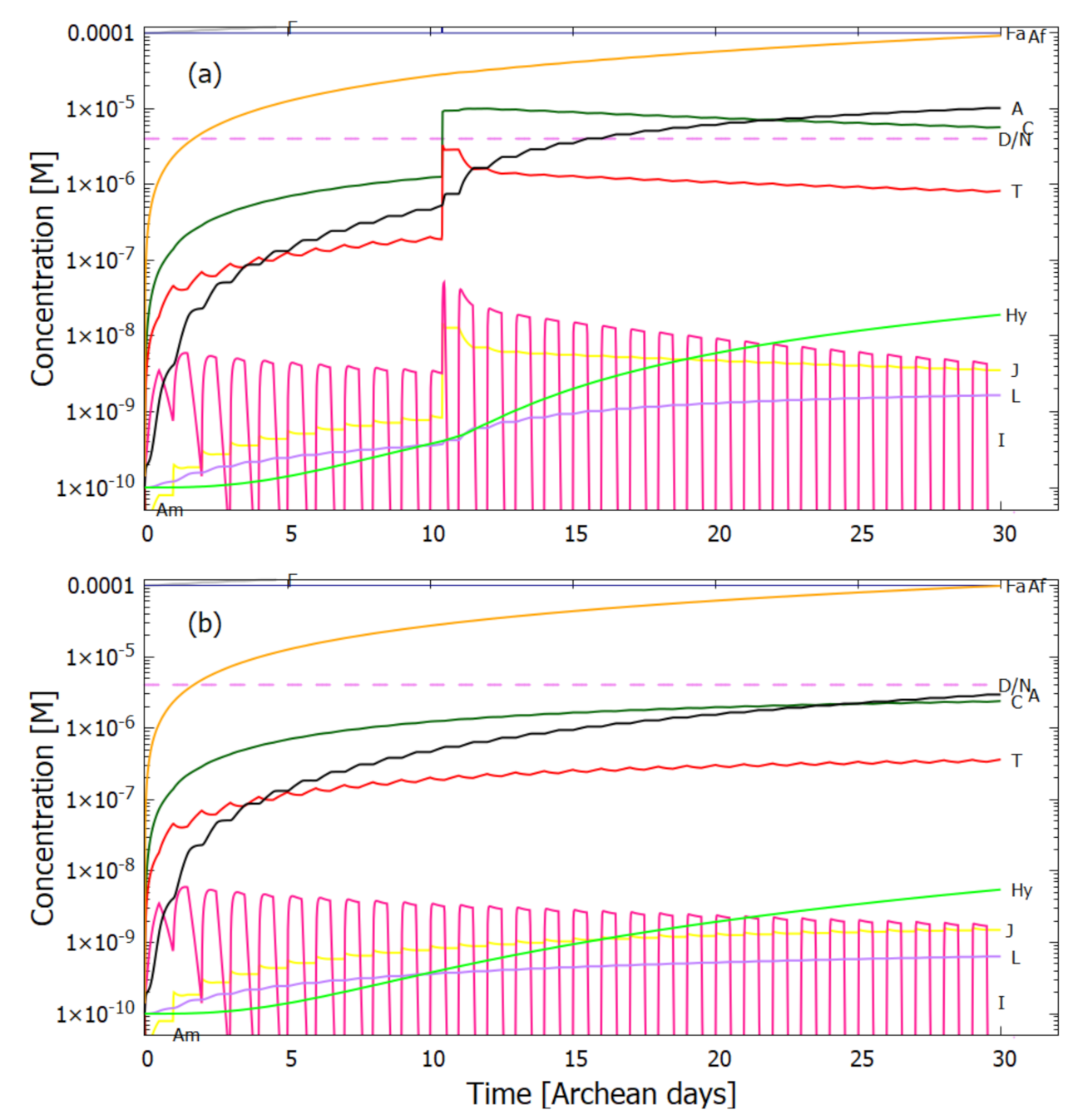
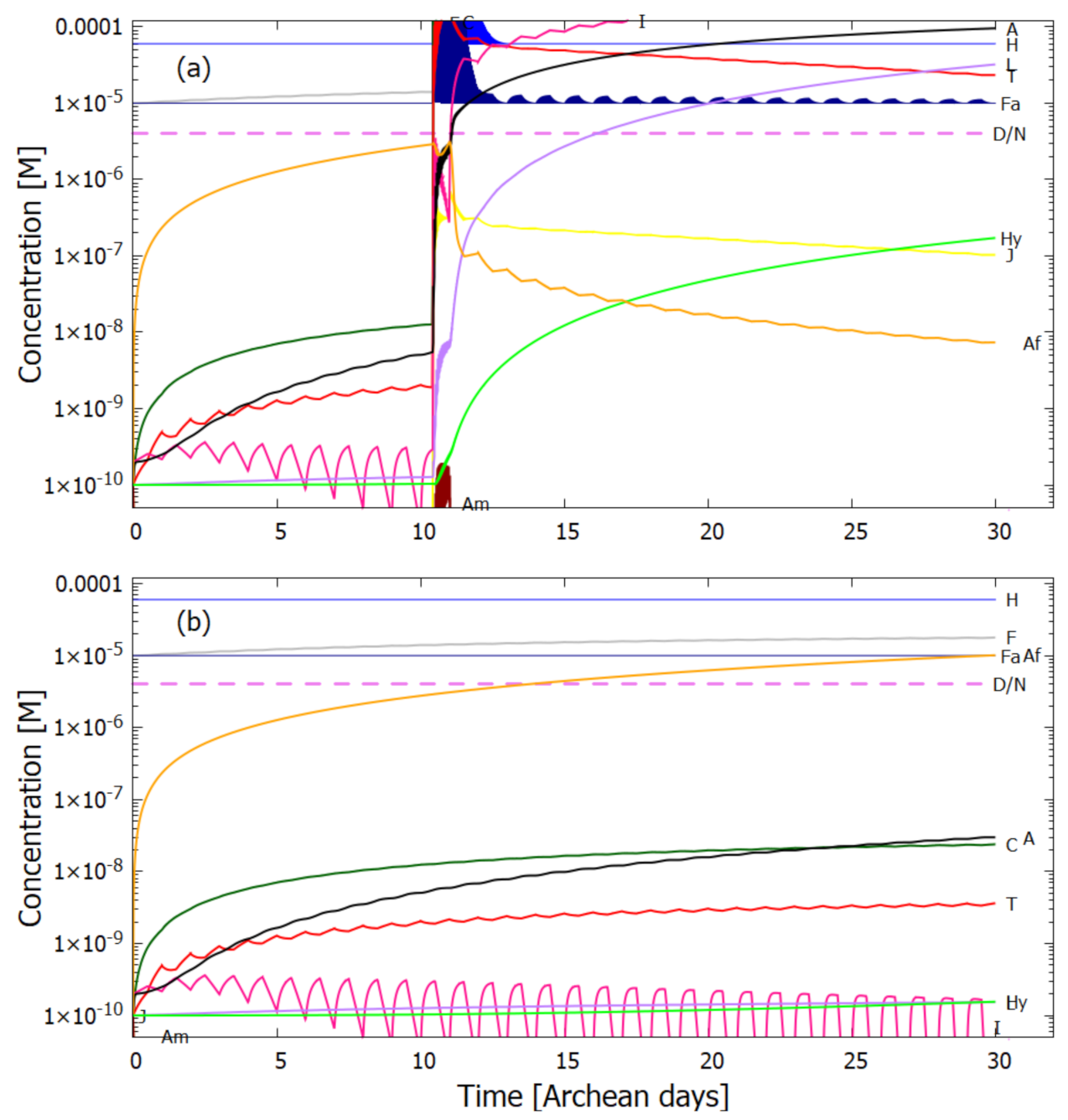
5. Results
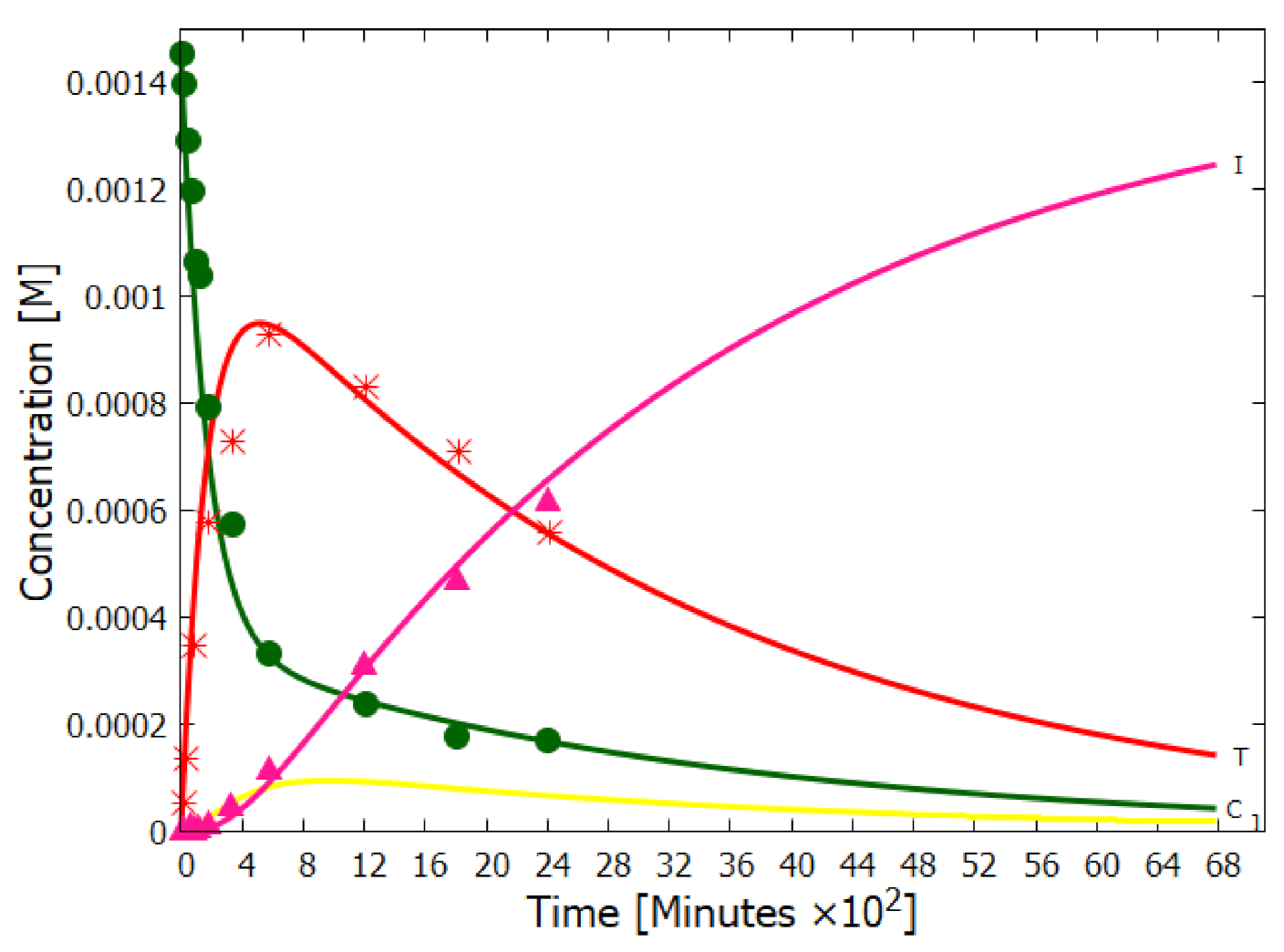
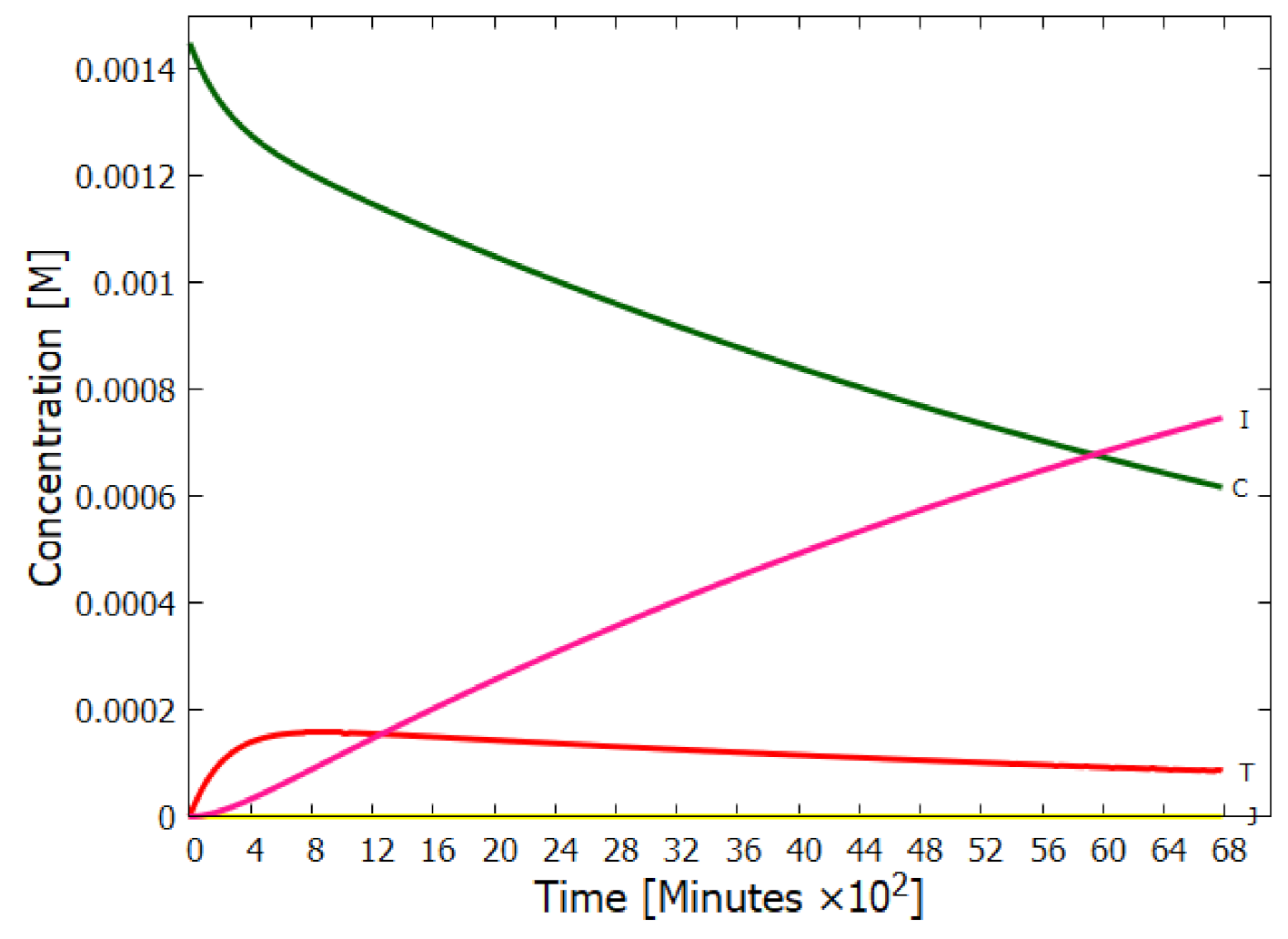
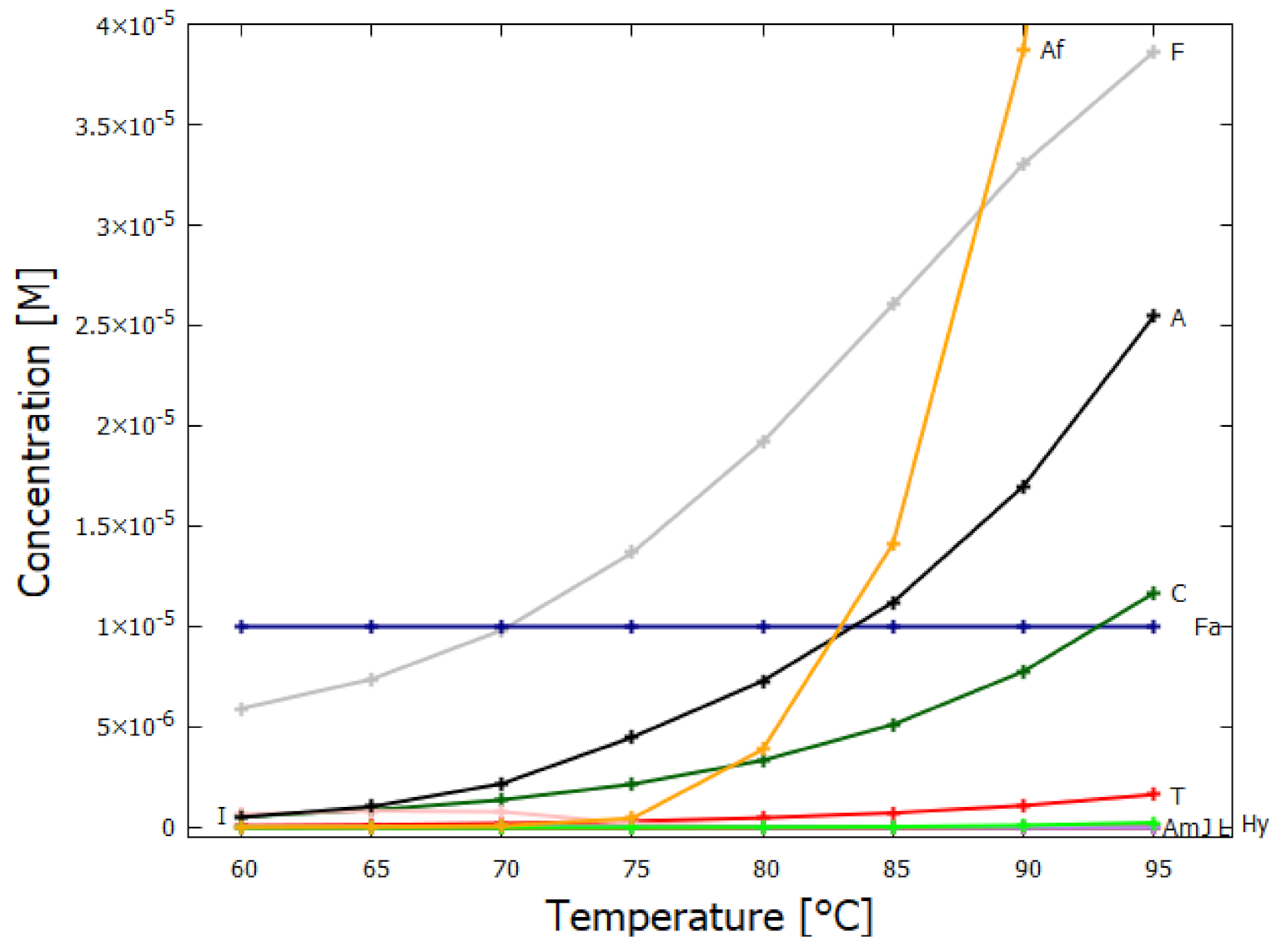
5.1. Validation of Model
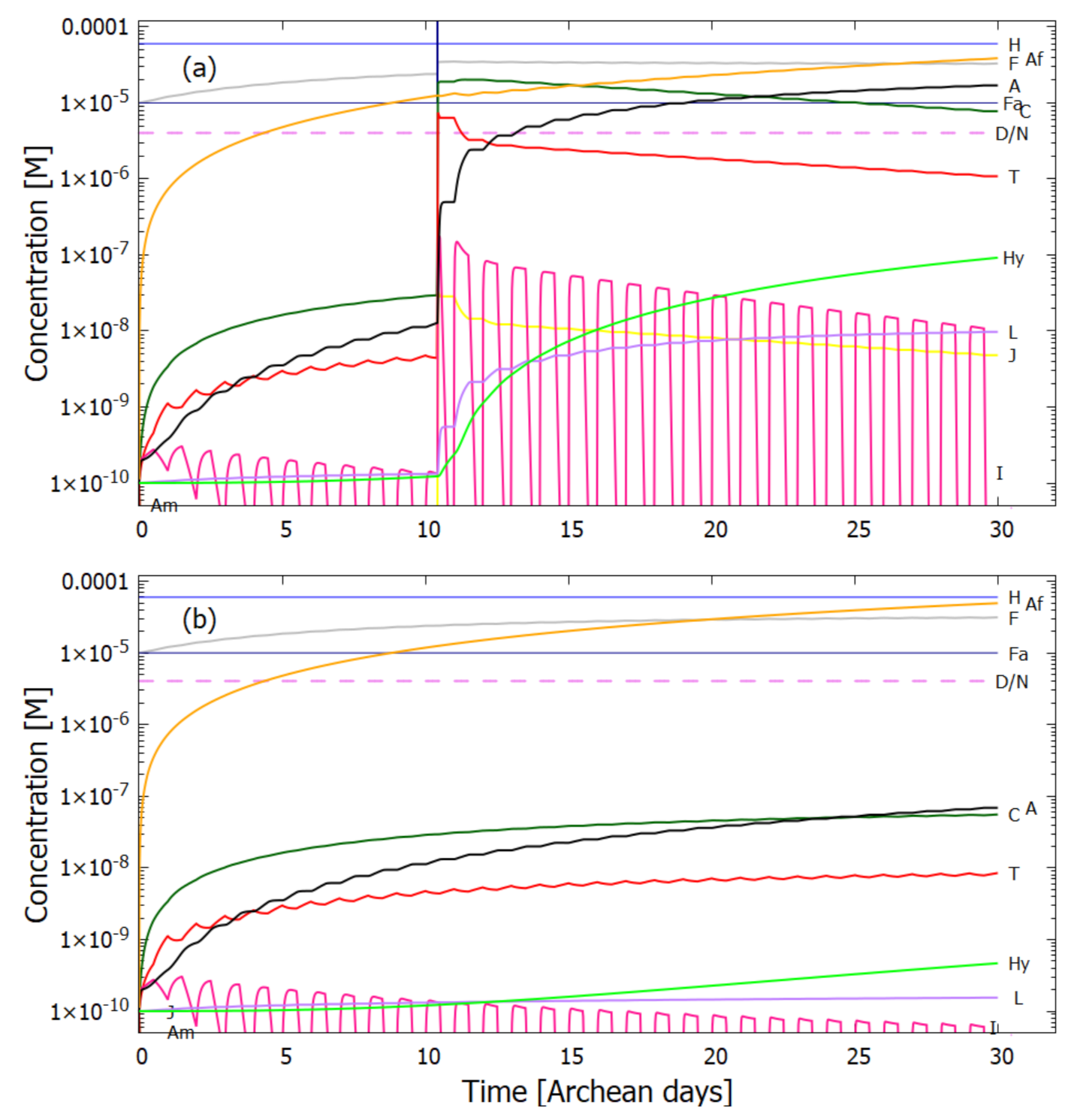
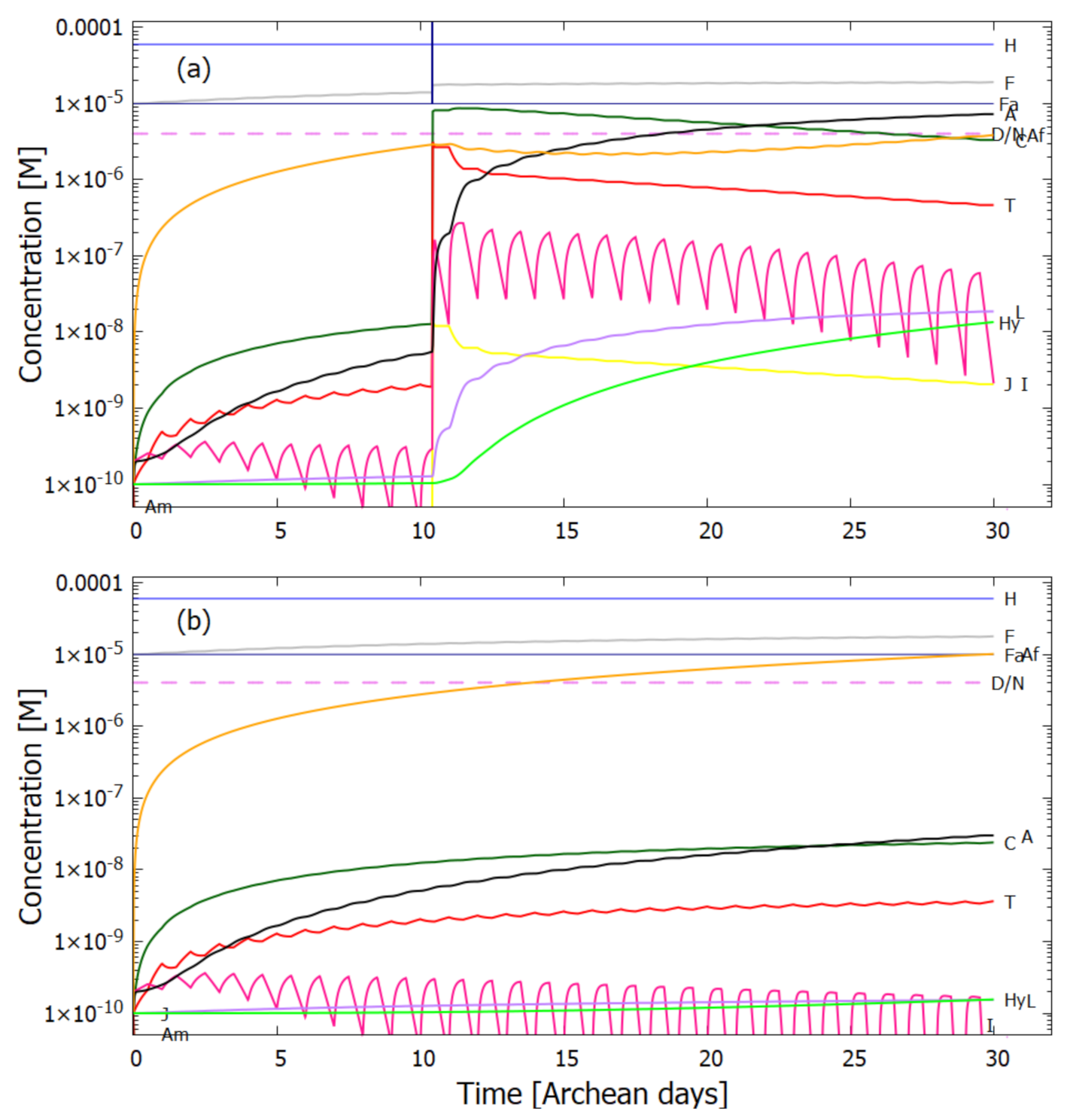
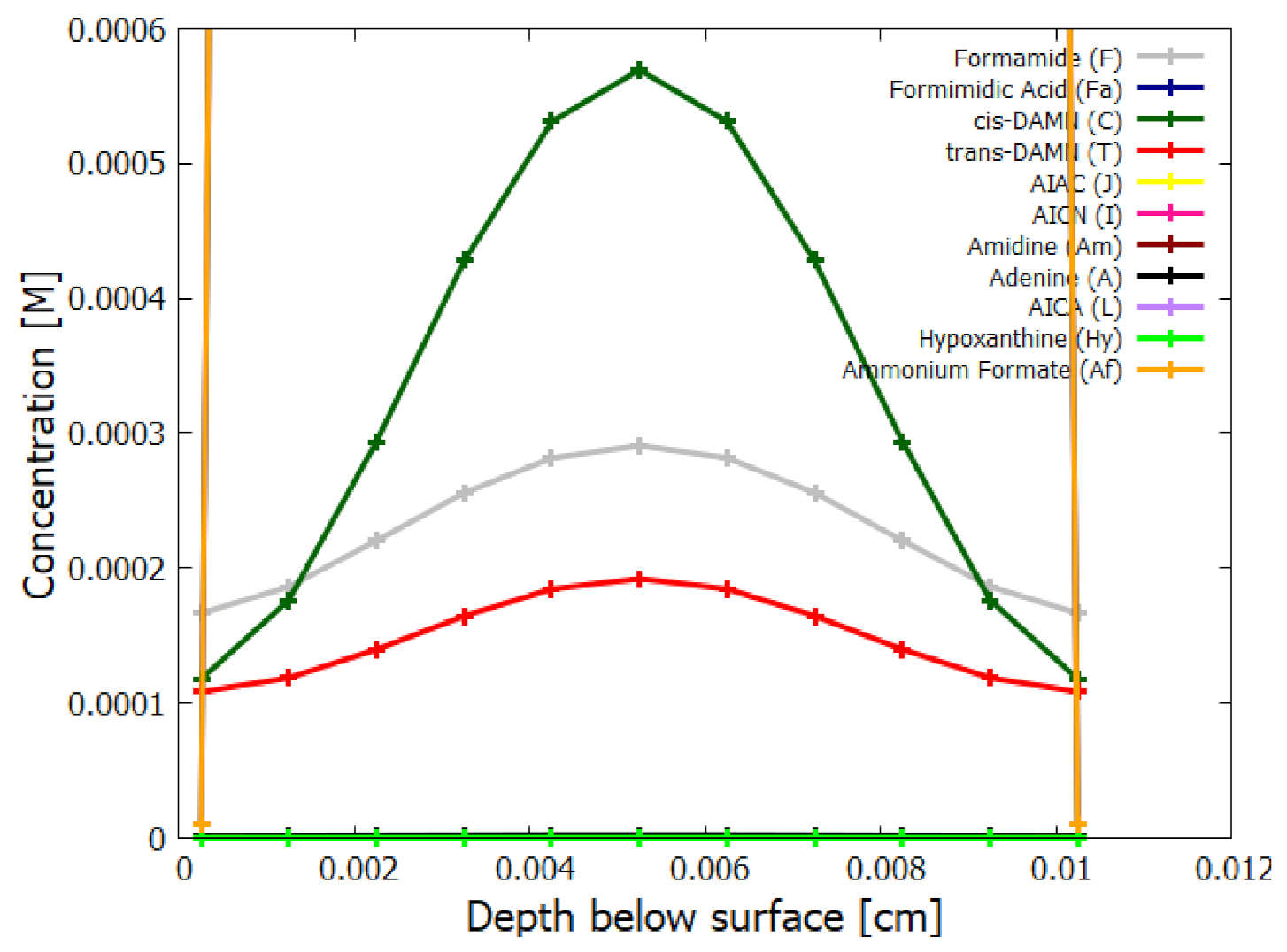
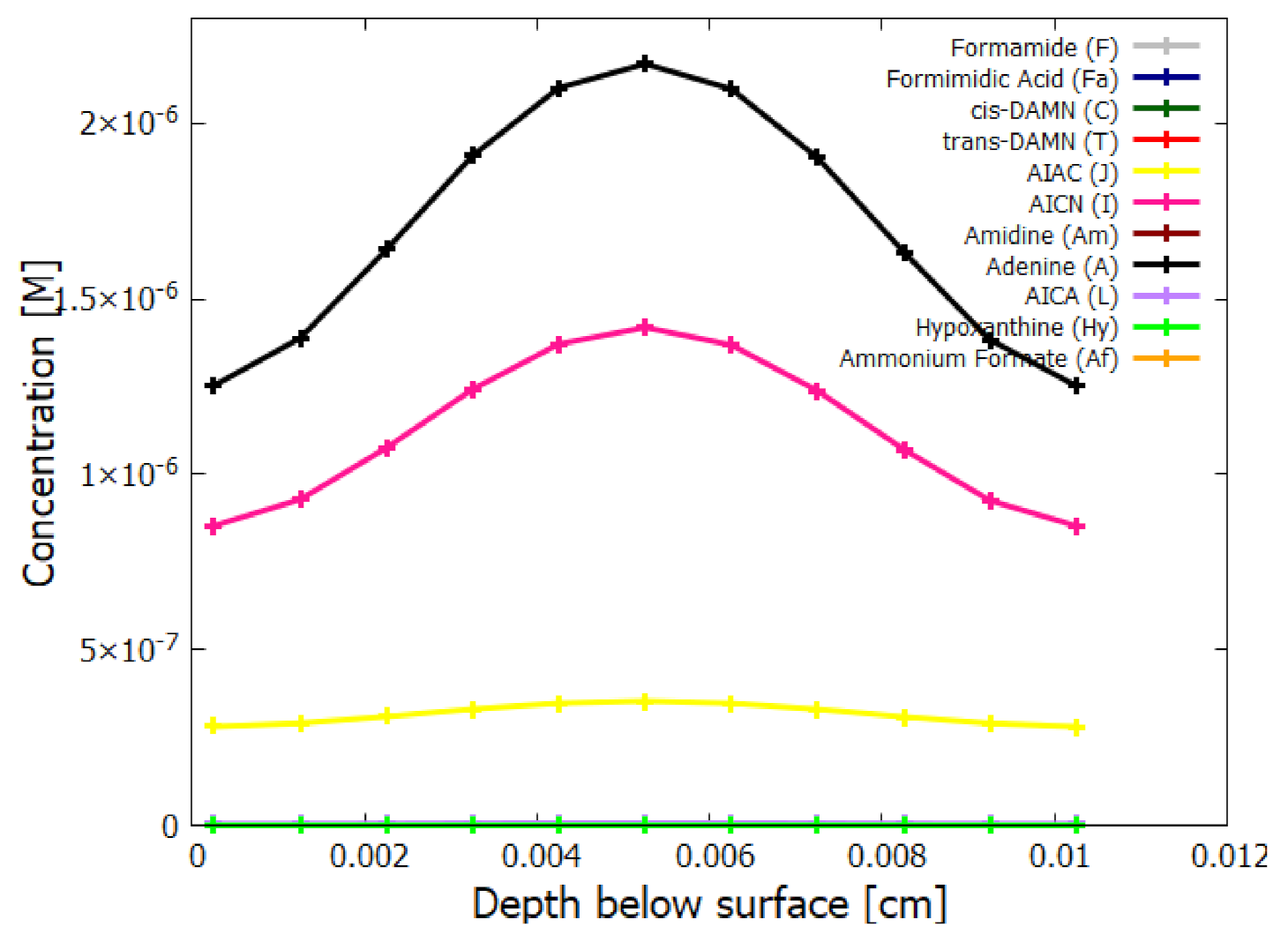
5.2. Evolution of the Concentration Profile
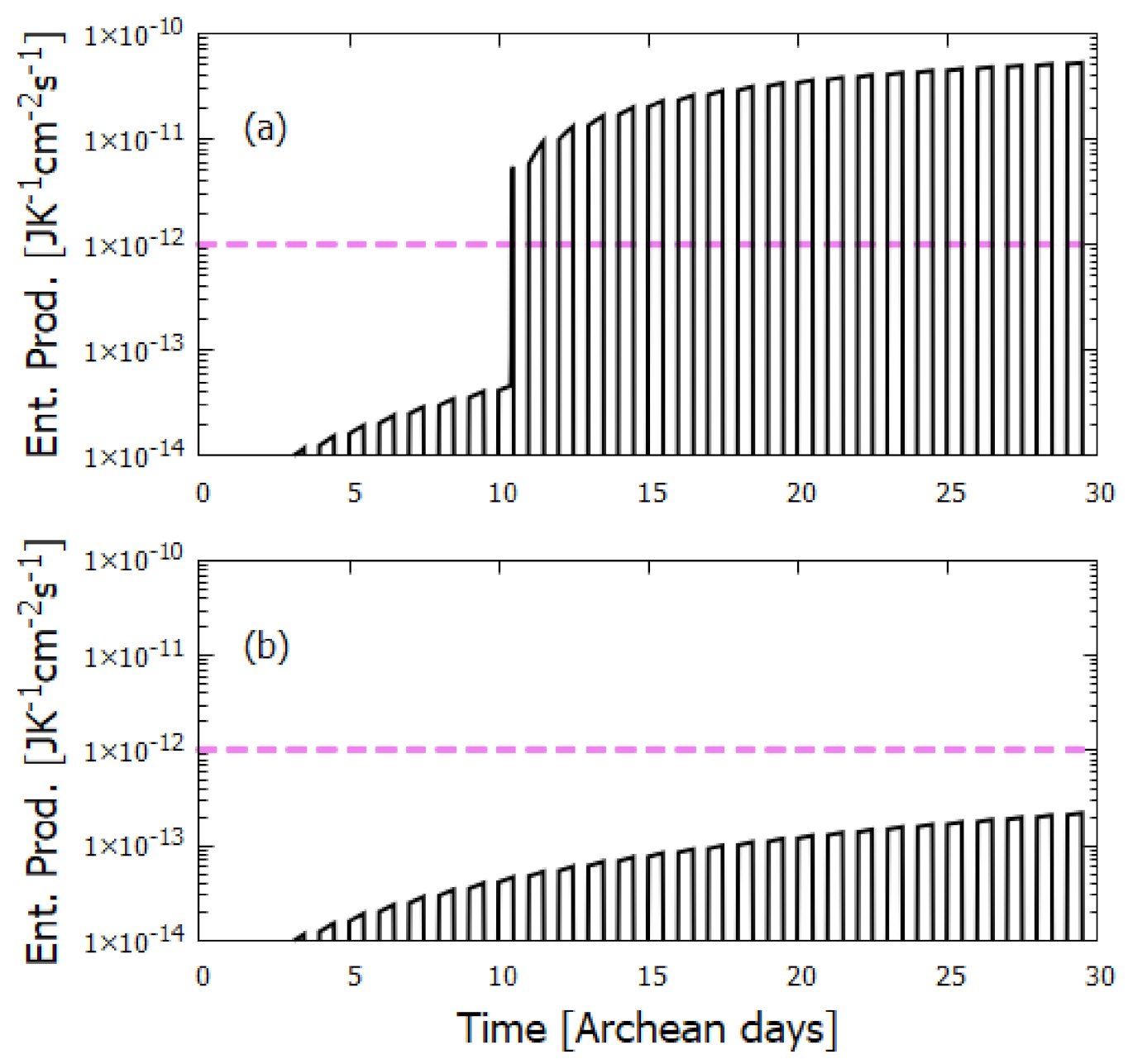
6. Discussion
7. Summary and Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIAC | 2-amino-3-iminoacrylimidoyl cyanide |
| AICA | 4-aminoimidazole-5-carboxamide |
| AICN | 4-aminoimidazole-5-carbonitrile |
| CIT | Classical Irreversible Thermodynamics |
| DAMN | diaminomaleonitrile |
| DAFN | diaminofumaronitrile |
| PAHs | Poly-Aromatic Hydrocarbons |
| UVA | light in the region 360–400 nm |
| UVB | light in the region 285–360 nm (only the region 310–360 nm is relevant here) |
| UVC | light in the region 100–285 nm (only the region 205–285 nm is relevant here) |
| UVTAR | Ultraviolet and Temperature Assisted Replication |
Appendix A. Thermodynamic Foundations of the Origin of Life
Appendix A.1. Classical Irreversible Thermodynamics
Appendix A.2. Stability, Evolution, and Entropy Production in the Non-Linear Regime
Appendix B. Relation to Statistical Mechanical Fluctuation Theorems Employing Linear Stability Theory for Evolution Analysis
References
- Anders, E. Pre-biotic organic matter from comets and asteroids. Nature 1989, 342, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Chyba, C.F.; Thomas, P.; Brookshaw, L.; Sagan, C. Cometary delivery of organic molecules to the early Earth. Science 1990, 249, 366–373. [Google Scholar] [CrossRef]
- Chyba, C.F.; Sagan, C. Endogenous production, exogenous delivery and impact-shock synthesis of organic molecules: An inventory for the origins of life. Nature 1992, 355, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Brack, A. (Ed.) The Molecular Origins of Life: Assembling Pieces of the Puzzle; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar] [CrossRef]
- Sutherland, J. Opinion: Studies on the origin of life—The end of the beginning. Nat. Rev. Chem. 2017, 1, 0012. [Google Scholar] [CrossRef]
- Miller, S.L.; Urey, H.C.; Oró, J. Origin of Organic Compounds on the Primitive Earth and in Meteorites. J. Mol. Evol. 1976, 9, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Oró, J.; Miller, S.L.; Lazcano, A. The origin and early evolution of life on Earth. Ann. Rev. Earth Planet. Sci. 1990, 18, 317–356. [Google Scholar] [CrossRef]
- Cleaves, H.; Miller, S. Prebiotic chemistry on the primitive Earth. In Systems Biology: Volume I: Genomics; Rigoutsos, I., Stephanopoulos, G., Eds.; Oxford University Press: New York, NY, USA, 2007; pp. 3–56. [Google Scholar]
- Glansdorff, P. Energetic Evolution of Complex Networks of Reactions. In Living Systems as Energy Converters; Buvet, R., Allen, M.J., Massué, J.P., Eds.; North-Holland Publishing Co.: Amsterdam, Holland, 1977; pp. 41–54. [Google Scholar]
- Michaelian, K. Thermodynamic origin of life. arXiv 2009, arXiv:physics.gen-ph/0907.0042. [Google Scholar]
- Michaelian, K. Thermodynamic dissipation theory for the origin of life. Earth Syst. Dynam. 2011, 224, 37–51. [Google Scholar] [CrossRef]
- Michaelian, K.; Simeonov, A. Fundamental molecules of life are pigments which arose and co-evolved as a response to the thermodynamic imperative of dissipating the prevailing solar spectrum. Biogeosciences 2015, 12, 4913–4937. [Google Scholar] [CrossRef]
