Thermoelectricity and Thermodiffusion in Magnetic Nanofluids: Entropic Analysis

 , ,
, ,  , , and
, , and 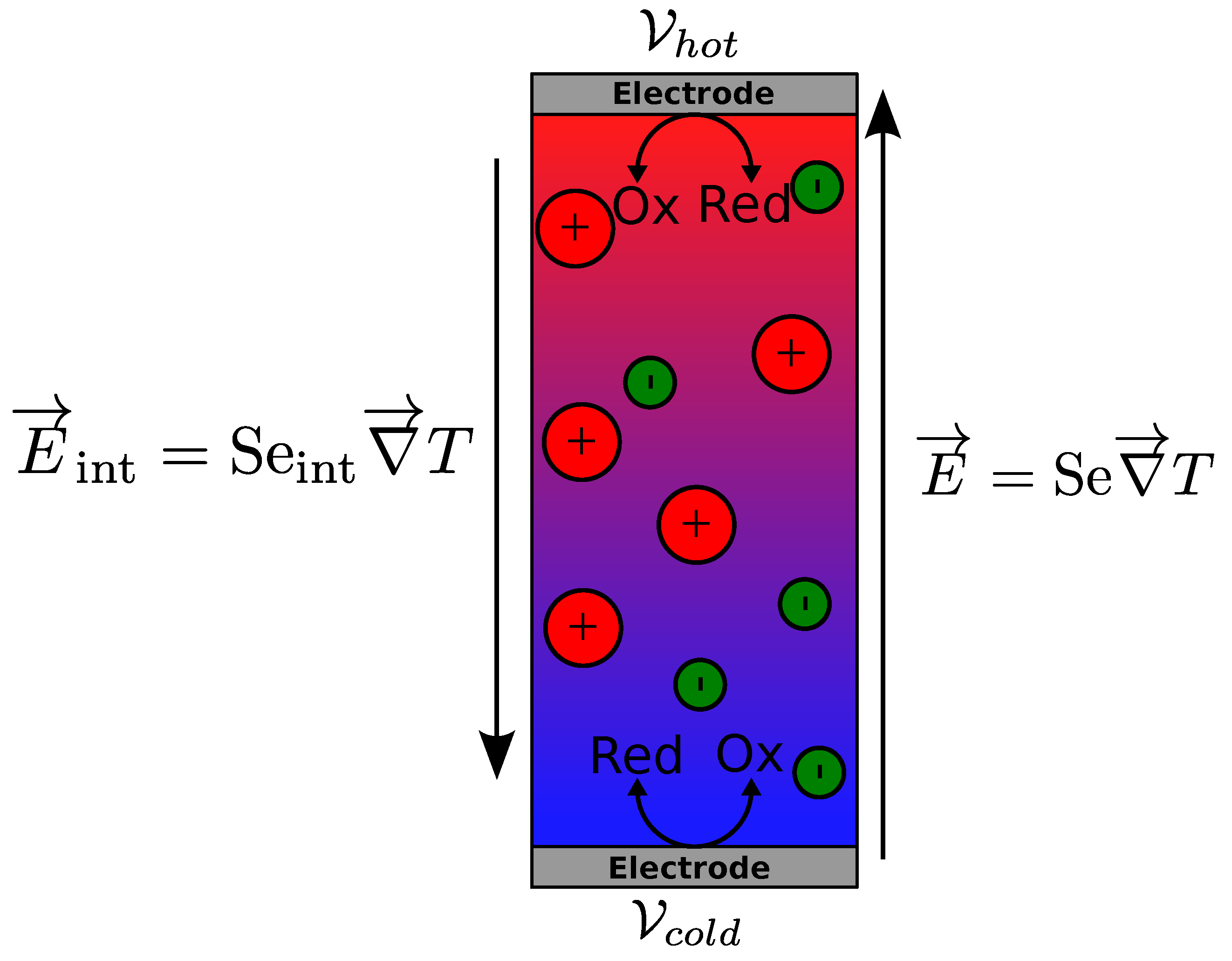{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
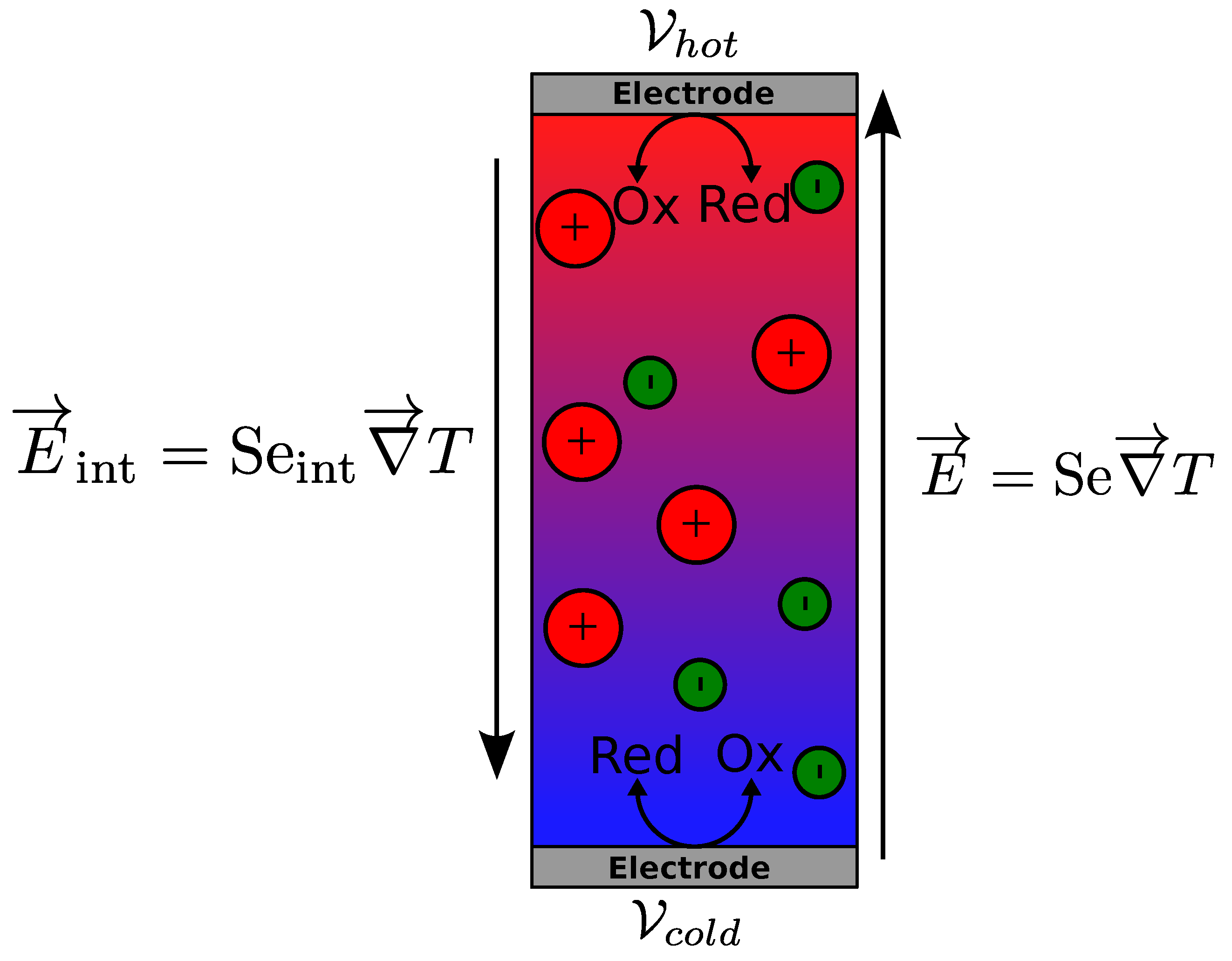
2. Thermogalvanic Cell
Thermogalvanic Seebeck Coefficient
3. Particle Flux
3.1. Chemical Potential of Magnetic Nanofluids
- Diamagnetic solutes, ions or neutral species, all less than nanometer in size. These solutes will be treated as an ideal gas.
- Charged magnetic particles whose characteristic sizes are in the order of ten nanometers. These particles will be described by an effective hard-sphere model derived from Carnahan-Starling equation of state and the inter-particle magnetic interactions are taken into account through a mean-field approach.
3.2. Electric Component
3.3. Magnetic Component
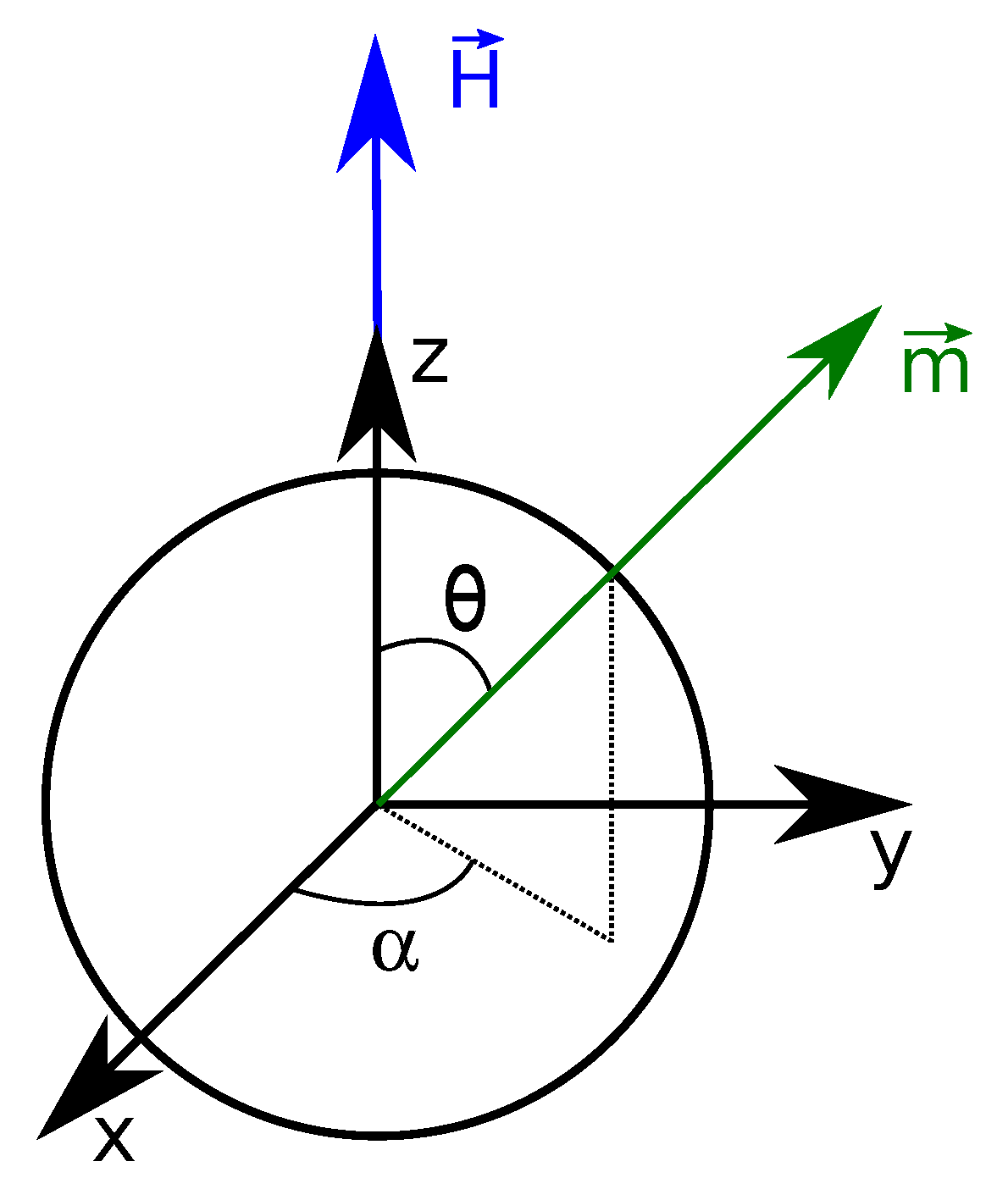
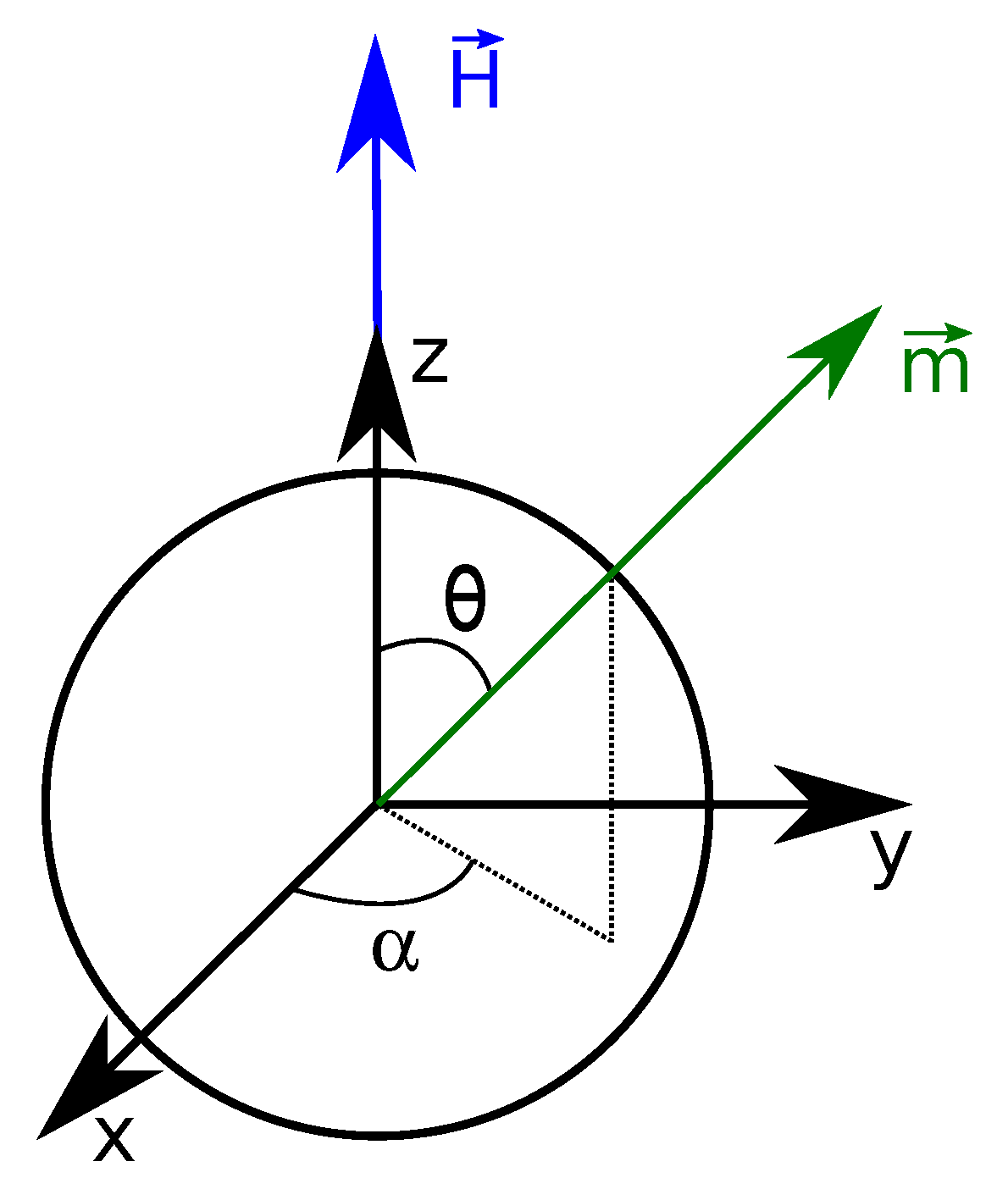
3.3.1. Single Particle Magnetization
3.3.2. Magnetization of Interacting Particles: Mean-Field Approach
3.3.3. Expression for
3.4. Total Chemical Potential
3.5. General Expression for Particle Flux
3.5.1. Chemical Potential Gradient: With Respect to N
3.5.2. Derivative of with Respect to Temperature
3.5.3. Derivative of with Respect to Magnetic Field
3.5.4. Electric Term of the Chemical Potential Gradient
3.5.5. General Expressions for Chemical Potential Gradient and Particle Flux
3.5.6. Local Field Perturbation Effect on Particle Flux
3.5.7. Final Expression of Particle Flux in directions parallel and perpendicular to
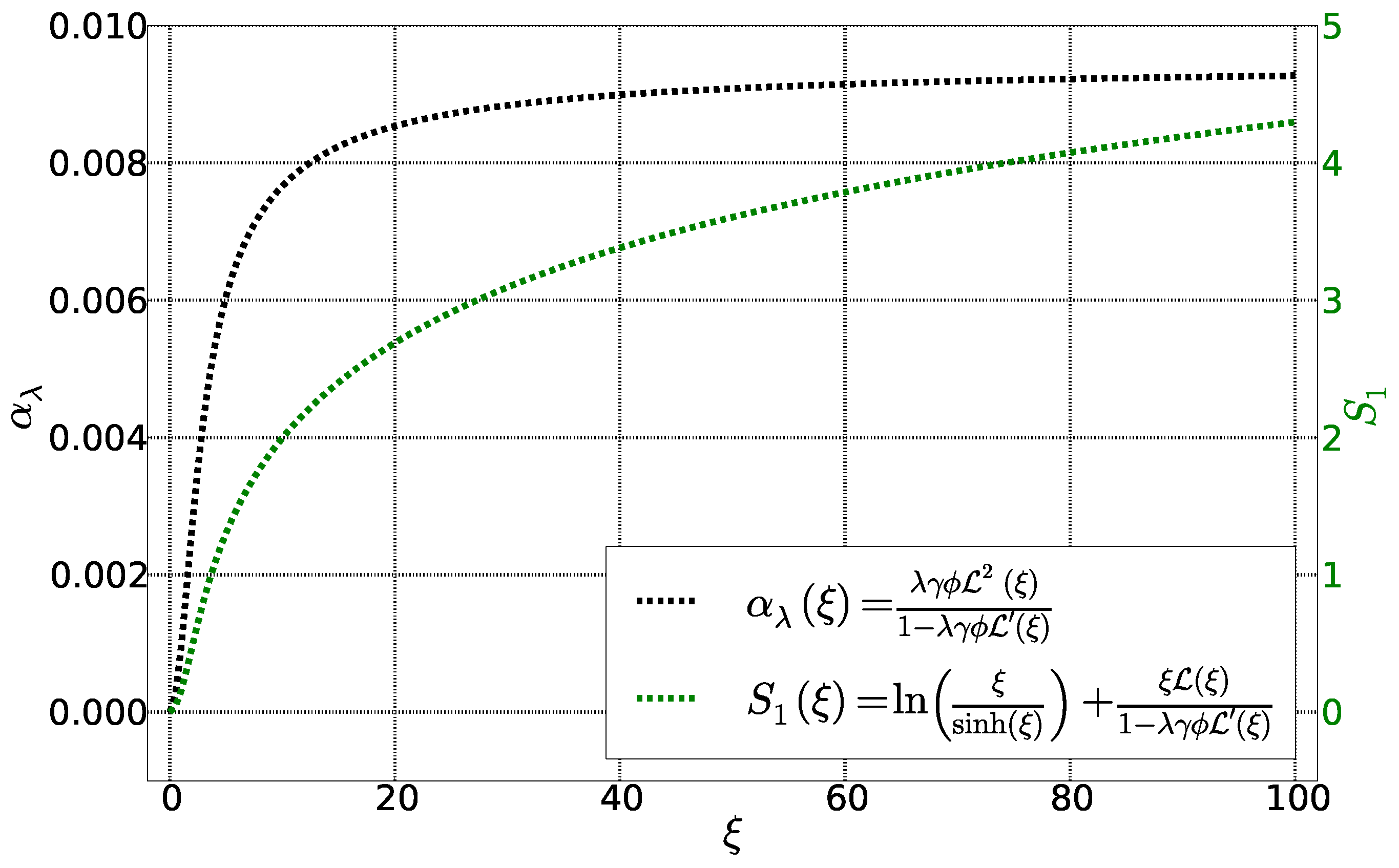
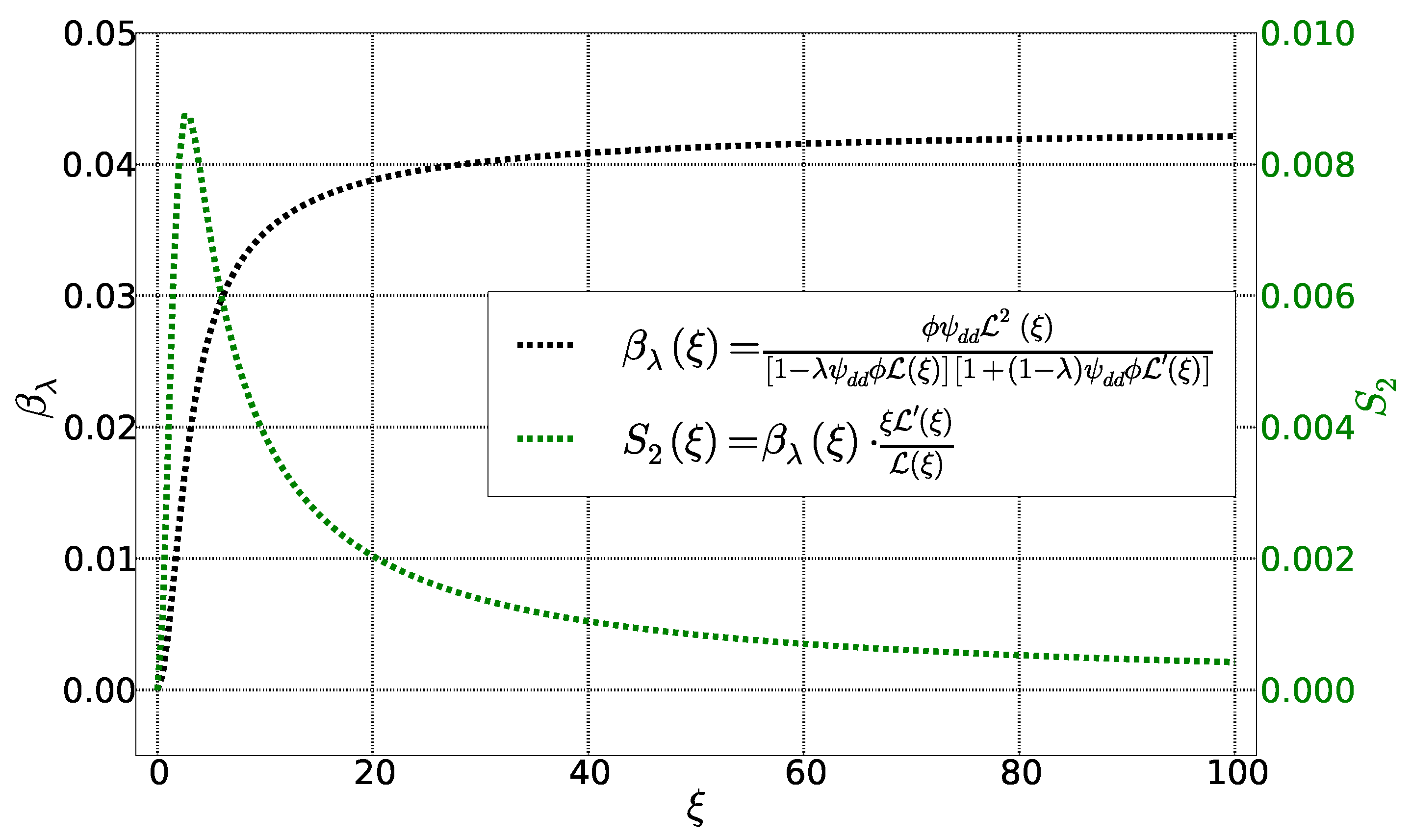
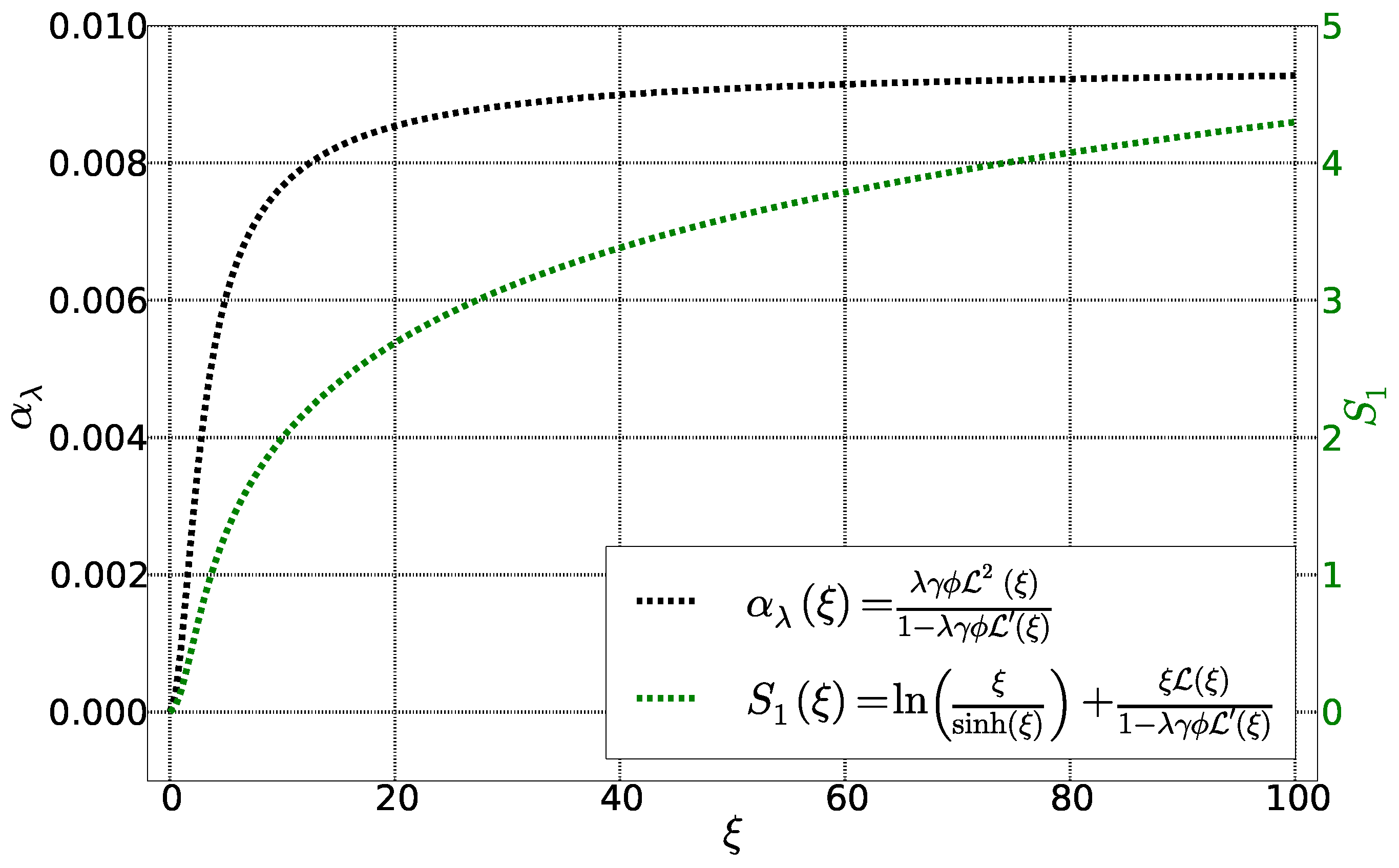
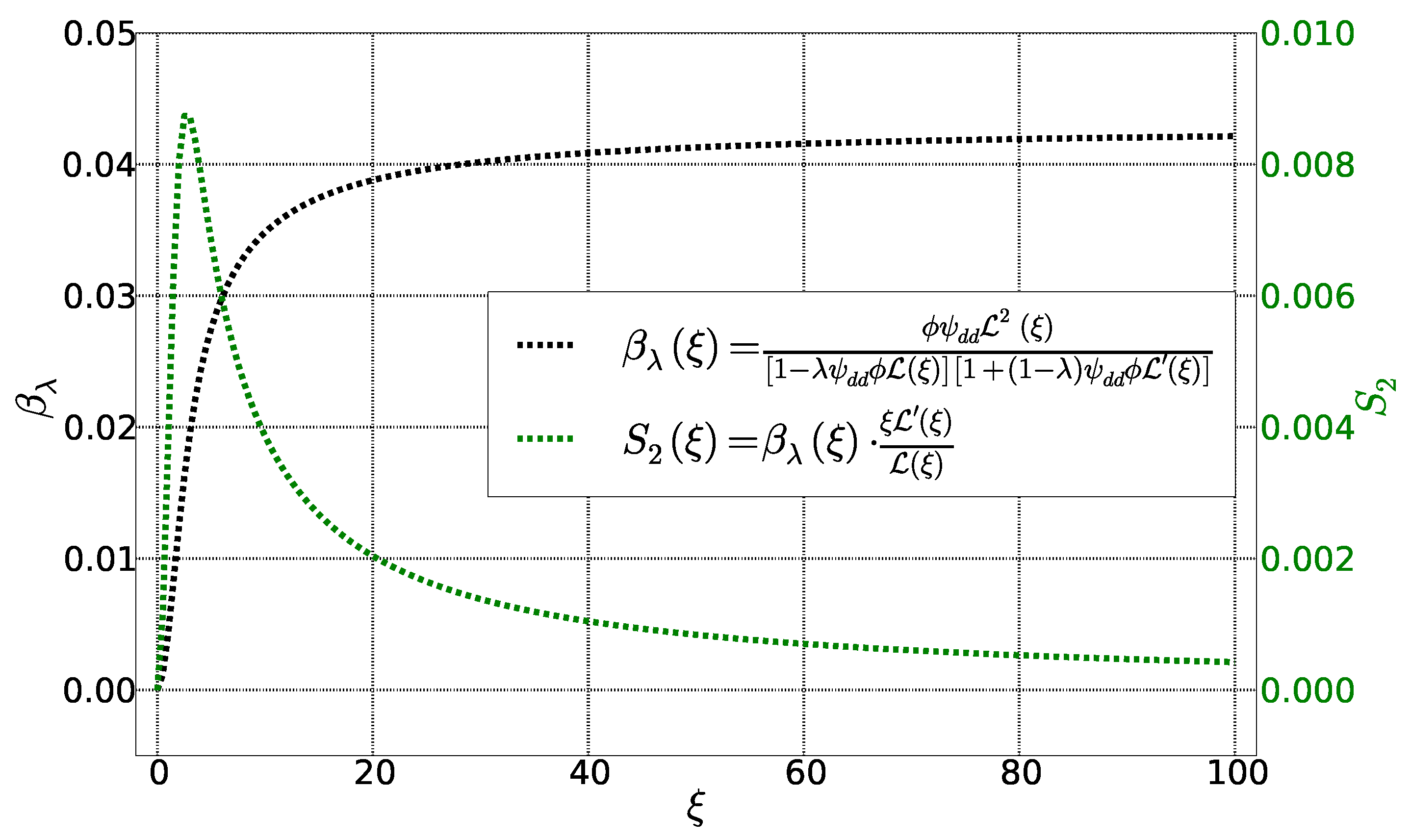
4. Calculation of Se and
4.1. Initial State
4.2. Stationary State: Soret Equilibrium
4.3. Comparison with Experiments in Ferrofluids
5. Summary
Author Contributions
Acknowledgments
