A Mechanistic, Enantioselective, Physiologically Based Pharmacokinetic Model of Verapamil and Norverapamil, Built and Evaluated for Drug–Drug Interaction Studies

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Software
2.2. Clinical Data
2.3. PBPK Model Building
2.4. PBPK Model Evaluation
2.5. DDI Modeling
2.6. DDI Modeling Evaluation
3. Results
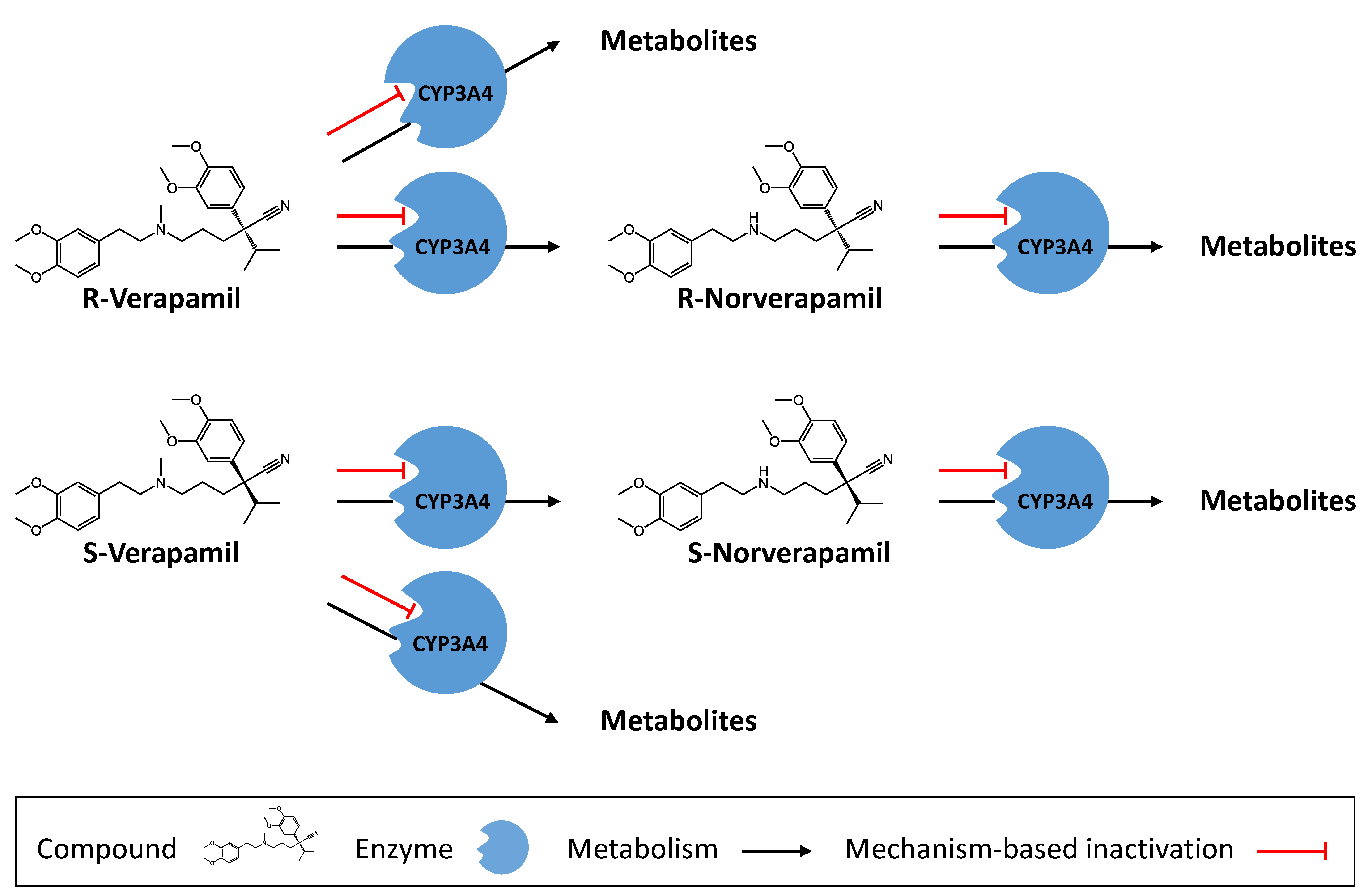
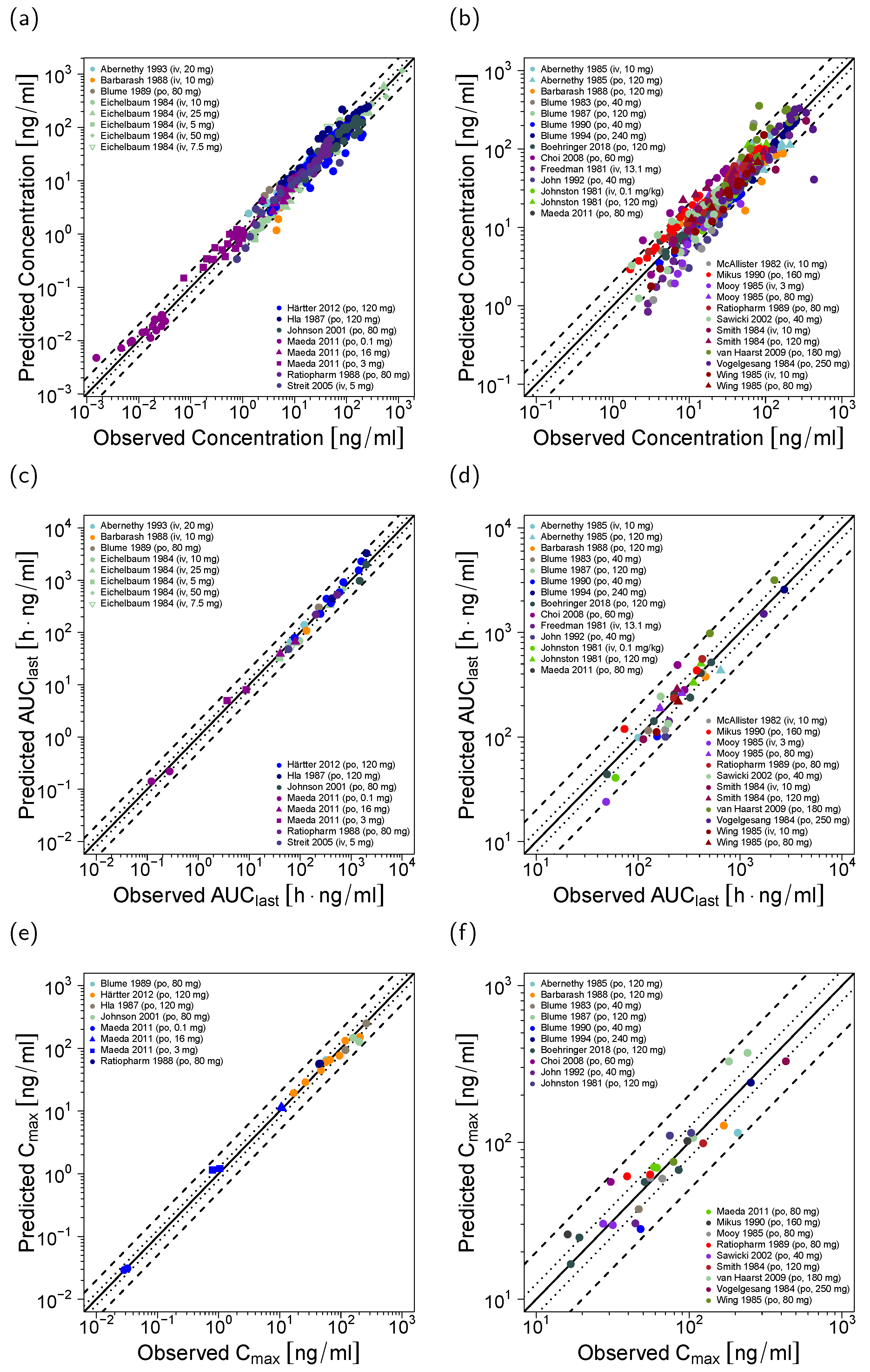
3.1. Verapamil PBPK Model Building and Evaluation
