
Cellular Mechanism Underlying Highly-Active or Antiretroviral Therapy-Induced Lipodystrophy: Atazanavir, a Protease Inhibitor, Compromises Adipogenic Conversion of Adipose-Derived Stem/Progenitor Cells through Accelerating ER Stress-Mediated Cell Death in Differentiating Adipocytes

 ,
,
Abstract
:1. Introduction
2. Results
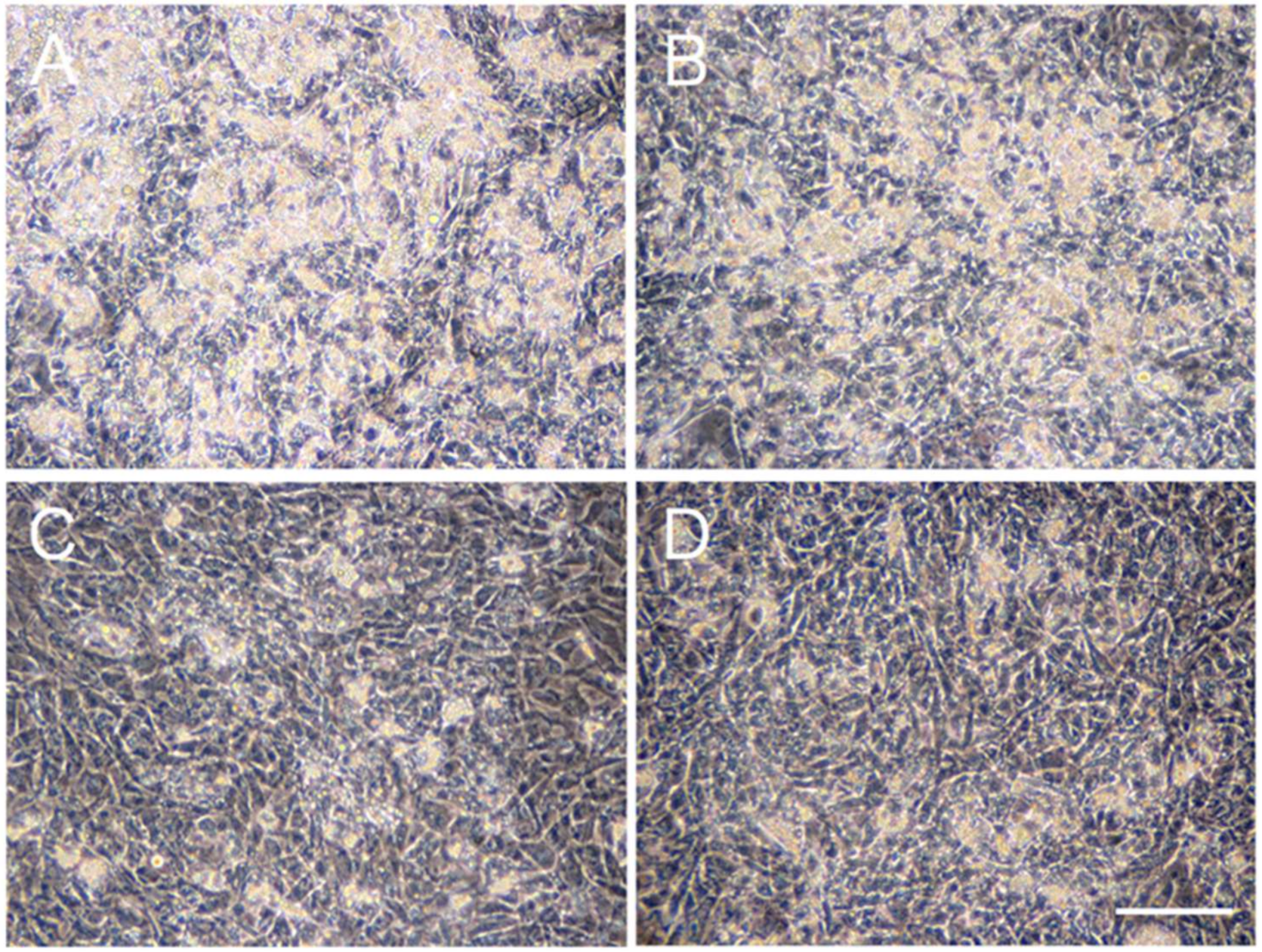
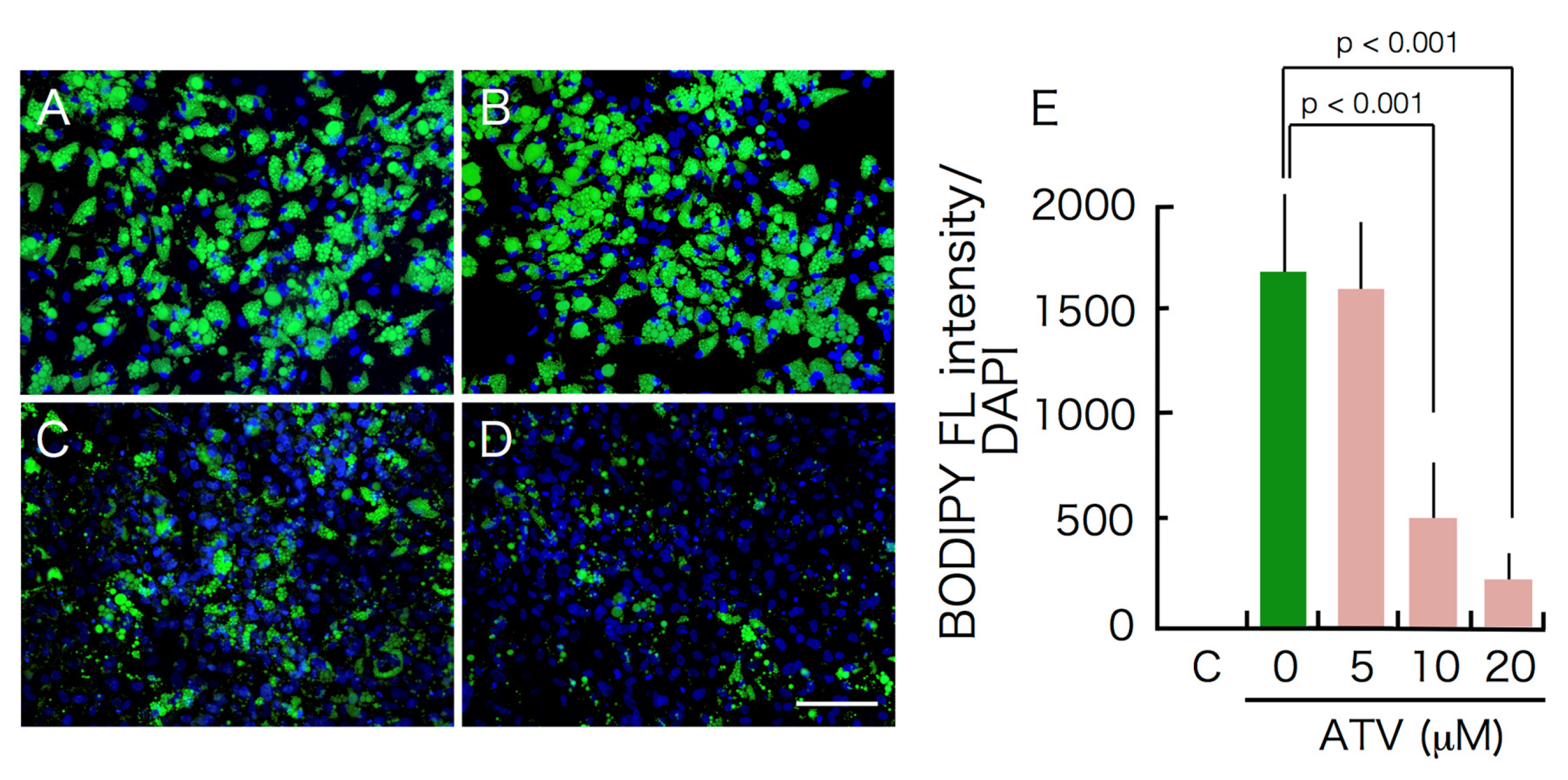
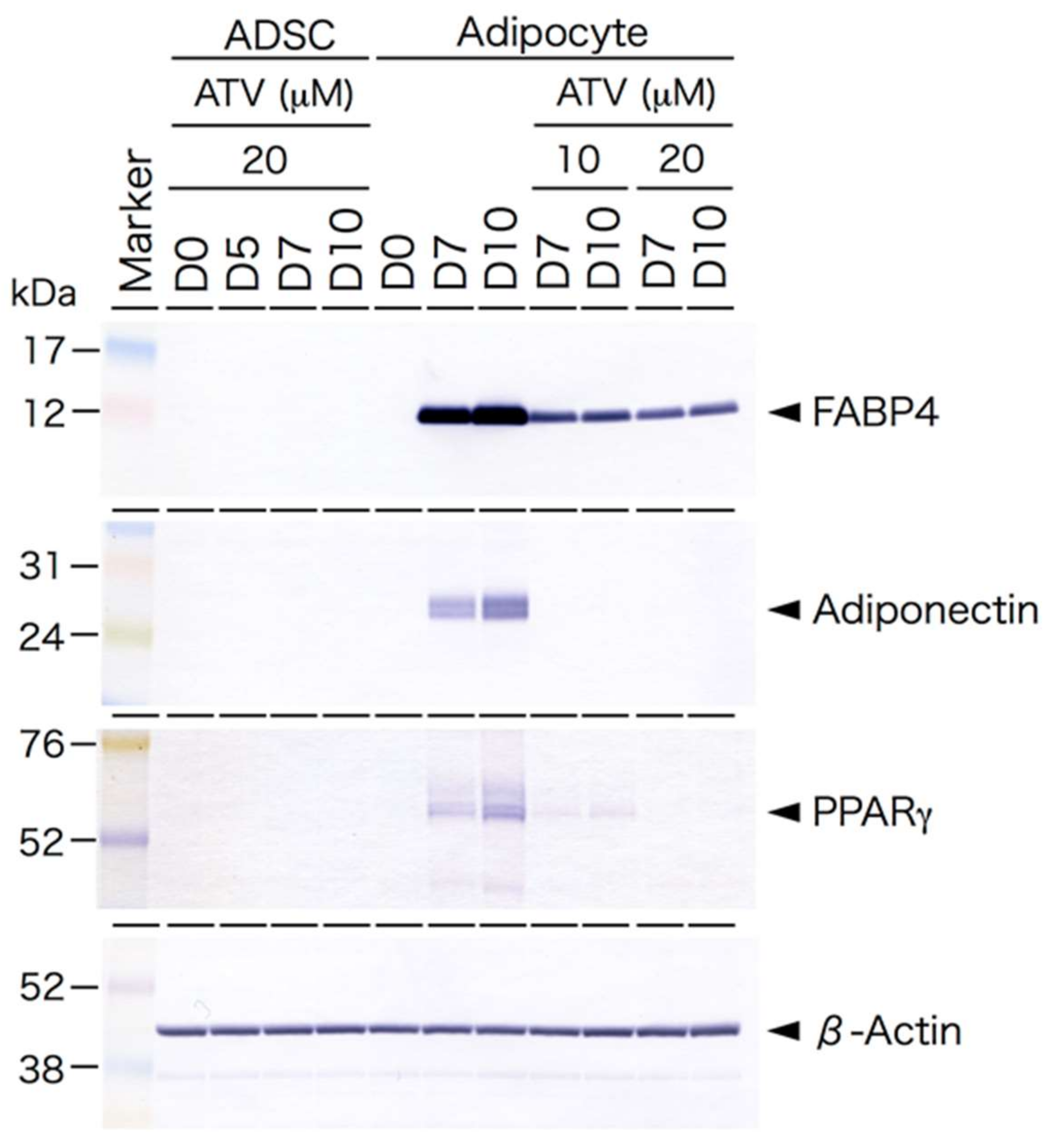
2.1. Effects of ATV Treatment on Adipogenic Differentiation of ADSCs
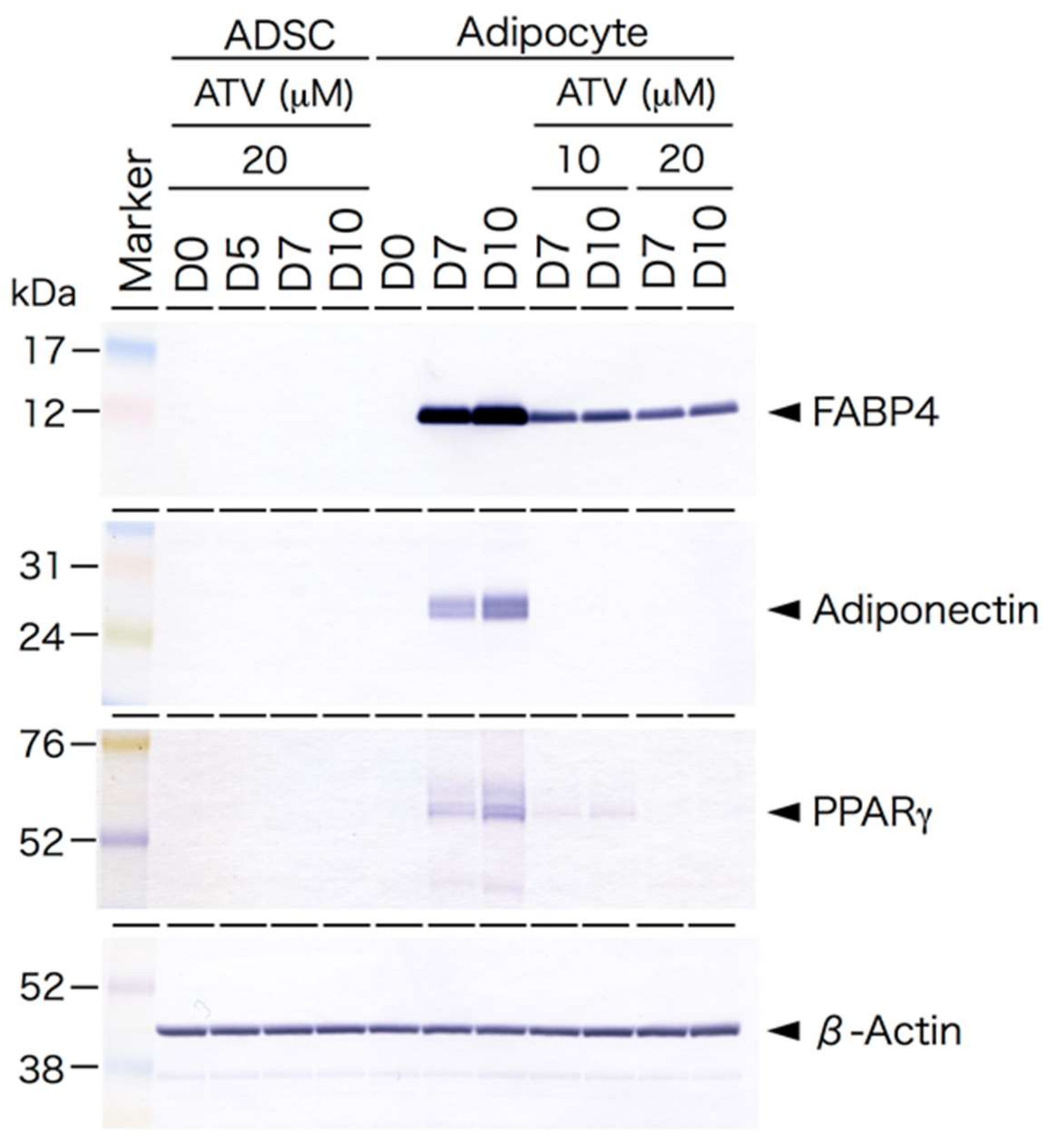
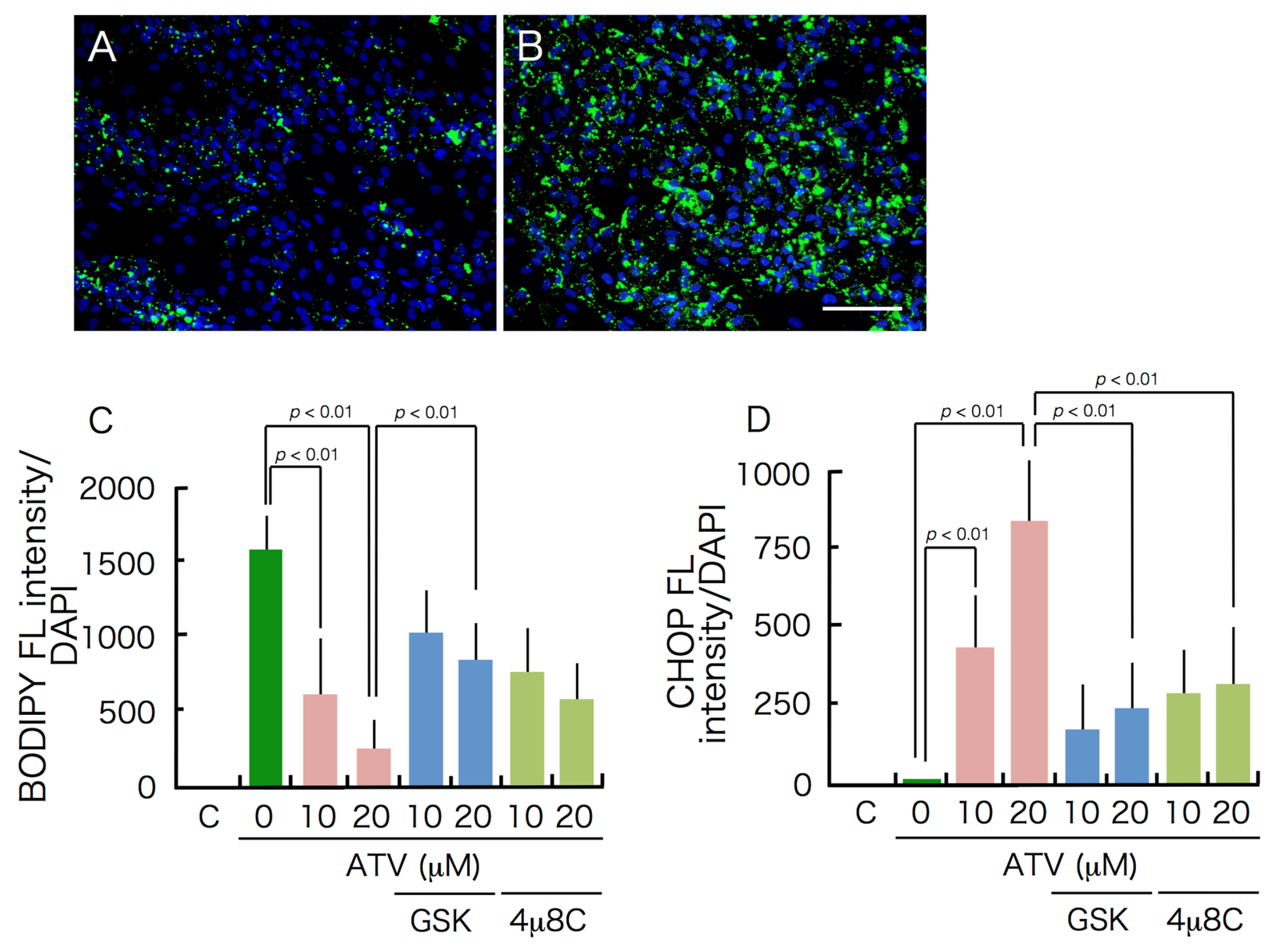
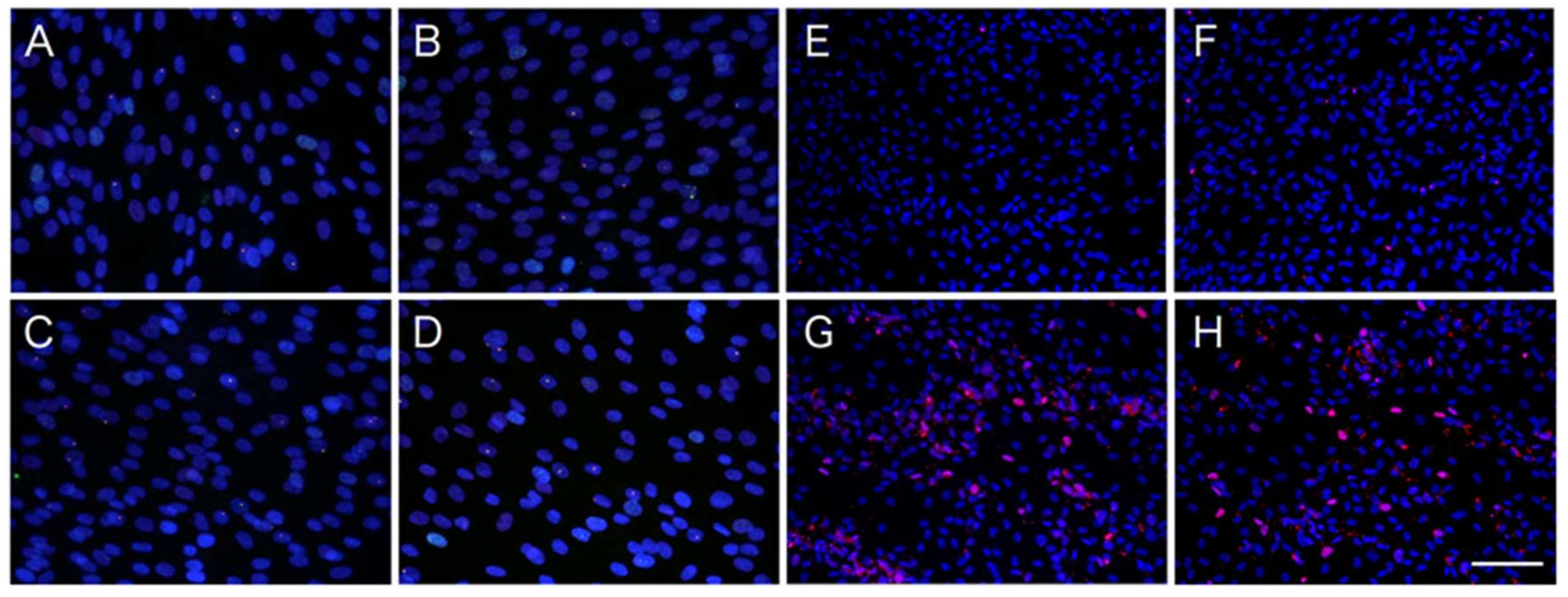
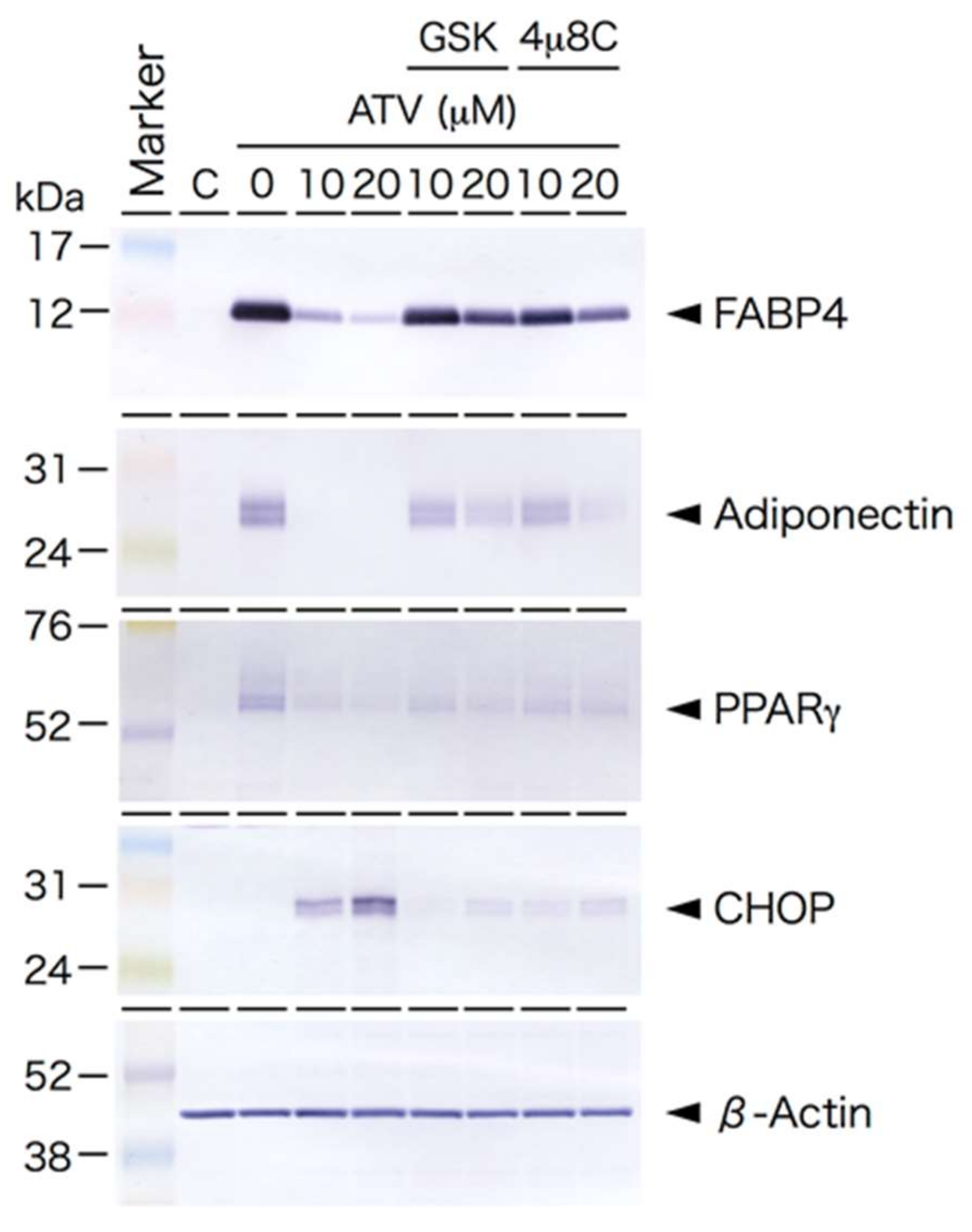
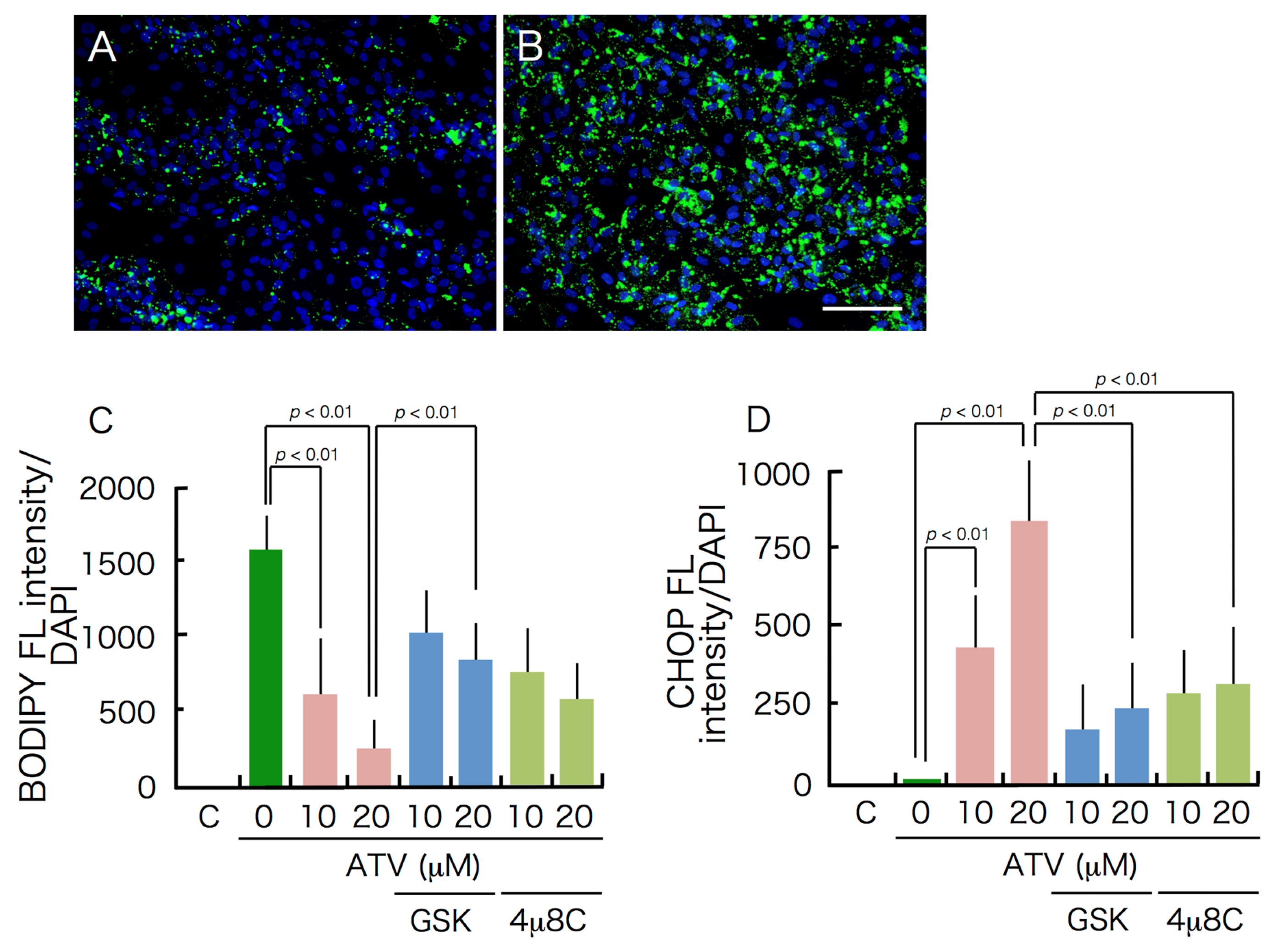
2.2. Differential Effects of ATV on ADSCs and Differentiating Adipocytes
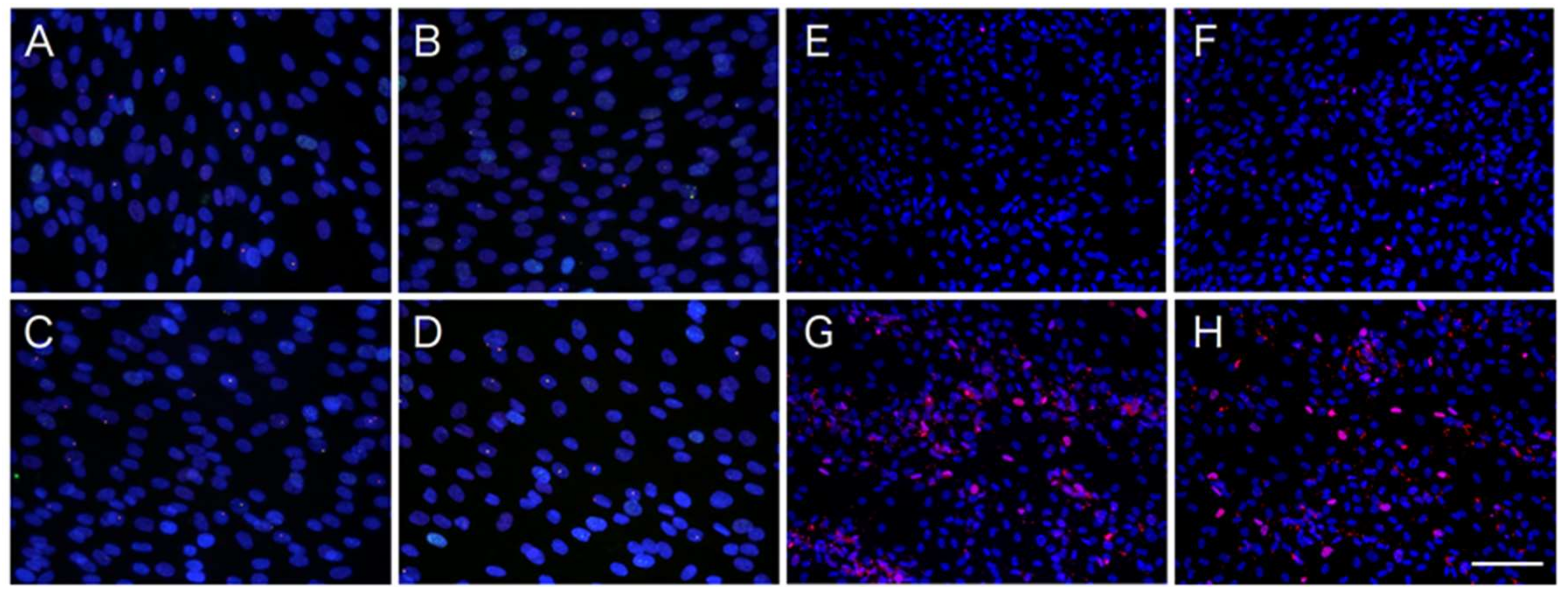
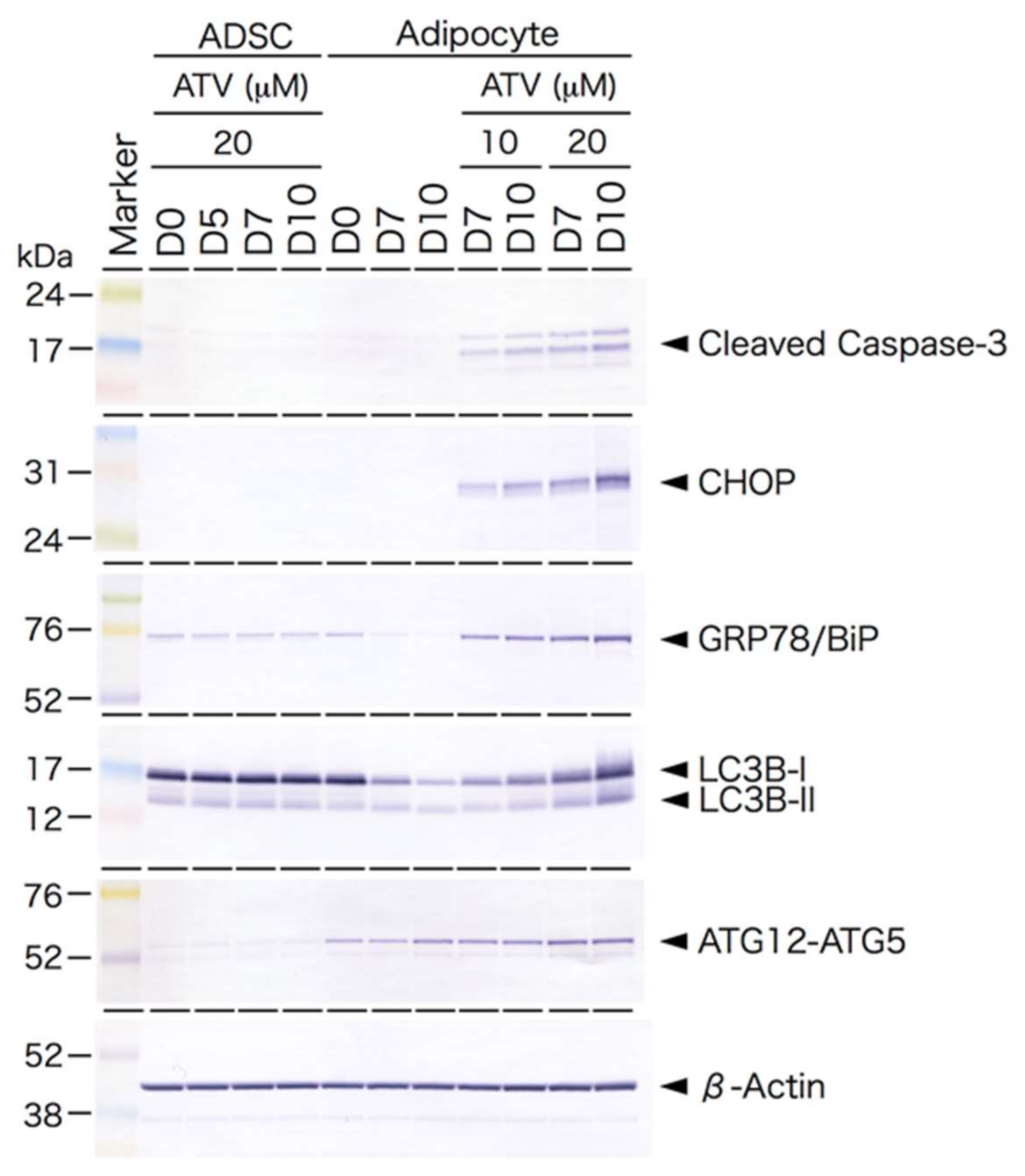
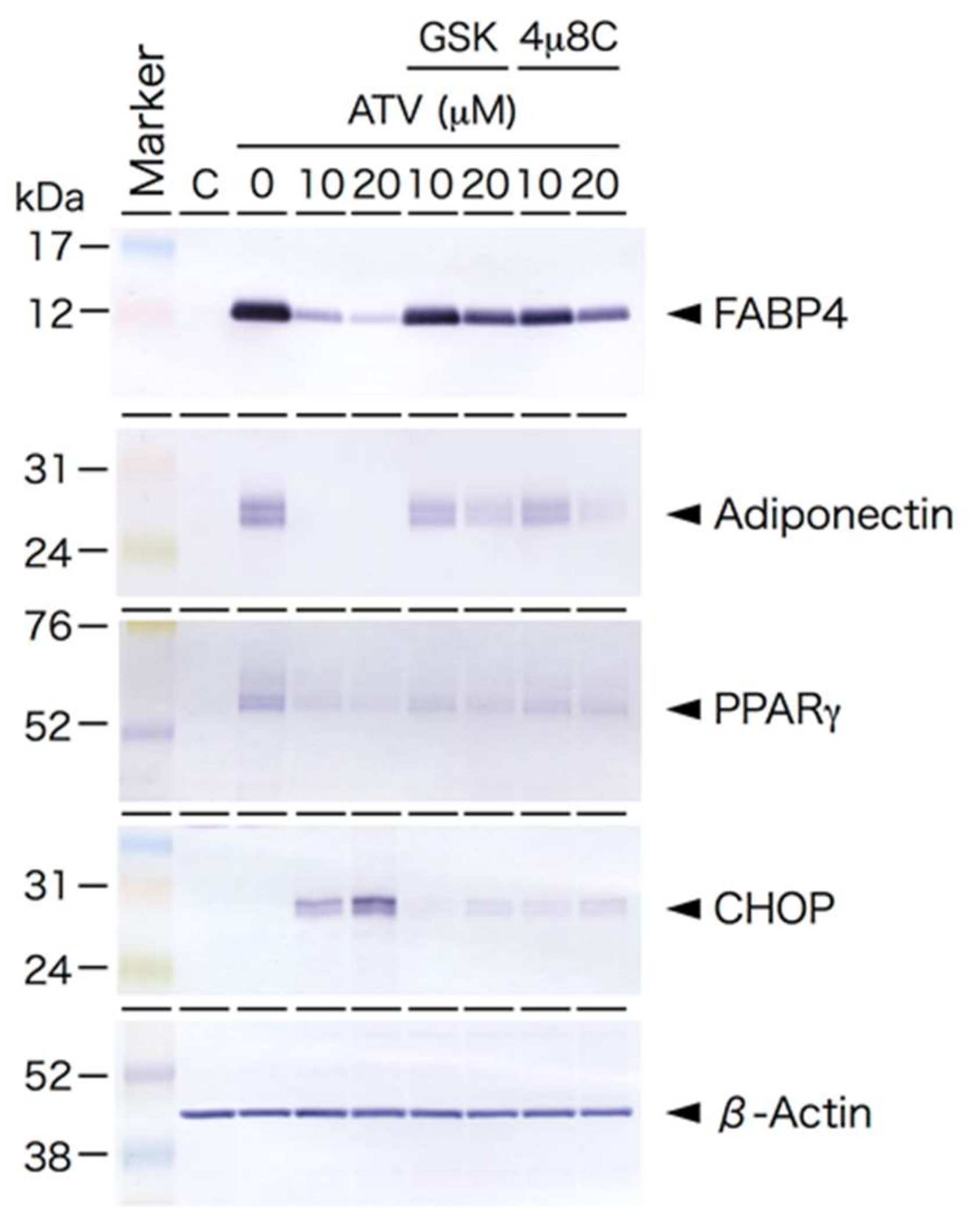
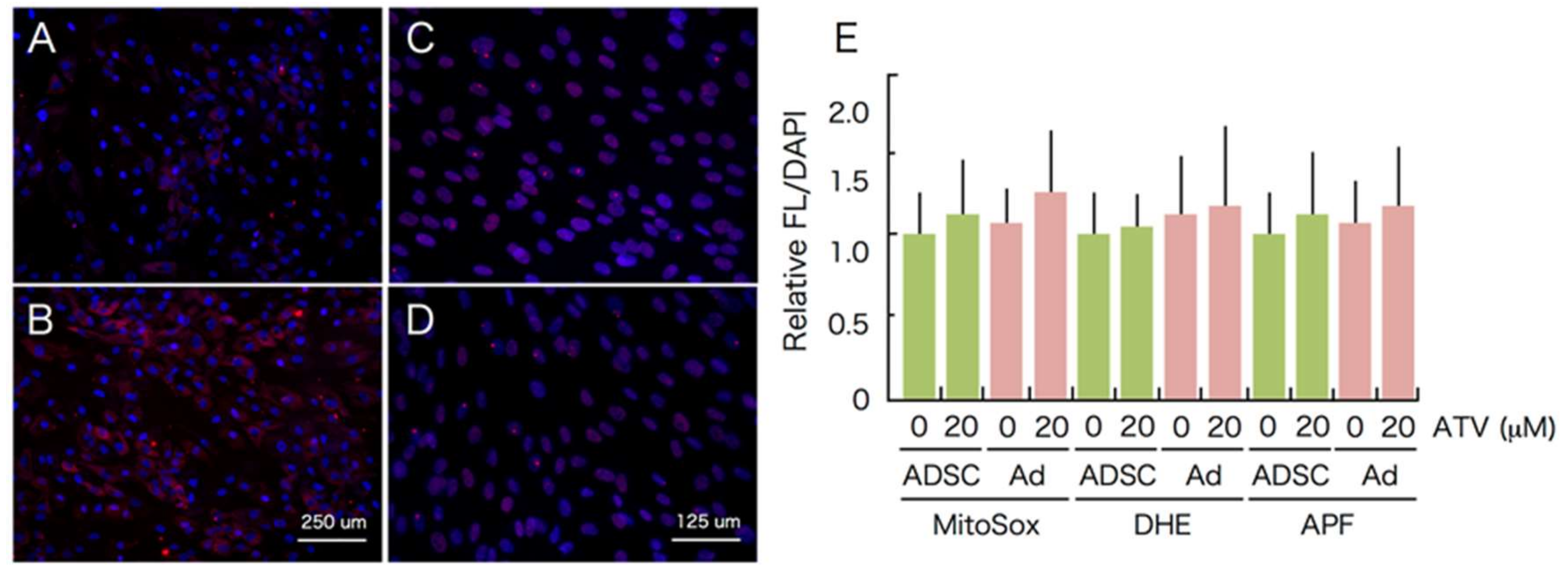
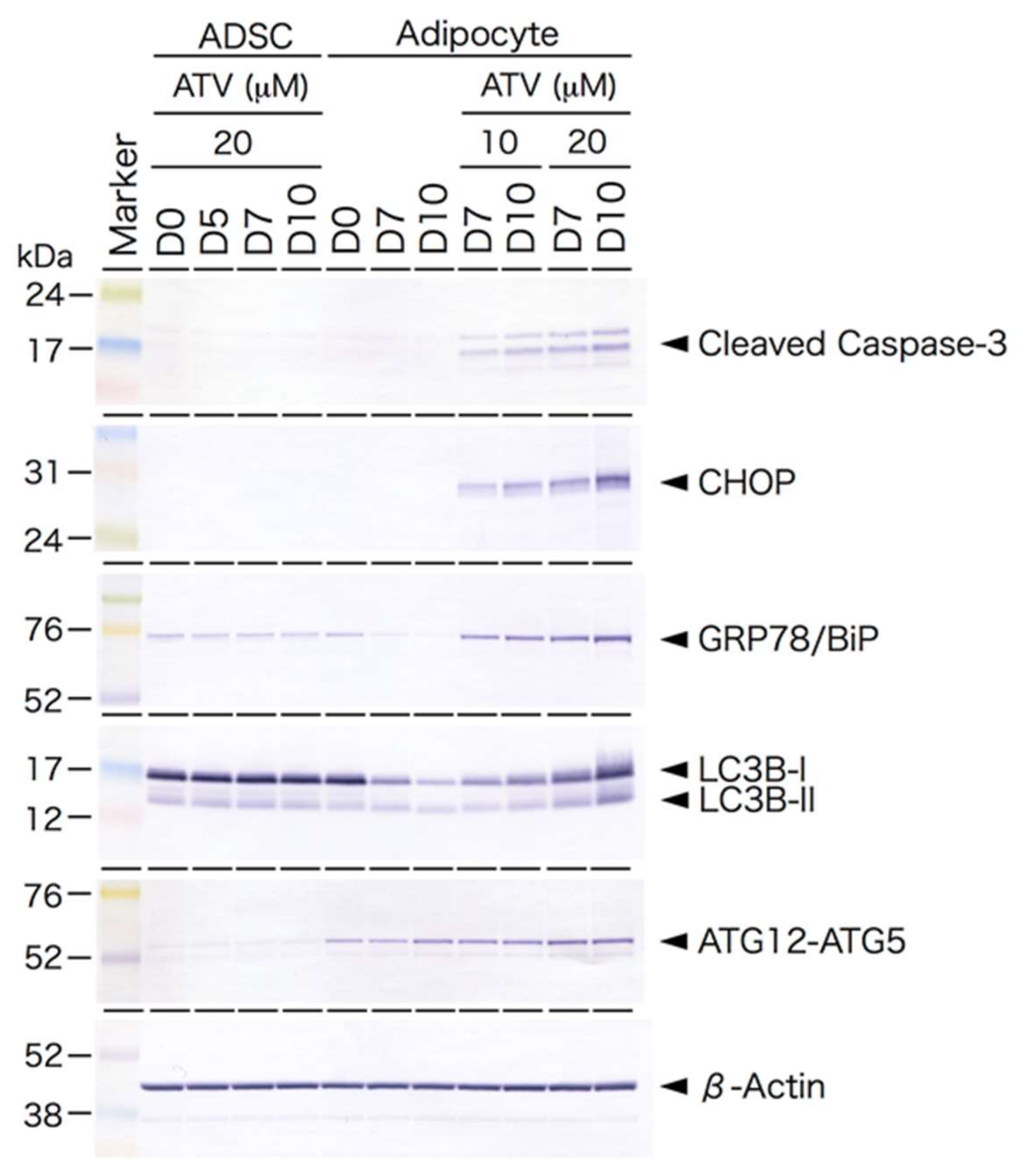
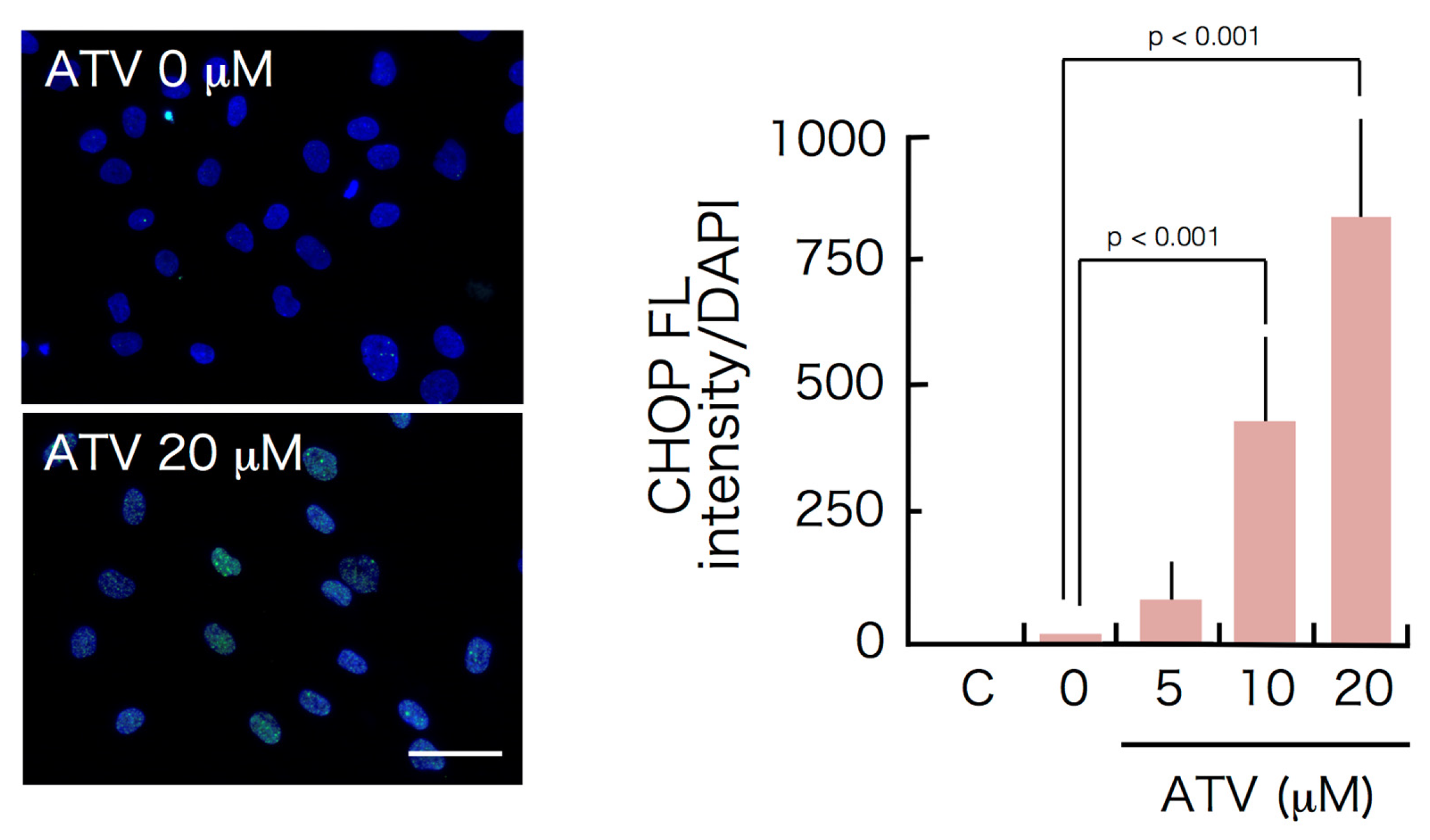
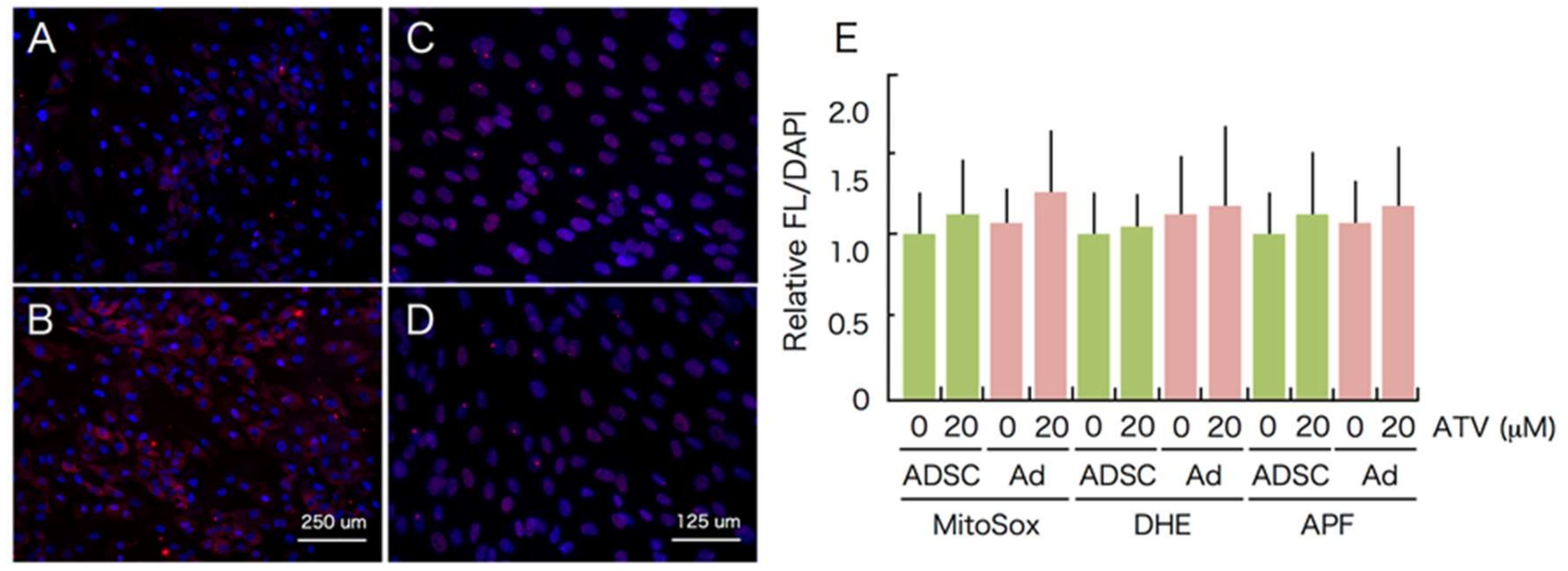
2.3. Induction of ER Stress and Oxidative Stress by ATV Treatment
3. Discussion
4. Materials and Methods
4.1. ADSCs and Culture
4.2. Adipocyte Differentiation and ATV Treatment
4.3. Immunofluorescence
4.4. DNA Damage and Apoptosis Detection
4.5. Western Blotting
4.6. Measurement of ROS
4.7. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADSC | adipose-derived stem/progenitor cells |
| HIV | human immunodeficiency virus |
| HAART | highly active antiretroviral therapy |
| ART | antiretroviral therapy |