Conflicts of Interest
References
- deBethune, A.J.; Licht, T.S.; Swendeman, N. The Temperature Coefficients of Electrode Potentials— The Isothermal and Thermal CoefficientsmThe Standard Ionic Entropy of Electrochemical Transport of the Hydrogen Ion. J. Electrochem. Soc. 1959, 106, 616–625. [Google Scholar] [CrossRef]
- Quickenden, T.I.; Mua, Y. A Review of Power Generation in Aqueous Thermogalvanic Cells. J. Electrochem. Soc. 1995, 142, 3985–3994. [Google Scholar] [CrossRef]
- Dupont, M.F.; MacFarlane, D.R.; Pringle, J.M. Thermo-electrochemical cells for waste heat harvesting— Progress and perspectives. Chem. Commun. 2017, 53, 6288–6302. [Google Scholar] [CrossRef] [PubMed]
- Anari, E.H.B.; Romano, M.; Teh, W.X.; Black, J.J.; Jiang, E.; Chen, J.; To, T.Q.; Panchompoo, J.; Aldous, L. Substituted ferrocenes and iodine as synergistic thermoelectrochemical heat harvesting redox couples in ionic liquids. Chem. Commun. 2016, 52, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Al-Masri, D.; Dupont, M.; Yunis, R.; MacFarlane, D.R.; Pringle, J.M. The electrochemistry and performance of cobalt-based redox couples for thermoelectrochemical cells. Electrochim. Acta 2018, 269, 714–723. [Google Scholar] [CrossRef]
- Laux, E.; Uhl, S.; Jeandupeux, L.; López, P.P.; Sanglard, P.; Vanoli, E.; Marti, R.; Keppner, H. Thermoelectric Generators Based on Ionic Liquids. J. Electron. Mater. 2018, 47, 3193–3197. [Google Scholar] [CrossRef]
- Aldous, L.; Black, J.J.; Elias, M.C.; Gélinas, B.; Rochefort, D. Enhancing thermoelectrochemical properties by tethering ferrocene to the anion or cation of ionic liquids: Altered thermodynamics and solubility. Phys. Chem. Chem. Phys. 2017, 19, 24255–24263. [Google Scholar] [CrossRef] [PubMed]
- Im, H.; Kim, T.; Song, H.; Choi, J.; Park, J.S.; Ovalle-Robles, R.; Yang, H.D.; Kihm, K.D.; Baughman, R.H.; Lee, H.H.; et al. High-efficiency electrochemical thermal energy harvester using carbon nanotube aerogel sheet electrodes. Nat. Commun. 2016, 7, 10600. [Google Scholar] [CrossRef] [PubMed]
- Kazim, A.H.; Booeshagghi, A.S.; Stephens, S.T.; Cola, B.A. Thermo-electrochemical generator: energy harvesting and thermoregulation for liquid cooling applications. Sustain. Energy Fuels 2017, 1, 1381–1389. [Google Scholar] [CrossRef]
- Zhou, H.; Yamada, T.; Kimizuka, N. Supramolecular Thermo-Electrochemical Cells: Enhanced Thermoelectric Performance by Host–Guest Complexation and Salt-Induced Crystallization. J. Am. Chem. Soc. 2016, 138, 10502–10507. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, M.; Straub, A.P.; Zhang, F.; Zhu, X.; Elimelech, M.; Gorski, C.A.; Logan, B.E. Emerging electrochemical and membrane-based systems to convert low-grade heat to electricity. Energy Environ. Sci. 2018, 11, 276–285. [Google Scholar] [CrossRef]
- Kim, T.; Lee, J.S.; Lee, G.; Yoon, H.; Yoon, J. High thermopower of ferri/ferrocyanide redox couple in organic-water solutions. Nano Energy 2017, 31, 160–167. [Google Scholar] [CrossRef]
- Jia, H.; Ju, Z.; Tao, X.; Yao, X.Q.; Wang, Y. P-N Conversion in a Water-Ionic Liquid Binary System for Non-Redox Thermocapacitive Converters. Langmuir 2017, 33, 7600–7605. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Black, J.J.; Aldous, L. Thermoelectrochemistry using conventional and novel gelled electrolytes in heat-to-current thermocells. Electrochim. Acta 2017, 225, 482–492. [Google Scholar] [CrossRef]
- Yang, P.; Liu, K.; Chen, Q.; Mo, X.; Zhou, Y.; Li, S.; Feng, G.; Zhou, J. Wearable Thermocells Based on Gel Electrolytes for the Utilization of Body Heat. Angew. Chem. 2016, 55, 12050–12053. [Google Scholar] [CrossRef] [PubMed]
- Salez, T.J.; Huang, B.T.; Rietjens, M.; Bonetti, M.; Wiertel-Gasquet, C.; Roger, M.; Filomeno, C.L.; Dubois, E.; Perzynski, R.; Nakamae, S. Can charged colloidal particles increase the thermoelectric energy conversion efficiency? Phys. Chem. Chem. Phys. 2017, 19, 9409–9416. [Google Scholar] [CrossRef] [PubMed]