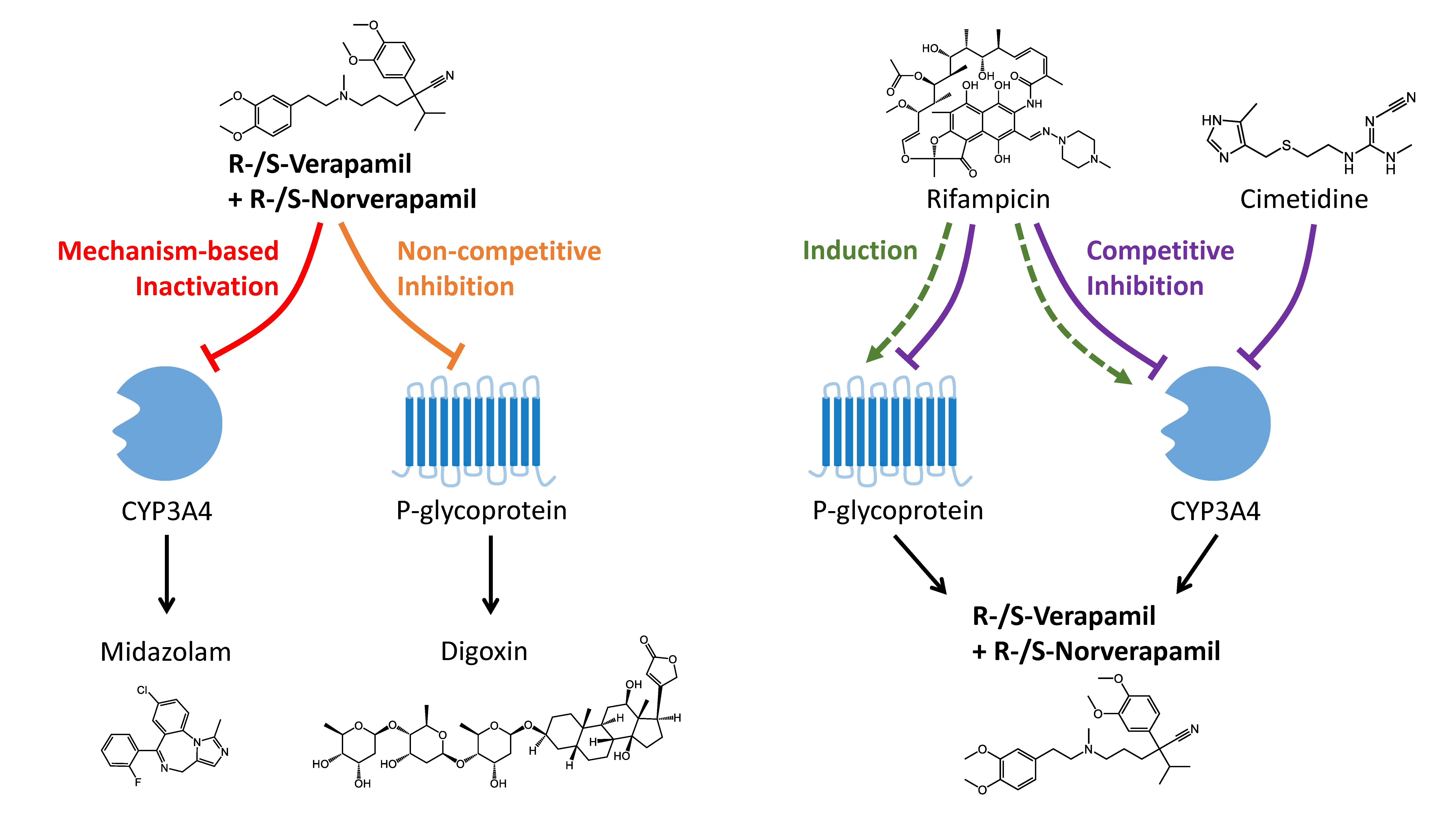
3.2. Verapamil DDI Modeling and Evaluation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- ClinCalc LLC. ClinCalc DrugStats Database. Available online: https://clincalc.com/DrugStats/ (accessed on 25 March 2020).