| NRTI | nucleoside reverse transcriptase inhibitor |
| PI | protease inhibitor |
| UPR | unfolded protein response |
| ER | endoplasmic reticulum |
References
- Carr, A. HIV lipodystrophy: Risk factors, pathogenesis, diagnosis and management. AIDS 2003, 17, S141–S148. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Garg, A. Lipodystrophy syndromes. Endocrinol. Metab. Clin. North Am. 2010, 85, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Nakahara, M.; Oka, M.; Saeki, K. Additional attention to combination antiretroviral therapy-related lipodystrophy. World J Virol. 2017, 85, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Koethe, J.R.; Lagathu, C.; Lake, J.E.; Domingo, P.; Calmy, A.; Falutz, J.; Brown, T.T.; Capeau, J. HIV and antiretroviral therapy-related fat alterations. Nat. Rev. Dis. Primers 2020, 6, 48. [Google Scholar] [CrossRef]
- Mallon, P.W.; Cooper, D.A.; Carr, A. HIV-associated lipodystrophy. HIV Med. 2001, 2, 166–173. [Google Scholar] [CrossRef]
- Shevitz, A.; Wanke, C.A.; Falutz, J.; Kotler, D.P. Clinical perspectives on HIV-associated lipodystrophy syndrome: An update. AIDS 2001, 15, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Mallon, P.W.G. Pathogenesis of lipodystrophy and lipid abnormalities in patients taking antiretroviral therapy. Aids Rev. 2007, 9, 3–15. [Google Scholar]
- Fiorenza, C.G.; Chou, S.H.; Mantzoros, C.S. Lipodystrophy: Pathophysiology and advances in treatment. Nat. Rev. Endocrinol. 2010, 7, 137–150. [Google Scholar] [CrossRef]
- Brinkman, K.; Smeitink, J.A.; Romijin, J.A.; Reiss, P. Mitochondrial toxicity induced by nucleotide-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet 1999, 354, 1112–1115. [Google Scholar] [CrossRef]