- Michaelian, K. Thermodynamic Dissipation Theory of the Origina and Evolution of Life: Salient Characteristics of RNA and DNA and Other Fundamental Molecules Suggest an Origin of Life Driven by UV-C Light; Self-Published, Printed by CreateSpace; Mexico City, Mexico; 2016; ISBN 9781541317482. [Google Scholar]
- Michaelian, K. Microscopic Dissipative Structuring and Proliferation at the Origin of Life. Heliyon 2017, 3, e00424. [Google Scholar] [CrossRef]
- Michaelian, K. Homochirality through Photon-Induced Denaturing of RNA/DNA at the Origin of Life. Life 2018, 8, 21. [Google Scholar] [CrossRef]
- Michaelian, K.; Santillan, N. UVC photon-induced denaturing of DNA: A possible dissipative route to Archean enzyme-less replication. Heliyon 2019, 5, e01902. [Google Scholar] [CrossRef] [PubMed]
- Michaelian, K.; Rodriguez, O. Prebiotic fatty acid vesicles through photochemical dissipative structuring. Rev. Cuba. Química 2019, 31, 354–370. [Google Scholar]
- Mejía Morales, J.; Michaelian, K. Photon Dissipation as the Origin of Information Encoding in RNA and DNA. Entropy 2020, 22, 940. [Google Scholar] [CrossRef]
- Prigogine, I.; Nicolis, G.; Babloyantz, A. Thermodynamics of evolution. Phys. Today 1972, 25, 23–44. [Google Scholar] [CrossRef]
- Babloyantz, A.; Hiernaux, J. Models for cell differentiation and generation of polarity in diffusion-governed morphogenetic fields. Bull. Math. Biol. 1975, 37, 637–657. [Google Scholar] [CrossRef]
- Michaelian, K. Thermodynamic stability of ecosystems. J. Theor. Biol. 2005, 237, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Kleidon, A.; Malhi, Y.; Cox, P.M. Maximum entropy production in environmental and ecological systems. Philos. Trans. R. Soc. Biol. Sci. 2010, 365, 1297–1302. Available online: http://xxx.lanl.gov/abs/https://royalsocietypublishing.org/doi/pdf/10.1098/rstb.2010.0018 (accessed on 9 February 2021). [CrossRef]
- Prigogine, I.; Nicolis, G. Biological order, structure and instabilities. Q. Rev. Biophys. 1971, 4, 107–144. [Google Scholar] [CrossRef]
- Kleidon, A. Maximum entropy production and general trends in biospheric evolution. Paleontol. J. 2009, 43, 980–985. [Google Scholar] [CrossRef]
- Michaelian, K. Biological catalysis of the hydrological cycle: Lifes thermodynamic function. Hydrol. Earth Syst. Sci. 2012, 16, 2629–2645. [Google Scholar] [CrossRef]
- Michaelian, K. The Biosphere: A Thermodynamic Imperative. In The Biosphere; Natarajan, I., Ed.; IntechOpen: London, UK, 2012; pp. 51–60. [Google Scholar]
- Michaelian, K.; Simeonov, A. Thermodynamic explanation of the cosmic ubiquity of organic pigments. Astrobiol. Outreach 2017, 5, 156. [Google Scholar] [CrossRef]
- Schuurman, M.S.; Stolow, A. Dynamics at Conical Intersections. Annu. Rev. Phys. Chem. 2018, 69, 427–450. [Google Scholar] [CrossRef]
- Orr-Ewing, A. Reaction Dynamics–Relaxation Pathways. Lect. Notes 2014, 1–36. Available online: http://iramis.cea.fr/meetings/MTS2/publies/Orr-Ewing%20MOLIM%20lecture%202%20for%20publication.pdf (accessed on 8 February 2021).
- Roberts, G.M.; Marroux, H.J.B.; Grubb, M.P.; Ashfold, M.N.R.; Orr-Ewing, A.J. On the Participation of Photoinduced N–H Bond Fission in Aqueous Adenine at 266 and 220 nm: A Combined Ultrafast Transient Electronic and Vibrational Absorption Spectroscopy Study. J. Phys. Chem. A 2014, 118, 11211–11225. [Google Scholar] [CrossRef] [PubMed]
- Kleinermanns, K.; Nachtigallová, D.; de Vries, M.S. Excited state dynamics of DNA bases. Int. Rev. Phys. Chem. 2013, 32, 308–342. [Google Scholar] [CrossRef]