- Echizen, H.; Brecht, T.; Niedergesäss, S.; Vogelgesang, B.; Eichelbaum, M. The effect of dextro-, levo-, and racemic verapamil on atrioventricular conduction in humans. Am. Heart J. 1985, 109, 210–217. [Google Scholar] [CrossRef]
- Echizen, H.; Vogelgesang, B.; Eichelbaum, M. Effects of D,L-verapamil on atrioventricular conduction in relation to its stereoselective first-pass metabolism. Clin. Pharmacol. Ther. 1985, 38, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Belpomme, D.; Gauthier, S.; Pujade-Lauraine, E.; Facchini, T.; Goudier, M.J.; Krakowski, I.; Netter-Pinon, G.; Frenay, M.; Gousset, C.; Marié, F.N.; et al. Verapamil increases the survival of patients with anthracycline-resistant metastatic breast carcinoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2000, 11, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Cowan, K.H.; Solomon, D.; Ognibene, F.; Goldspiel, B.; Chang, R.; Noone, M.H.; Denicoff, A.M.; Barnes, C.S.; Gossard, M.R.; et al. Phase I crossover study of paclitaxel with r-verapamil in patients with metastatic breast cancer. J. Clin. Oncol. 1996, 14, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.-H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef] [PubMed][Green Version]
- U.S. Food and Drug Administration. Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 25 February 2020).
- Wang, Y.-H.; Jones, D.R.; Hall, S.D. Prediction of cytochrome P450 3A inhibition by verapamil enantiomers and their metabolites. Drug Metab. Dispos. 2004, 32, 259–266. [Google Scholar] [CrossRef]
- Wang, J.; Xia, S.; Xue, W.; Wang, D.; Sai, Y.; Liu, L.; Liu, X. A semi-physiologically-based pharmacokinetic model characterizing mechanism-based auto-inhibition to predict stereoselective pharmacokinetics of verapamil and its metabolite norverapamil in human. Eur. J. Pharm. Sci. 2013, 50, 290–302. [Google Scholar] [CrossRef]
- Ito, S.; Woodland, C.; Harper, P.A.; Koren, G. The mechanism of the verapamil-digoxin interaction in renal tubular cells (LLC-PK1). Life Sci. 1993, 53, PL399–PL403. [Google Scholar] [CrossRef]
- Pauli-Magnus, C.; von Richter, O.; Burk, O.; Ziegler, A.; Mettang, T.; Eichelbaum, M.; Fromm, M.F. Characterization of the major metabolites of verapamil as substrates and inhibitors of P-glycoprotein. J. Pharmacol. Exp. Ther. 2000, 293, 376–382. [Google Scholar]
- Woodland, C.; Koren, G.; Wainer, I.W.; Batist, G.; Ito, S. Verapamil metabolites: Potential P-glycoprotein-mediated multidrug resistance reversal agents. Can. J. Physiol. Pharmacol. 2003, 81, 800–805. [Google Scholar] [CrossRef]
- Ledwitch, K.V.; Barnes, R.W.; Roberts, A.G. Unravelling the complex drug–drug interactions of the cardiovascular drugs, verapamil and digoxin, with P-glycoprotein. Biosci. Rep. 2016, 36, e00309. [Google Scholar] [CrossRef]
- Backman, J.T.; Olkkola, K.T.; Aranko, K.; Himberg, J.J.; Neuvonen, P.J. Dose of midazolam should be reduced during diltiazem and verapamil treatments. Br. J. Clin. Pharmacol. 1994, 37, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Rodin, S.M.; Johnson, B.F.; Wilson, J.; Ritchie, P.; Johnson, J. Comparative effects of verapamil and isradipine on steady-state digoxin kinetics. Clin. Pharmacol. Ther. 1988, 43, 668–672. [Google Scholar] [CrossRef] [PubMed]
- G.D. Searle LLC CALAN®—Verapamil Hydrochloride Tablet, Film Coated—Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/018817s021lbl.pdf (accessed on 25 March 2020).
- Vogelgesang, B.; Echizen, H.; Schmidt, E.; Eichelbaum, M. Stereoselective first-pass metabolism of highly cleared drugs: Studies of the bioavailability of L- and D-verapamil examined with a stable isotope technique. Br. J. Clin. Pharmacol. 1984, 18, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Schomerus, M.; Spiegelhalder, B.; Stieren, B.; Eichelbaum, M. Physiological disposition of verapamil in man. Cardiovasc. Res. 1976, 10, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Eichelbaum, M.; Ende, M.; Remberg, G.; Schomerus, M.; Dengler, H.J. The metabolism of DL-[14C]verapamil in man. Drug Metab. Dispos. 1979, 7, 145–148. [Google Scholar]
- Echizen, H.; Eichelbaum, M. Clinical pharmacokinetics of verapamil, nifedipine and diltiazem. Clin. Pharmacokinet. 1986, 11, 425–449. [Google Scholar] [CrossRef]
- Wojtyniak, J.-G.; Britz, H.; