Sequence Versus Composition: What Prescribes IDP Biophysical Properties?


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Protein Datasets
2.2. Bioinformatics Analysis
2.3. Survey of PDB-DisProt Overlap
2.4. Sequence Permutations
2.5. Statistical Analysis
3. Results
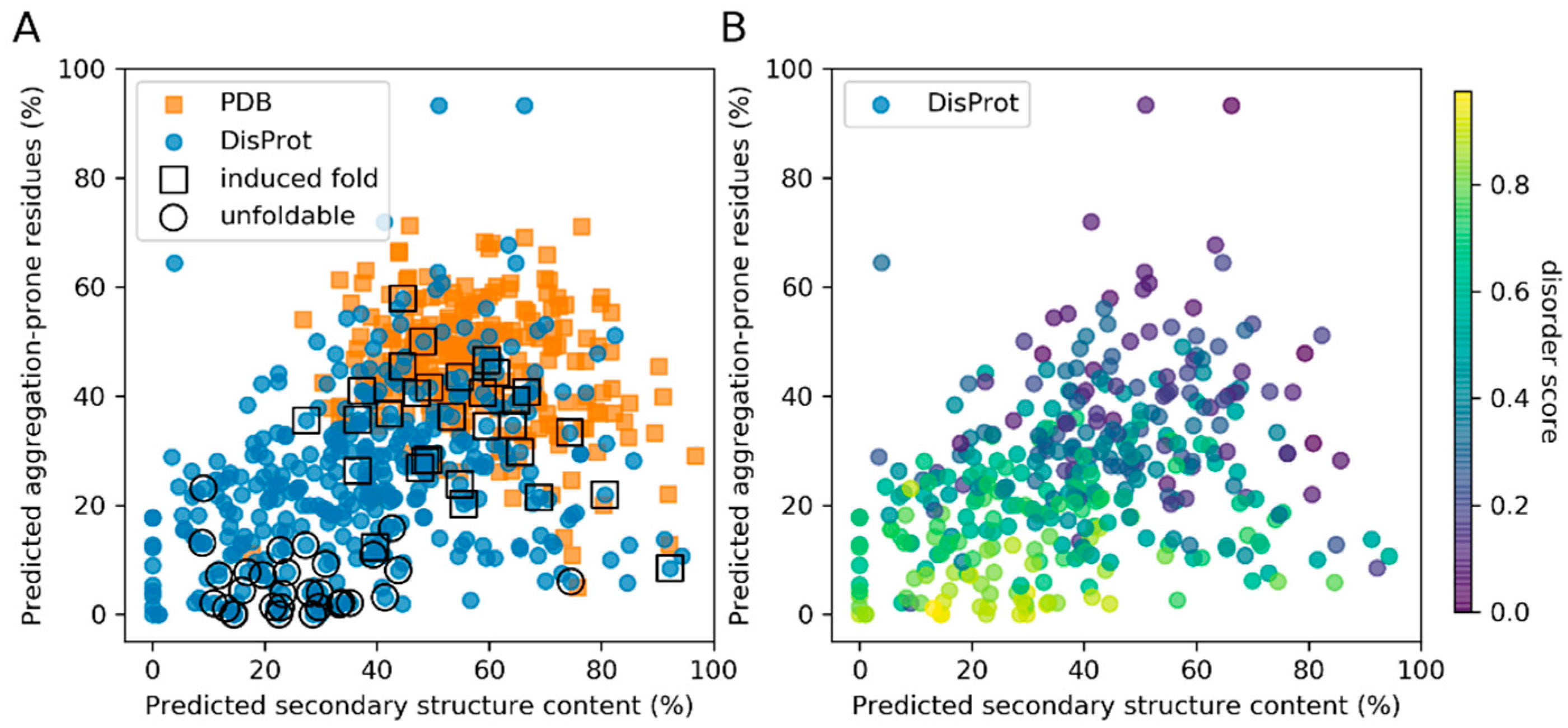
3.1. Secondary Structure and Aggregation Propensity Can Distinguish Between Induced Fold and Unfoldable IDPs
3.2. The Composition of IDPs Partly Overlaps with Narrower Globular Protein Composition
3.3. Sequence Permutation Experiments Suggest a Dominant Role of Composition
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Babu, M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef]
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Theillet, F.X.; Kalmar, L.; Tompa, P.; Han, K.H.; Selenko, P.; Dunker, A.K.; Daughdrill, G.W.; Uversky, V.N. The alphabet of intrinsic disorder: I. Act like a Pro: On the abundance and roles of proline residues in intrinsically disordered proteins. Intrinsically Disord. Proteins 2013, 1, e24360. [Google Scholar] [CrossRef]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins Struct. Funct. Bioinform. 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Uversky, V.N. Paradoxes and wonders of intrinsic disorder: Complexity of simplicity. Intrinsically Disord. Proteins 2016, 4, e1135015. [Google Scholar] [CrossRef]
- Piovesan, D.; Tabaro, F.; Mičetić, I.; Necci, M.; Quaglia, F.; Oldfield, C.J.; Aspromonte, M.C.; Davey, N.E.; Davidović, R.; Dosztányi, Z.; et al. DisProt 7.0: a major update of the database of disordered proteins. Nucleic Acids Res. 2016, 45, D219–D227. [Google Scholar] [CrossRef]
- Heffernan, R.; Dehzangi, A.; Lyons, J.; Paliwal, K.; Sharma, A.; Wang, J.; Sattar, A.; Zhou, Y.; Yang, Y. Highly accurate sequence-based prediction of half-sphere exposures of amino acid residues in proteins. Bioinformatics 2015, 32, 843–849. [Google Scholar] [CrossRef]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef]
- Frishman, D.; Argos, P. Seventy-five percent accuracy in protein secondary structure prediction. Proteins Struct. Funct. Bioinform. 1997, 27, 329–335. [Google Scholar] [CrossRef]
- Cuff, J.A.; Barton, G.J. Evaluation and improvement of multiple sequence methods for protein secondary structure prediction. Proteins Struct. Funct. Bioinform. 1999, 34, 508–519. [Google Scholar] [CrossRef]
- Levine, J.M.; Pascarella, S.; Argos, P.; Garnier, J. Quantification of secondary structure prediction improvement using multiple alignments. Prot. Eng. 1993, 6, 849–854. [Google Scholar] [CrossRef]
- Garnier, J. GOR secondary structure prediction method version IV. Meth. Enzym. 1998, 266, 540–553. [Google Scholar]
- Fang, Y.; Gao, S.; Tai, D.; Middaugh, C.R.; Fang, J. Identification of properties important to protein aggregation using feature selection. Bmc Bioinform. 2013, 14, 314. [Google Scholar] [CrossRef]
- Necci, M.; Piovesan, D.; Dosztányi, Z.; Tosatto, S.C. MobiDB-lite: fast and highly specific consensus prediction of intrinsic disorder in proteins. Bioinformatics 2017, 33, 1402–1404. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Naranjo, Y.; Pons, M.; Konrat, R. Meta-structure correlation in protein space unveils different selection rules for folded and intrinsically disordered proteins. Mol. Biosyst. 2012, 8, 411–416. [Google Scholar] [CrossRef]
- Linding, R.; Schymkowitz, J.; Rousseau, F.; Diella, F.; Serrano, L. A comparative study of the relationship between protein structure and β-aggregation in globular and intrinsically disordered proteins. J. Mol. Biol. 2004, 342, 345–353. [Google Scholar] [CrossRef]
- Uversky, V.N. The alphabet of intrinsic disorder: II. Various roles of glutamic acid in ordered and intrinsically disordered proteins. Intrinsically Disord. Proteins 2013, 1, e24684. [Google Scholar] [CrossRef]
- Vucetic, S.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Flavors of protein disorder. Proteins Struct. Funct. Bioinform. 2003, 52, 573–584. [Google Scholar] [CrossRef]
- Mao, A.H.; Lyle, N.; Pappu, R.V. Describing sequence–ensemble relationships for intrinsically disordered proteins. Biochem. J. 2013, 449, 307–318. [Google Scholar] [CrossRef]
- Das, R.K.; Ruff, K.M.; Pappu, R.V. Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2015, 32, 102–112. [Google Scholar] [CrossRef]
- Bastolla, U.; Moya, A.; Viguera, E.; van Ham, R.C. Genomic determinants of protein folding thermodynamics in prokaryotic organisms. J. Mol. Biol. 2004, 343, 1451–1466. [Google Scholar] [CrossRef]
- Monsellier, E.; Ramazzotti, M.; De Laureto, P.P.; Tartaglia, G.G.; Taddei, N.; Fontana, A.; Vendruscolo, M.; Chiti, F. The distribution of residues in a polypeptide sequence is a determinant of aggregation optimized by evolution. Biophys. J. 2007, 93, 4382–4391. [Google Scholar] [CrossRef]
- English, L.R.; Tischer, A.; Demeler, A.K.; Demeler, B.; Whitten, S.T. Sequence Reversal Prevents Chain Collapse and Yields Heat-Sensitive Intrinsic Disorder. Biophys. J. 2018, 115, 328–340. [Google Scholar] [CrossRef]
- Tretyachenko, V.; Vymětal, J.; Bednárová, L.; Kopecký, V.; Hofbauerová, K.; Jindrová, H.; Hubálek, M.; Souček, R.; Konvalinka, J.; Vondrášek, J.; et al. Random protein sequences can form defined secondary structures and are well-tolerated in vivo. Sci. Rep. 2017, 7, 15449. [Google Scholar] [CrossRef]
- Ángyán, A.F.; Perczel, A.; Gáspári, Z. Estimating intrinsic structural preferences of de novo emerging random-sequence proteins: Is aggregation the main bottleneck? Febs Lett. 2012, 586, 2468–2472. [Google Scholar] [CrossRef]
- Moesa, H.A.; Wakabayashi, S.; Nakai, K.; Patil, A. Chemical composition is maintained in poorly conserved intrinsically disordered regions and suggests a means for their classification. Mol. Biosyst. 2012, 8, 3262–3273. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vymětal, J.; Vondrášek, J.; Hlouchová, K. Sequence Versus Composition: What Prescribes IDP Biophysical Properties? Entropy 2019, 21, 654. https://doi.org/10.3390/e21070654
Vymětal J, Vondrášek J, Hlouchová K. Sequence Versus Composition: What Prescribes IDP Biophysical Properties? Entropy. 2019; 21(7):654. https://doi.org/10.3390/e21070654
Chicago/Turabian StyleVymětal, Jiří, Jiří Vondrášek, and Klára Hlouchová. 2019. "Sequence Versus Composition: What Prescribes IDP Biophysical Properties?" Entropy 21, no. 7: 654. https://doi.org/10.3390/e21070654
APA StyleVymětal, J., Vondrášek, J., & Hlouchová, K. (2019). Sequence Versus Composition: What Prescribes IDP Biophysical Properties? Entropy, 21(7), 654. https://doi.org/10.3390/e21070654