- Barbatti, M.; Aquino, A.; Szymczak, J.; Nachtigallová, D.; Hobza, P.; Lischka, H. Relaxation mechanisms of UV-photoexcited DNA and RNA nucleobases. Proc. Natl. Acad. Sci. USA 2010, 107, 21453–21458. [Google Scholar] [CrossRef]
- Polli, D.; Altoè, P.; Weingart, O.; Spillane, K.M.; Manzoni, C.; Brida, D.; Tomasello, G.; Orlandi, G.; Kukura, P.; Mathies, R.A.; et al. Conical intersection dynamics of the primary photoisomerization event in vision. Nature 2010, 467, 440–443. [Google Scholar] [CrossRef]
- Serrano-Perez, J.J.; de Vleeschouwer, F.; de Proft, F.; Mendive-Tapia, D.; Bearpark, M.J.; Robb, M.A. How the Conical Intersection Seam Controls Chemical Selectivity in the Photocycloaddition of Ethylene and Benzene. J. Org. Chem. 2013, 78, 1874–1886. [Google Scholar] [CrossRef]
- Sagan, C. Ultraviolet Selection Pressure on the Earliest Organisms. J. Theor. Biol. 1973, 39, 195–200. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y.; Cherepanov, D.A.; Galperin, M.Y. Survival of the fittest before the beginning of life: Selection of the first oligonucleotide-like polymers by UV light. BMC Evol. Biol. 2003, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Prigogine, I. Introduction to Thermodynamics of Irreversible Processes, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 1967. [Google Scholar]
- Boltzmann, L. Ludwig Boltzmann: Theoretical Physics and Philosophical Problems: Selected Writings; Springer: Berlin/Heidelberg, Germany, 1974. [Google Scholar]
- Boltzmann, L. Populäre Schriften; Engelbert Broda, Ed.; Friedr. Vieweg & Sohn: Braunschweig/Wiesbaden, Germany, 1979. [Google Scholar]
- Berkner, L.; Marshall, L. Origin and Evolution of the Oceans and Atmosphere; John Wiley and Sons: Hoboken, NJ, USA, 1964; pp. 102–126. [Google Scholar]
- Cnossen, I.; Sanz-Forcada, J.; Favata, F.; Witasse, O.; Zegers, T.; Arnold, N.F. The habitat of early life: Solar X-ray and UV radiation at Earth’s surface 4–3.5 billion years ago. J. Geophys. Res. 2007, 112, E02008. [Google Scholar] [CrossRef]
- Ferris, J.P.; Orgel, L.E. An Unusual Photochemical Rearrangement in the Synthesis of Adenine from Hydrogen Cyanide. J. Am. Chem. Soc. 1966, 88, 1074. [Google Scholar] [CrossRef]
- Sagan, C.; Khare, B.N. Long-Wavelength Ultraviolet Photoproduction of Amino Acids on the Primitive Earth. Science 1971, 173, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Bermejo, M.; Zorzano, M.P.; Osuna-Esteban, S. Simple Organics and Biomonomers Identified in HCN Polymers: An Overview. Life 2013, 3, 421–448. [Google Scholar] [CrossRef]
- Cleaves, H.J.; Miller, S.L. Oceanic protection of prebiotic organic compounds from UV radiation. Proc. Natl. Acad. Sci. USA 1998, 95, 7260–7263. [Google Scholar] [CrossRef] [PubMed]
- Cockell, C.S. The ultraviolet history of the terrestrial planets—Implications for biological evolution. Planet. Space Sci. 2000, 48, 203–214. [Google Scholar] [CrossRef]
- Oparin, A.I. The origin of life. In The Origin of Life; Bernal, J.D., Ed.; Weidenfeld & Nicolson: London, UK, 1967; pp. 199–234. [Google Scholar]
- Haldane, J.B.S. Origin of life. Ration. Annu. 1929, 148, 3–10. [Google Scholar]
- Urey, H.C. On the Early Chemical History of the Earth and the Origin of Life. Proc. Natl. Acad. Sci. USA 1952, 38, 351–363. Available online: https://www.pnas.org/content/38/4/351.full.pdf (accessed on 9 February 2021). [CrossRef]
- Sagan, C. Radiation and the Origin of the Gene. Evolution 1957, 11, 40–55. [Google Scholar] [CrossRef]