- Villarroya, F.; Domingo, P.; Giralt, M. Lipodystrophy associated with highly active anti-retroviral therapy for HIV infection: The adipocyte as a target of anti-retroviral-induced mitochondrial toxicity. Trends Pharm. Sci. 2005, 26, 88–93. [Google Scholar] [CrossRef]
- Mallewa, J.E.; Wilkins, E.; Vilar, J.; Mallewa, M.; Doran, D.; Back, D.; Pirmohamed, M. HIV-associated lipodystrophy: A review of understanding mechanisms and therapeutic options. J. Antimicrob. Chemother. 2008, 62, 648–660. [Google Scholar] [CrossRef] [Green Version]
- McComsey, G.A.; Kitch, D.; Sax, P.E.; Tebas, T.; Tierney, C.; Jahed, N.C.; Myers, L.; Melbourne, K.; Ha, B.; Daar, E.S. Peripheral and central fat changes in subjects randomized to abacavir-lamivudine or tenofovir-emtricitabine with atazanavir-ritonavir or efavirenz: ACTG Study A5224s. Clin. Dis. 2011, 53, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Domingo, P.; Matias-Guiu, X.; Pujol, R.M.; Francia, E.; Sambeat, M.A.; Vazquez, G. Subcutaneous adipocyte apoptosis in HIV-1 protease inhibitor-associated lipodystrophy. AIDS 1999, 13, 2261–2267. [Google Scholar] [CrossRef]
- Zhou, H.; Pandak, W.M.; Hylemon, P.B. Cellular mechanisms of lipodystrophy induction by HIV protease inhibitors. Future Med. 2006, 1, 163–172. [Google Scholar] [CrossRef]
- De Waal, R.; Cohen, K.; Maartens, G. Systematic review of antiretroviral-associated lipodystrophy: Lipoatrophy, but not central fat gain, is an antiretroviral adverse drug reaction. PLoS ONE 2013, 5, e63623. [Google Scholar] [CrossRef] [Green Version]
- Parker, R.A.; Flint, O.P.; Mulvey, R.; Elosua, C.; Wang, F.; Fenderson, W.; Wang, S.; Yang, W.P.; Noor, M.A. Endoplasmic reticulum stress links dyslipidemia to inhibition of proteasome activity and glucose transport by HIV protease inhibitors. Mol. Pharm. 2005, 67, 1909–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Kitamura, M. Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am. J. Physiol. Ren. Physiol. 2008, 295, F323–F334. [Google Scholar] [CrossRef] [Green Version]
- Kopp, M.C.; Larburu, N.; Dutairaj, V.; Adams, C.J.; Ali, M.M.U. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Struct. Mol. Biol. 2019, 26, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akita, S.; Akino, K.; Hirano, A.; Ohtsuru, A.; Yamashita, S. Mesenchymal stem cell therapy for cutaneous radiation syndrome. Health Phys. 2010, 98, 858–862. [Google Scholar] [CrossRef] [Green Version]
- Zuk, P.; Zhu, M.; Ashjian, P.; De Ugarte, D.A.; Huang, J.I.; Mizuno, H.; Alfonso, Z.C.; Fraser, J.K.; Benhaim, P.; Hedrick, M.H. Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell 2002, 13, 4279–4295. [Google Scholar] [CrossRef]
- Bunnell, B.A.; Flaat, M.; Gagliardi, C.; Patel, B.; Ripoll, C. Adipose-derived stem cells: Isolation, expansion, and differentiation. Methods 2008, 45, 115–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moseti, D.; Regassa, A.; Kim, W.K. Molecular regulation of adipogenesis and potential anti-adipogenic bioactive molecules. Int. J. Mol. Sci. 2016, 17, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Akita, S.; Yoshimoto, H.; Ohtsuru, A.; Hirano, A.; Yamashita, S. Biological Features Implies Potential Use of Autologous Adipose-Derived Stem/Progenitor Cells in Wound Repair and Regenerations for the Patients with Lipodystrophy. Int. J. Mol. Sci. 2019, 20, 5505. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.J.; Wilson, C.G.; Wabotsch, M.; Lazar, M.A.; Steppen, C.M. HIV protease inhibitor-specific alterations in human adipocyte differentiation and metabolism. Obesity 2006, 14, 994–1002. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.P.; Waitt, C.; Sutton, R.; Back, D.J.; Pirmohamed, M. Effect of atazanavir and ritonavir on the differentiation and adipokine secretion of human subcutaneous and omental preadipocytes. AIDS 2008, 22, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Pyrko, P.; Kardosh, A.; Wang, W.; Xiong, W.; Schonthal, A.H. HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res. 2007, 67, 10920–10928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Gurley, E.C.; Jarujaron, S.; Ding, H.; Fang, Y.; Xu, Z.; Pandak, W.M., Jr.; Hylemon, P.B. HIV protease inhibitors activate the unfolded protein response and disrupt lipid metabolism in primary hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G1071–G1080. [Google Scholar] [CrossRef]
- Zha, B.S.; Zhou, H. ER stress and lipid metabolism in adipocytes. Biochem. Res. Int. 2012, 2012, 312943. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Murthy, R.; Wood, B.; Song, B.; Wang, S.; Sun, B.; Malhi, H.; Kaufman, R.J. ER stress signalling through eIF2alpha and CHOP, but not IRE1alpha, attenuates adipogenesis in mice. Diabetologia 2013, 56, 911–924. [Google Scholar] [CrossRef] [Green Version]
- Longo, M.; Spinelli, R.; D’Esposito, V.; Zatterale, F.; Fiory, F.; Nigro, C.; Raciti, G.A.; Miele, C.; Formisano, P.; Beguinot, F.; et al. Pathologic endoplasmic reticulum stress induced by glucotoxic insults inhibits adipocyte differentiation and induces an inflammatory phenotype. Biochim. Biophys. Acta 2016, 1863, 1146–1156. [Google Scholar] [CrossRef]
- Obacz, J.; Avril, T.; Le Reste, P.J.; Urra, H.; Quillien, V.; Hetz, C.; Chevet, E. Endoplasmic reticulum proteostasis in glioblastoma-From molecular mechanisms to therapeutic perspectives. Sci. Signal. 2017, 10, eaal2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zha, B.S.; Wan, X.; Zhang, X.; Zha, W.; Zhou, J.; Wabitsch, M.; Wang, G.; Lyall, V.; Hylemon, P.B.; Zhou, H. HIV protease inhibitors disrupt lipid metabolism by activating endoplasmic reticulum stress and inhibiting autophagy activity in adipocytes. PLoS ONE 2013, 8, e59514. [Google Scholar] [CrossRef]
- Pacenti, M.; Barzon, L.; Favaretto, F.; Fincati, K.; Romano, S.; Milan, G.; Vettor, R.; Palù, G. Microarray analysis during adipogenesis identifies new genes altered by antiretroviral drugs. AIDS 2006, 20, 1691–1705. [Google Scholar] [CrossRef] [PubMed]
- Squillace, N.; Bresciani, E.; Torsello, A.; Bandera, A.; Sabbatini, F.; Giovannetti, C.; Giunta, G.; Rovati, L.; Del Bene, M.; Locatelli, V.; et al. Changes in subcutaneous adipose tissue microRNA expression in HIV-infected patients. J. Antimicrob. Chemother. 2014, 69, 3067–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresciani, E.; Saletti, C.; Squillace, N.; Rizzi, L.; Molteni, L.; Meanti, R.; Omeljaniuk, R.J.; Biagini, G.; Gori, A.; Locatelli, V.; et al. miRNA-218 Targets Lipin-1 and Glucose Transporter Type 4 Genes in 3T3-L1 Cells Treated With Lopinavir/Ritonavir. Front. Pharm. 2019, 10, 461. [Google Scholar] [CrossRef] [Green Version]
- Goldman, S.; Zhang, Y.; Jin, S. Autophagy and adipogenesis: Implications in obesity and type II diabetes. Autophagy 2010, 6, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Bartke, A.; Yuan, R. Genetic manupulations of autophagy regulate adipocyte differentiation and metabolism. Cur. Opin. Endocr. Metab. Res. 2019, 5, 74–81. [Google Scholar] [CrossRef]
- Romero, M.; Zorzano, A. Role of autophagy in the regulation of adipose tissue biology. Cell Cycle 2019, 18, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Postigo, M.; Tinahones, A.; El Bekay, R.; Malagon, M.M.; Tenahones, F.J. The role of autophagy in white adipose tissue function: Implications for metabolic health. Metabolites 2020, 10, 179. [Google Scholar] [CrossRef] [PubMed]
- Hoyer-Hansen, M.; Jaattela, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.-O.; Yadav, R.K.; Kim, H.-R.; Chae, H.-J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; De Biasi, S.; Pinti, M.; Nasi, M.; Riccio, M.; Carnevale, G.; Cavallini, G.M.; Sala de Oyanguren, F.J.; O’Connor, J.E.; Mussini, C.; et al. The protease inhibitor atazanavir triggers autophagy and mitophagy in human preadipocytes. AIDS 2012, 26, 2017–2026. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells. | ATV Dose | % Apoptotic Cells | p-Value * |
|---|---|---|---|
| ADSC | 0.0 | NS | |
| 0 μM | 0.0 | NS | |
| 5 μM | 0.0 | NS | |
| 10 μM | 0.0 | NS | |