- Würger, A. Transport in Charged Colloids Driven by Thermoelectricity. Phys. Rev. Lett. 2008, 101, 108302. [Google Scholar] [CrossRef] [PubMed]
- Majee, A. Effet ThermoéLectrique Dans Les Dispersions ColloïDale. Ph.D. Thesis, Université Bordeaux I, Talence, France, 2012. [Google Scholar]
- Eslahian, K.A.; Majee, A.; Maskos, M.; Wurger, A. Specific salt effects on thermophoresis of charged colloids. Soft Matter 2014, 10, 1931–1936. [Google Scholar] [CrossRef] [PubMed]
- Burelbach, J.; Frenkel, D.; Pagonabarraga, I.; Elser, E. A unified description of colloidal thermophoresis. Eur. Phys. J. E 2018, 41, 7. [Google Scholar] [CrossRef] [PubMed]
- Eslahian, K.A.; Maskos, M. Hofmeister effect in thermal field-flow fractionation of colloidal aqueous dispersions. Colloids Surf. A Physicochem. Eng. Asp. 2012, 413, 65–70. [Google Scholar] [CrossRef]
- Goupil, C.; Seifert, W.; Zabrocki, K.; Müller, E.; Snyder, G.J. Thermodynamics of Thermoelectric Phenomena and Applications. Entropy 2011, 13, 1481–1517. [Google Scholar] [CrossRef]
- Agar, J.N.; Turner, J.C.R. Thermal Diffusion in Solutions of Electrolytes. Proc. R. Soc. Lond. A Math. Phys. Eng. Sci. 1960, 255, 307–330. [Google Scholar] [CrossRef]
- Di Lecce, S.; Bresme, F. Thermal Polarization of Water Influences the Thermoelectric Response of Aqueous Solutions. J. Phys. Chem. B 2018, 122, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Agar, J.N.; Breck, W.G. Thermal Diffusion in Non-Isothermal Cells. Part 1.—Theoretical Relations and Experiments on Solutions of Thallous Salts. Trans. Faraday Soc. 1957, 53, 167–178. [Google Scholar] [CrossRef]
- Snyder, G.J.; Ursell, T.S. Thermoelectric efficiency and compatibility. Phys. Rev. Lett. 2003, 91, 148301. [Google Scholar] [CrossRef] [PubMed]
- Kuzminskii, Y.; Zasukha, V.; Kuzminskaya, G. Thermoelectric effects in electrochemical systems. Nonconventional thermogalvanic cells. J. Power Sources 1994, 52, 231–242. [Google Scholar] [CrossRef]
- Ikeshoji, T.; de Nahui, F.N.B.; Kimura, S.; Yoneya, M. Computer analysis on natural convection in thin-layer thermocells with a soluble redox couple. J. Electroanal. Chem. Interfacial Electrochem. 1991, 312, 43–56. [Google Scholar] [CrossRef]
- Zhang, L.; Kim, T.; Li, N.; Kang, T.J.; Chen, J.; Pringle, J.M.; Zhang, M.; Kazim, A.H.; Fang, S.; Haines, C.; et al. High Power Density Electrochemical Thermocells for Inexpensively Harvesting Low-Grade Thermal Energy. Adv. Mater. 2017, 29, 1605652. [Google Scholar] [CrossRef] [PubMed]
- Majee, A.; Würger, A. Charging of Heated Colloidal Particles Using the Electrolyte Seebeck Effect. Phys. Rev. Lett. 2012, 108, 118301. [Google Scholar] [CrossRef] [PubMed]
- Majee, A.; Würger, A. Collective thermoelectrophoresis of charged colloids. Phys. Rev. E 2011, 83, 061403. [Google Scholar] [CrossRef] [PubMed]
- Würger, A. Hydrodynamic Boundary Effects on Thermophoresis of Confined Colloids. Phys. Rev. Lett. 2016, 116, 138302. [Google Scholar] [CrossRef] [PubMed]
- Würger, A. Thermal non-equilibrium transport in colloids. Rep. Prog. Phys. 2010, 73, 126601. [Google Scholar] [CrossRef]
- Onsager, L. Reciprocal Relations in Irreversible Processes. I. Phys. Rev. 1931, 37, 405–426. [Google Scholar] [CrossRef]
- Onsager, L. Reciprocal Relations in Irreversible Processes. II. Phys. Rev. 1931, 38, 2265–2279. [Google Scholar] [CrossRef]
- de Groot, S.R. Théorie Phénoménologique De L’effet Soret. Physica 1942, 9, 699–708. [Google Scholar] [CrossRef]
- Carnahan, N.F.; Starling, K.E. Equation of State for Nonattracting Rigid Spheres. J. Chem. Phys. 1969, 51, 635–636. [Google Scholar] [CrossRef]
- Kittel, C. Physique de L’éTat Solide, 8th ed.; Dunod: Malakoff, France, 2007. [Google Scholar]
- Blums, E.; Cebers, A.; Maiorov, M.M. Magnetic Fluids; Walter de Gruyter: Berlin, Germany, 1997. [Google Scholar]
- Bacri, J.C.; Cebers, A.; Bourdon, A.; Demouchy, G.; Heegaard, B.M.; Perzynski, R. Forced Rayleigh Experiment in a Magnetic Fluid. Phys. Rev. Lett. 1995, 74, 5032–5035. [Google Scholar] [CrossRef] [PubMed]
- Bacri, J.C.; Cebers, A.; Bourdon, A.; Demouchy, G.; Heegaard, B.M.; Kashevsky, B.; Perzynski, R. Transient grating in a ferrofluid under magnetic field: Effect of magnetic interactions on the diffusion coefficient of translation. Phys. Rev. E 1995, 52, 3936–3942. [Google Scholar] [CrossRef]
- Gazeau, F.; Dubois, E.; Bacri, J.C.; Boué, F.; Cebers, A.; Perzynski, R. Anisotropy of the structure factor of magnetic fluids under a field probed by small-angle neutron scattering. Phys. Rev. E 2002, 65, 031403. [Google Scholar] [CrossRef] [PubMed]
- Mériguet, G.; Cousin, F.; Dubois, E.; Boué, F.; Cebers, A.; Farago, B.; Perzynski, R. What tunes the structural anisotropy of magnetic fluids under a magnetic field? J. Phys. Chem. B 2006, 110, 4378–4386. [Google Scholar] [CrossRef] [PubMed]
- Wandersman, E.; Dubois, E.; Cousin, F.; Dupuis, V.; Mériguet, G.; Perzynski, R.; Cēbers, A. Relaxation of the field-induced structural anisotropy in a rotating magnetic fluid. EPL (Europhys. Lett.) 2009, 86, 10005. [Google Scholar] [CrossRef]
- Mériguet, G.; Dubois, E.; Jardat, M.; Bourdon, A.; Demouchy, G.; Dupuis, V.; Farago, B.; Perzynski, R.; Turq, P. Understanding the structure and the dynamics of magnetic fluids: Coupling of experiment and simulation. J. Phys. Condens. Matter 2006, 18, S2685. [Google Scholar] [CrossRef]
- Mériguet, G.; Jardat, M.; Turq, P. Brownian dynamics investigation of magnetization and birefringence relaxations in ferrofluids. J. Chem. Phys. 2005, 123, 144915. [Google Scholar] [CrossRef] [PubMed]
- Würger, A. Is Soret equilibrium a non-equilibrium effect? C. R. Mécanique 2013, 341, 438–448. [Google Scholar] [CrossRef]
- De Groot, S. Sur la thermodynamique de quelques processus irréversibles. II. Diffusion thermique et phénomènes connexes. J. Phys. Radium 1947, 8, 193–200. [Google Scholar] [CrossRef]
- Agar, J. Thermogalvanic cells. In Advances in Electrochemistry and Electrochemical Engineering; Delahay, P., Ed.; Interscience: New York, NY, USA, 1963; pp. 31–121. [Google Scholar]
- Eastman, E. Theory of the Soret effect. J. Am. Chem. Soc. 1928, 50, 283–291. [Google Scholar] [CrossRef]
- Ikeda, T. Absolute Estimation of the Ionic Entropies of Transfer. Bull. Chem. Soc. Jpn. 1959, 32, 96–97. [Google Scholar] [CrossRef]
- Ikeda, T. Transported Entropies and Conventional Eastman Entropies of the Transfer of Some Univalent Ions in Aqueous Solutions at 25C. Bull. Chem. Soc. Jpn. 1964, 37, 1485–1489. [Google Scholar] [CrossRef]
- Snowdon, P.; Turner, J. The Soret effect in some 0.01 normal aqueous electrolytes. Trans. Faraday Soc. 1960, 56, 1409–1418. [Google Scholar] [CrossRef]
- Agar, J.N.; Mou, C.Y.; Lin, J.L. Single-ion heat of transport in electrolyte solutions: A hydrodynamic theory. J. Phys. Chem. 1989, 93, 2079–2082. [Google Scholar] [CrossRef]
- Nernst, W. Die elektromotorische wirksamkeit der jonen. Z. Phys. Chem. 1889, 4, 129–181. [Google Scholar] [CrossRef]
- Debye, P.; Hückel, E. Zur Theorie der Elektrolyte. I. Gefrierpunktserniedrigung und verwandte Erscheinungen. Phys. Z. 1923, 24, 185–206. [Google Scholar]
- Huang, B.T.; Roger, M.; Bonetti, M.; Salez, T.J.; Wiertel-Gasquet, C.; Dubois, E.; Cabreira Gomes, R.; Demouchy, G.; Mériguet, G.; Peyre, V.; et al. Thermoelectricity and thermodiffusion in charged colloids. J. Chem. Phys. 2015, 143, 054902. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M. Thermophoresis of Polymers in Electrolyte Solutions. Ph.D. Thesis, Ludwig-Maximilians Universität, München, Germany, 2016. [Google Scholar]
- Würger, A. Temperature Dependence of the Soret Motion in Colloids. Langmuir 2009, 25, 6696–6701. [Google Scholar] [CrossRef] [PubMed]
- Filomeno, C.L.; Kouyaté, M.; Cousin, F.; Demouchy, G.; Dubois, E.; Michot, L.; Mériguet, G.; Perzynski, R.; Peyre, V.; Sirieix-Plénet, J.; et al. Ionic magnetic fluids in polar solvents with tuned counter-ions. J. Magn. Magn. Mater. 2017, 431, 2–7. [Google Scholar] [CrossRef]
- Vigolo, D.; Buzzaccaro, S.; Piazza, R. Thermophoresis and Thermoelectricity in Surfactant Solutions. Langmuir 2010, 26, 7792–7801. [Google Scholar] [CrossRef] [PubMed]
- Cabreira Gomes, R.; Ferreira da Silva, A.; Kouyaté, M.; Demouchy, G.; Mériguet, G.; Aquino, R.; Dubois, E.; Nakamae, S.; Roger, M.; Depeyrot, J.; et al. Thermodiffusion od repulsive charged nanoparticles-Interplay between single-particle and thermoelectric contributions. Phys. Chem. Chem. Phys. 2018. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salez, T.J.; Nakamae, S.; Perzynski, R.; Mériguet, G.; Cebers, A.; Roger, M. Thermoelectricity and Thermodiffusion in Magnetic Nanofluids: Entropic Analysis. Entropy 2018, 20, 405. https://doi.org/10.3390/e20060405
Salez TJ, Nakamae S, Perzynski R, Mériguet G, Cebers A, Roger M. Thermoelectricity and Thermodiffusion in Magnetic Nanofluids: Entropic Analysis. Entropy. 2018; 20(6):405. https://doi.org/10.3390/e20060405
Chicago/Turabian StyleSalez, Thomas J., Sawako Nakamae, Régine Perzynski, Guillaume Mériguet, Andrejs Cebers, and Michel Roger. 2018. "Thermoelectricity and Thermodiffusion in Magnetic Nanofluids: Entropic Analysis" Entropy 20, no. 6: 405. https://doi.org/10.3390/e20060405
APA StyleSalez, T. J., Nakamae, S., Perzynski, R., Mériguet, G., Cebers, A., & Roger, M. (2018). Thermoelectricity and Thermodiffusion in Magnetic Nanofluids: Entropic Analysis. Entropy, 20(6), 405. https://doi.org/10.3390/e20060405