- Baly, E.C.C.; Stephen, W.E.; Hood, N.R. The photosynthesis of naturally occurring compounds—II. The photosynthesis of carbohydrates from carbonic acid by means of visible light. Proc. R. Soc. Lond. Ser. A Contain. Pap. Math. Phys. Character 1927, 116, 212–219. Available online: https://royalsocietypublishing.org/doi/pdf/10.1098/rspa.1927.0132 (accessed on 9 February 2021). [CrossRef]
- Miller, S.L. A Production of Amino Acids Under Possible Primitive Earth Conditions. Science 1953, 117, 528–529. Available online: https://science.sciencemag.org/content/117/3046/528.full.pdf (accessed on 9 February 2021). [CrossRef] [PubMed]
- Oró, J.; Kimball, A. Synthesis of purines under possible primitive earth conditions: II. Purine intermediates from hydrogen cyanide. Arch. Biochem. Biophys. 1962, 96, 293–313. [Google Scholar] [CrossRef]
- Ponnamperuma, C.; Sagan, C.; Mariner, R. Synthesis of adenosine triphosphate under possible primitive Earth conditions. Nature 1963, 199, 222–226. [Google Scholar] [CrossRef]
- Ponnamperuma, C.; Mariner, R. Formation of Ribose and Deoxyribose by Ultraviolet Irradiation of Formaldehyde in Water. Rad. Res. 1963, 19, 183. [Google Scholar]
- Ponnamperuma, C.; Mariner, R.; Sagan, C. Formation of Adenosine by Ultraviolet Irradiation of a Solution of Adenine and Ribose. Nature 1963, 198, 1199–1200. [Google Scholar] [CrossRef]
- Mejía, J.; Michaelian, K. Information Encoding in Nucleic Acids through a Dissipation-Replication Relation. arXiv 2018, arXiv:physics.bio-ph/1804.05939. [Google Scholar]
- Yarus, M.; Widmann, J.; Knight, R. RNA-Amino Acid Binding: A Stereochemecal Era for the Genetic Code. J. Mol. Evol. 2009, 69, 406–429. [Google Scholar] [CrossRef]
- Vitas, M.; Dobovišek, A. In the Beginning was a Mutualism—On the Origin of Translation. Orig. Life Evol Biosph. 2018. [Google Scholar] [CrossRef] [PubMed]
- Trainer, M.G.; Jimenez, J.L.; Yung, Y.L.; Toon, O.B.; Tolbert, M.A. Nitrogen Incorporation in CH4-N2 Photochemical Aerosol Produced by Far UV Irradiation. NASA Arch. 2012. Available online: https://ntrs.nasa.gov/archive/nasa/casi.ntrs.nasa.gov/20120009529.pdf (accessed on 9 February 2021).
- Ritson, D.; Sutherland, J. Prebiotic synthesis of simple sugars by photoredox systems chemistry. Nat. Chem. 2012, 4, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Ghule, S.; Vanka, K. Insights Into the Origin of Life: Did It Begin from HCN and H2O? ACS Cent. Sci. 2019, 5, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Pflüger, E. Beitragë zur Lehre von der Respiration. I. Ueber die physiologische Verbrennung in den lebendigen organismen. Arch. Ges. Physiol. 1875, 10, 641–644. [Google Scholar] [CrossRef]
- Minard, R.D.; Matthews, C.N. HCN World: Establishing Proteininucleic Acid Life via Hydrogen Cyanide Polymers. Abstr. Pap. Am. Chem. Soc. 2004, 228, U963. [Google Scholar]
- Matthews, C.N. Series: Cellular Origin and Life in Extreme Habitats and Astrobiology. In Origins: Genesis, Evolution and Diversity of Life; Seckbach, J., Ed.; Kluwer: Dordrecht, The Netherlands, 2004; Volume 6, pp. 121–135. [Google Scholar]
- Neveu, M.; Kim, H.J.; Benner, S.A. The “Strong” RNA World Hypothesis: Fifty Years Old. Astrobiology 2013, 13, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Oró, J. Synthesis of adenine from ammonium cyanide. Biochem. Biophys. Res. Commun. 1960, 2, 407–412. [Google Scholar] [CrossRef]
- Sanchez, R.A.; Ferris, J.P.; Orgel, L.E. Studies in Prebiodc Synthesis II: Synthesis of Purine Precursors and Amino Acids from Aqueous Hydrogen Cyanide. J. Mol. Biol. 1967, 80, 223–253. [Google Scholar]
- Sanchez, R.A.; Ferris, J.P.; Orgel, L.E. Studies in Prebiodc Synthesis IV: Conversion of 4-Aminoimidazole-5-carbonitrile Derivatives to Purines. J. Mol. Biol. 1968, 38, 121–128. [Google Scholar] [CrossRef]
- Roy, D.; Najafian, K.; von Rague Schleyer, P. Chemical evolution: The mechanism of the formation of adenine under prebiotic conditions. Proc. Natl. Acad. Sci. USA 2007, 104, 17272–17277. [Google Scholar] [CrossRef]
- Boulanger, E.; Anoop, A.; Nachtigallova, D.; Thiel, W.; Barbatti, M. Photochemical Steps in the Prebiotic Synthesis of Purine Precursors from HCN. Angew. Chem. Int. 2013, 52, 8000–8003. [Google Scholar] [CrossRef]
- Stribling, R.; Miller, S.L. Energy Yields for Hydrogen Cyanide and Formaldehyde Syntheses: The HCN and Amino Acid Concentrations in the Primitive Ocean. Orig. Life 1986, 17, 261–273. [Google Scholar] [CrossRef]
- Sanchez, R.; Ferris, J.; Orgel, L.E. Conditions for Purine Synthesis: Did Prebiotic Synthesis Occur at Low Temperatures? Science 1966, 153, 72–73. Available online: https://science.sciencemag.org/content/153/3731/72.full.pdf (accessed on 9 February 2021). [CrossRef]
- Miller, S.L.; Lazcano, A. The Origin of Life—Did It Occur at High Temperatures? Mol. Evol. 1995, 41, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Bada, J.L.; Lazcano, A. Some Like It Hot, But Not the First Biomolecules. Science 2002, 296, 1982–1983. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, S.; Cleaves, H.J.; Miller, S.L. The cold origin of life: B. Implications based on pyrimidines and purines produced from frozen ammonium cyanide solutions. Orig. Life Evol Biosph. 2002, 32, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.T. The sea-surface Microlayer (1982) Biology, Chemistry and Anthropogenic Enrichment. Prog. Oceanogr. 1982, 11, 307–328. [Google Scholar] [CrossRef]
- Grammatika, M.; Zimmerman, W.B. Microhydrodynamics of flotation processes in the sea-surface layer. Dynam. Atmos. Ocean. 2001, 34, 327–348. [Google Scholar] [CrossRef]
- Fábián, B.; Szőri, M.; Jedlovszky, P. Floating Patches of HCN at the Surface of Their Aqueous Solutions—Can They Make “HCN World” Plausible? J. Phys. Chem. C 2014, 118, 21469–21482. [Google Scholar] [CrossRef]
- Oró, J. Chemical synthesis of lipids and the origin of life. J. Biol. Phys. 1995, 20, 135–147. [Google Scholar] [CrossRef]
- Walde, P.; Wick, R.; Fresta, M.; Mangone, A.; Luisi, P.L. Autopoietic Self-Reproduction of Fatty Acid Vesicles. J. Am. Chem. Soc. 1994, 116, 11649–11654. [Google Scholar] [CrossRef]
- Deamer, D. The Role of Lipid Membranes in Life’s Origin. Life 2017, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Fang, Y.; Ma, L. The self-crosslinked ufasome of conjugated linoleic acid: Investigation of morphology, bilayer membrane and stability. Colloids Surf. B Biointerfaces 2014, 123, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Calvin, M. Occurrence of fatty acids and aliphatic hydrocarbons in a 3.4 billion-year-old sediment. Nature 1969, 224, 576–577. [Google Scholar] [CrossRef] [PubMed]
- Van Hoeven, W.; Maxwell, J.; Calvin, M. Fatty acids and hydrocarbons as evidence of life processes in ancient sediments and crude oils. Geochim. Cosmochim. Acta 1969, 33, 877–881. [Google Scholar] [CrossRef]
- Karhu, J.; Epstein, S. The Implication of the Oxygen isotope Records in Coexisting Cherts and Phosphates. Geochim. Cosmochim. Acta 1986, 50, 1745–1756. [Google Scholar] [CrossRef]
- Knauth, L.P. Lecture Notes in Earth Sciences #43; Chapter Isotopic Signatures and Sedimentary Records; Springer: Berlin, Germany, 1992; pp. 123–152. [Google Scholar]
- Knauth, L.P.; Lowe, D.R. High Archean climatic temperature inferred from oxygen isotope geochemistry of cherts in the 3.5 Ga Swaziland group, South Africa. Geol. Soc. Am. Bull. 2003, 115, 566–580. [Google Scholar] [CrossRef]
- Petersen, C.; Dahl, N.H.; Jensen, S.K.; Poulsen, J.A.; Thøgersen, J.; Keiding, S.R. Femtosecond Photolysis of Aqueous Formamide. J. Phys. Chem. A 2008, 112, 3339–3344. [Google Scholar] [CrossRef] [PubMed]
- Basch, H.; Robin, M.B.; Kuebler, N.A. Electronic Spectra of Isoelectronic Amides, Acids, and Acyl Fluorides. J. Chem. Phys. 1968, 49, 5007–5018. [Google Scholar] [CrossRef]
- Lelj, F.; Adamo, C. Solvent effects on isomerization equilibria: An energetic analysis in the framework of density functional theory. Theor. Chim. Acta 1995, 91, 199–214. [Google Scholar] [CrossRef]
- Koch, T.; Rodehorst, R. Quantitative investigation of the photochemical conversion of diaminomaleonitrile to diaminofumaronitrile and 4-amino-5-cyanoimidazole. J. Am. Chem. Soc. 1974, 96, 6707–6710. [Google Scholar] [CrossRef]
- Gupta, V.; Tandon, P. Conformational and vibrational studies of isomeric hydrogen cyanide tetramers by quantum chemical methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 89, 55–66. [Google Scholar] [CrossRef]
- Ferris, J.; Joshi, P.; Edelson, E.; Lawless, J. HCN: A plausible source of purines, pyrimidines and amino acids on the primitive Earth. J. Mol. Evol. 1978, 11, 293–311. [Google Scholar] [CrossRef]
- Glaser, R.; Hodgen, B.; Farrelly, D.; McKee, E. Adenine synthesis in interstellar space: Mechanisms of prebiotic pyrimidine-ring formation of monocyclic HCN-pentamers. Astrobiology 2007, 7, 455–470. [Google Scholar] [CrossRef]
- Cavaluzzi, M.J.; Borer, P.N. Revised UV extinction coefficients for nucleoside-5’- monophosphates and unpaired DNA and RNA. Nucleic Acids Res. 2004, 32, e13. [Google Scholar] [CrossRef]
- Franz, J.; Gianturco, F. Low-energy positron scattering from DNA nucleobases: The effects from permanent dipoles. Eur. Phys. J. D 2014, 68. [Google Scholar] [CrossRef]
- Stimson, M.M.; Reuter, M.A. Ultraviolet Absorption Spectra of Nitrogenous Heterocycles. VII. The Effect of Hydroxy Substitutions on the Ultraviolet Absorption of the Series: Hypoxanthine, Xanthine and Uric Acid1. J. Am. Chem. Soc. 1943, 65, 153–155. [Google Scholar] [CrossRef]
- Miyakawa, S.; Cleaves, H.J.; Miller, S.L. The Cold Origin of Life: A. Implications Based On The Hydrolytic Stabilities Of Hydrogen Cyanide And Formamide. Orig. Life Evol Biosph. 2002, 32, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Kua, J.; Thrush, K.L. HCN, Formamidic Acid, and Formamide in Aqueous Solution: A Free-Energy Map. J. Phys. Chem. B 2016, 120, 8175–8185. [Google Scholar] [CrossRef] [PubMed]
- Maier, G.; Endres, J. Isomerization of Matrix-Isolated Formamide: IR-Spectroscopic Detection of Formimidic Acid. Eur. J. Org. Chem. 2000, 2000, 1061–1063. Available online: https://chemistry-europe.onlinelibrary.wiley.com/doi/pdf/10.1002 (accessed on 9 February 2021). [CrossRef]
- Duvernay, F.; Trivella, A.; Borget, F.; Coussan, S.; Aycard, J.P.; Chiavassa, T. Matrix Isolation Fourier Transform Infrared Study of Photodecomposition of Formimidic Acid. J. Phys. Chem. A 2005, 109, 11155–11162. [Google Scholar] [CrossRef]
- Barks, H.L.; Buckley, R.; Grieves, G.A.; Di Mauro, E.; Hud, N.V.; Orlando, T.M. Guanine, Adenine, and Hypoxanthine Production in UV-Irradiated Formamide Solutions: Relaxation of the Requirements for Prebiotic Purine Nucleobase Formation. ChemBioChem 2010, 11, 1240–1243. Available online: https://chemistry-europe.onlinelibrary.wiley.com/doi/pdf/10.1002/cbic.201000074 (accessed on 9 February 2021). [CrossRef] [PubMed]
- Gingell, J.; Mason, N.; Zhao, H.; Walker, I.; Siggel, M. VUV optical-absorption and electron-energy-loss spectroscopy of formamide. Chem. Phys. 1997, 220, 191–205. [Google Scholar] [CrossRef]
- Yonemitsu, E.; Isshiki, T.; Kijima, Y. Process for Preparing Adenine. U.S. Patent 4,059,582, 31 July 1974. [Google Scholar]
- Zubay, G.; Mui, T. Prebiotic Synthesis of Nucleotides. Orig. Life Evol Biosph. 2001, 31, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, J.; Nguyen, M.T.; Springsteen, G.; Leszczynski, J. From Formamide to Purine: A Self-Catalyzed Reaction Pathway Provides a Feasible Mechanism for the Entire Process. J. Phys. Chem. B 2013, 117, 9333–9342. [Google Scholar] [CrossRef]
- Levy, M.; Miller, S.L. The stability of the RNA bases: Implications for the origin of life. Proc. Natl. Acad. Sci. USA 1998, 95, 7933–7938. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hu, A. Comparative study of spontaneous deamination of adenine and cytosine in unbuffered aqueous solution at room temperature. Chem. Phys. Lett. 2016, 653, 207–211. [Google Scholar] [CrossRef]
- Wang, X.C.; Nichols, J.; Feyereisen, M.; Gutowski, M.; Boatz, J.; Haymet, A.D.J.; Simons, J. Ab initio quantum chemistry study of formamide-formamidic acid tautomerization. J. Phys. Chem. 1991, 95, 10419–10424. [Google Scholar] [CrossRef]
- Cataldo, F.; Patanè, G.; Compagnini, G. Synthesis of HCN Polymer from Thermal Decomposition of Formamide. J. Macromol. Sci. Part A 2009, 46, 1039–1048. [Google Scholar] [CrossRef]
- Hill, A.; Orgel, L. Synthesis of Adenine from HCN Tetramer and Ammonium Formate. Orig. Life Evol Biosph. 2002, 32, 99–102. [Google Scholar] [CrossRef]
- Herbst, E. The chemistry of interstellar space. Chem. Soc. Rev. 2001, 30, 168–176. [Google Scholar] [CrossRef]
- Benallou, A. A new mechanistic insight of DNA base adenine formation from pentamer HCN in the gas phase of interstellar clouds. J. Taibah Univ. Sci. 2019, 13, 105–111. [Google Scholar] [CrossRef]
- Szabla, R.; Šponer, J.E.; Šponer, J.; Sobolewski, A.L.; Góra, R.W. Solvent effects on the photochemistry of 4-aminoimidazole-5-carbonitrile, a prebiotically plausible precursor of purines. Phys. Chem. Chem. Phys. 2014, 16, 17617–17626. [Google Scholar] [CrossRef]
- Frick, G. Formation of Amino-Acids in Hydrolysis of Adenine. Nature 1952, 169, 758–759. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Meng, F. Theoretical study of water-assisted hydrolytic deamination mechanism of adenine. Struct. Chem. 2009, 20, 943–949. [Google Scholar] [CrossRef]
- Yang, N.J.; Hinner, M.J. Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. Methods Mol. Biol. 2015, 1266, 29–53. [Google Scholar] [CrossRef]
- Agarwal, S.; Clancy, C.; Harvey, R. Mechanisms Restricting Diffusion of Intracellular cAMP. Sci. Rep. 2008, 6, 19577. [Google Scholar] [CrossRef]
- Bowen, W.J.; Martin, H.L. The diffusion of adenosine triphosphate through aqueous solutions. Arch. Biochem. Biophys. 1964, 107, 30–36. [Google Scholar] [CrossRef]
- Zahnle, K.J. Photochemistry of methane and the formation of hydrocyanic acid (HCN) in the Earth’s early atmosphere. J. Geophys. Res. Atmos. 1986, 91, 2819–2834. Available online: https://agupubs.onlinelibrary.wiley.com/doi/pdf/10.1029/JD091iD02p02819 (accessed on 9 February 2021). [CrossRef]
- Zhang, Z.; Liu, C.; Liu, L.; Yu, L.; Wang, Z. Study on Dissolved Trace Metals in Sea Surface Microlayer in Daya Bay. Chin. J. Oceanol. Limnol. 2004, 22, 54–63. [Google Scholar]
- Oksana Shvydkiv. Microphotochemistry—A New Resources-Efficient Synthesis Tool Approach. Ph.D. Thesis, Chemical Sciences Dublin City University, Dublin, Ireland, 2012.
- Turing, A.M. The Chemical Basis of Morphogenesis. Philos. Trans. R. Soc. Lond. Ser. B 1952, 237, 37–72. [Google Scholar] [CrossRef]
- Glansdorff, P.; Prigogine, I. Thermodynamic Theory of Structure, Stability and Fluctuations; Wiley-Interscience: London, UK, 1971. [Google Scholar]
- Schoffstall, A.M. Prebiotic phosphorylation of nucleosides in formamide. Orig. Life Evol Biosph. 1976, 7, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, G.; Saladino, R.; Crestini, C.; Ciciriello, F.; Di Mauro, E. Nucleoside phosphorylation by phosphate minerals. J. Biol. Chem. 2007, 282, 16729–16735. [Google Scholar] [CrossRef] [PubMed]
- Airey, P.L.; Dainton, F.S. The photochemistry of aqueous solutions of Fe(II) II. Processes in acidified solutions of potassium ferrocyanide at 25 ∘C. Proc. R. Soc. Lond. Ser. A. Math. Phys. Sci. 1966, 291, 478–486. Available online: https://royalsocietypublishing.org/doi/pdf/10.1098/rspa.1966.0109 (accessed on 9 February 2021). [CrossRef]
- Fox, S.W.; Harada, K. Synthesis of Uracil under Conditions of a Thermal Model of Prebiological Chemistry. Science 1961, 133, 1923–1924. Available online: https://science.sciencemag.org/content/133/3468/1923.full.pdf (accessed on 9 February 2021). [CrossRef]
- Ferris, J.P.; Sanchez, R.A.; Orgel, L.E. Studies in Prebiotic Synthesis III. Synthesis of Pyrimidines from Cyanoacetylene and Cyanate. J. Mol. Biol. 1968, 33, 693–704. [Google Scholar] [CrossRef]
- Choughuley, A.; Subbaraman, A.; Kazi, Z.; Chadha, M. A possible prebiotic synthesis of thymine: Uracil-formaldehyde-formic acid reaction. Biosystems 1977, 9, 73–80. [Google Scholar] [CrossRef]
- Luna, A.; Morizur, J.P.; Tortajada, J.; Alcamí, M.; Mó, O.; Yáñez, M. Role of Cu+ Association on the Formamide → Formamidic Acid → (Aminohydroxy)carbene Isomerizations in the Gas Phase. J. Phys. Chem. A 1998, 102, 4652–4659. [Google Scholar] [CrossRef]
- Gates, D.M. Biophysical Ecology; Springer: Berlin/Heidelberg, Germany, 1980. [Google Scholar]
- Ulanowicz, R.; Hannon, B. Life and the production of entropy. Proc. R. Soc. Lond. B 1987, 232, 181–192. [Google Scholar]
- Schneider, E.D.; Kay, J.J. Complexity and thermodynamics: Towards a new ecology. Futures 1994, 24, 626–647. [Google Scholar] [CrossRef]
- Kleidon, A. Entropy Production by Evapotranspiration and its Geographic Variation. Soil Water Res. 2008, 3, S89–S94. [Google Scholar] [CrossRef]
- Zotin, A.I. Bioenergetic trends of evolutionary progress of organisms. In Thermodynamics and Regulation of Biological Processes; Lamprecht, I., Zotin, A.I., Eds.; Walter de Gruyter: Berlin, Germany, 1984; pp. 451–458. [Google Scholar]
- Limaye, S.S.; Mogul, R.; Smith, D.J.; Ansari, A.H.; Słowik, G.P.; Vaishampayan, P. Venus’ Spectral Signatures and the Potential for Life in the Clouds. Astrobiology 2018, 18, 1181–1198. [Google Scholar] [CrossRef] [PubMed]
- Heinz, J.; Schulze-Makuch, D. Thiophenes on Mars: Biotic or Abiotic Origin? Astrobiology 2020, 20, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Pershin, S. Correlation of “chlorophyll” and water index on mars surface. Microsymposium 2002, 36, MS079. [Google Scholar]
- López-Puertas, M.; Dinelli, B.M.; Adriani, A.; Funke, B.; García-Comas, M.; Moriconi, M.L.; D’Aversa, E.; Boersma, C.; Allamandola, L.J. Large Abundances of Polycyclic Aromatic Hydrocarbons in Titan’s Upper Atmosphere. Astrophys. J. 2013, 770, 132. [Google Scholar] [CrossRef]
- Rayleigh, J.W. Some general theorems relating to vibrations. Proc. Math. Soc. Lond. 1873, 4, 357–368. [Google Scholar]
- Einstein, A. On the movement of small particles suspended in stationary liquids required by the molecular-kinetic theory of heat. Ann. Phys. 1905, 549. [Google Scholar] [CrossRef]
- Onsager, L. Reciprocal Relations in Irreversible Processes, I. Phys. Rev. 1931, 37, 405–426. [Google Scholar] [CrossRef]
- Onsager, L. Reciprocal Relations in Irreversible Processes, II. Phys. Rev. 1931, 38, 2265. [Google Scholar] [CrossRef]
- Callen, H.B.; Wellton, T.A. Irreversibility and Generalized Noise. Phys. Rev. 1951, 83, 34–40. [Google Scholar] [CrossRef]
- Onsager, L.; Machlup, S. Fluctuations and Irreversible Processes. Phys. Rev. 1953, 91, 1505–1512. [Google Scholar] [CrossRef]
- Kubo, R. The fluctuation-dissipation theorem. Rep. Prog. Phys. 1966, 29, 255–284. [Google Scholar] [CrossRef]
- Evans, D.J.; Cohen, E.G.D.; Morriss, G.P. Probability of second law violations in shearing steady states. Phys. Rev. Lett. 1993, 71, 2401–2404. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.J.; Cohen, E.G.D.; Morriss, G.P. The Fluctuation Theorem. Adv. Phys. 2002, 51, 1529–1585. [Google Scholar] [CrossRef]
- Gaspard, P. Self-Organization at the Nanoscale Scale in Far-From-Equilibrium Surface Reactions and Copolymerizations. In Engineering of Chemical Complexity; Mikhailov, A.S., Ertl, G., Eds.; World Scientific: Singapore, 2013; pp. 51–77. [Google Scholar]
- Gaspard, P. Fluctuation theorem for nonequilibrium reactions. J. Chem. Phys. 2004, 120, 8898–8905. [Google Scholar] [CrossRef]
- Andrieux, D.; Gaspard, P. Fluctuation Theorem for Currents and Schnakenberg Network Theory. J. Stat. Phys. 2007, 127, 107–131. [Google Scholar] [CrossRef]
- Andrieux, D.; Gaspard, P.; Monnai, T.; Tasaki, S. The fluctuation theorem for currents in open quantum systems. New J. Phys. 2009, 11, 043014. [Google Scholar] [CrossRef]
















| # | Reaction | Reaction Constants |
|---|---|---|
| 1 | H F | ; s; hydrolysis of HCN [68,99,100] |
| 2 | F → Fa | [89,90,101,102,103] |
| 3 | Fa → H + HO | [102,103,104] |
| 4 | F Af | ; s; hydrolysis of formamide [100,103] |
| 5 | 4H C | ; M s; kcal mol [68] |
| 6 | 4H T | ; M s; tetramization [68] |
| 7 | 4H + T C+T | ; M s [68] |
| 8 | 4H + T 2T | ; M s [68] |
| 9a | + C → T | [92] |
| 9b | + T → C | [68,71,92] |
| 10 | + T → J | [68,71,92] |
| 11 | + J → I | ; T ; [68,71] |
| 12 | I L | ; s; kcal mol; hydrolysis of AICN [69] |
| 13 | I:F + Af A + F | ; M s; kcal mol [105,106] |
| 14 | I:F + Fa Am + Fa +HO | ; M s; kcal mol [107] |
| 15 | + Am → A | [95] |
| 16 | A Hy | ; s; valid for pH within 5 to 8; hydrolysis of adenine [108,109] |
| 17 | + C → C | |
| 18 | + T → T | |
| 19 | + J → J | |
| 20 | + Am → Am | |
| 21 | + I → I | |
| 22 | + L → L | |
| 23 | + A → A | |
| 24 | + Hy → Hy |
| 7.752 | 2.988 | 11.190 | 6.689 | 0.892 | 4.073 | 4.073 | 1.908 | 1.532 | 1.000 | 1.000 | 2.482 |
| # | Reaction | Parameter | Nominal Value | −30% | Nominal | +30% |
|---|---|---|---|---|---|---|
| 9b | +T→ C | 0.020 | 8.222 | 7.292 | 6.528 | |
| 12 | I L hydrolysis of AICN | 19.93 kcal mol | 1.093 | 7.292 | 7.311 | |
| 13 | I:F + Af A + F | 6.68 kcal mol | 7.311 | 7.292 | 6.636 | |
| 14 | I:F + Fa Am + Fa +HO | 19.90 kcal mol | 7.292 | 7.292 | 7.292 | |
| 15 | +Am→ A | 0.060 | 7.292 | 7.292 | 7.292 | |
| 16 | A→ Hy hydrolysis of adenine | expn. of | −5902 | 1.586 | 7.292 | 7.306 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michaelian, K. The Dissipative Photochemical Origin of Life: UVC Abiogenesis of Adenine. Entropy 2021, 23, 217. https://doi.org/10.3390/e23020217
Michaelian K. The Dissipative Photochemical Origin of Life: UVC Abiogenesis of Adenine. Entropy. 2021; 23(2):217. https://doi.org/10.3390/e23020217
Chicago/Turabian StyleMichaelian, Karo. 2021. "The Dissipative Photochemical Origin of Life: UVC Abiogenesis of Adenine" Entropy 23, no. 2: 217. https://doi.org/10.3390/e23020217
APA StyleMichaelian, K. (2021). The Dissipative Photochemical Origin of Life: UVC Abiogenesis of Adenine. Entropy, 23(2), 217. https://doi.org/10.3390/e23020217