| 20 μM | 0.0 | NS | |
| Adipocytes | |||
| 0 μM | 0.52 ± 0.09 | NS | |
| 5 μM | 0.97 ± 0.10 | NS | |
| 10 μM | 2.52 ± 0.19 | p < 0.01 | |
| 20 μM | 4.34 ± 0.25 | p < 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akita, S.; Suzuki, K.; Yoshimoto, H.; Ohtsuru, A.; Hirano, A.; Yamashita, S. Cellular Mechanism Underlying Highly-Active or Antiretroviral Therapy-Induced Lipodystrophy: Atazanavir, a Protease Inhibitor, Compromises Adipogenic Conversion of Adipose-Derived Stem/Progenitor Cells through Accelerating ER Stress-Mediated Cell Death in Differentiating Adipocytes. Int. J. Mol. Sci. 2021, 22, 2114. https://doi.org/10.3390/ijms22042114
Akita S, Suzuki K, Yoshimoto H, Ohtsuru A, Hirano A, Yamashita S. Cellular Mechanism Underlying Highly-Active or Antiretroviral Therapy-Induced Lipodystrophy: Atazanavir, a Protease Inhibitor, Compromises Adipogenic Conversion of Adipose-Derived Stem/Progenitor Cells through Accelerating ER Stress-Mediated Cell Death in Differentiating Adipocytes. International Journal of Molecular Sciences. 2021; 22(4):2114. https://doi.org/10.3390/ijms22042114
Chicago/Turabian StyleAkita, Sadanori, Keiji Suzuki, Hiroshi Yoshimoto, Akira Ohtsuru, Akiyoshi Hirano, and Shunichi Yamashita. 2021. "Cellular Mechanism Underlying Highly-Active or Antiretroviral Therapy-Induced Lipodystrophy: Atazanavir, a Protease Inhibitor, Compromises Adipogenic Conversion of Adipose-Derived Stem/Progenitor Cells through Accelerating ER Stress-Mediated Cell Death in Differentiating Adipocytes" International Journal of Molecular Sciences 22, no. 4: 2114. https://doi.org/10.3390/ijms22042114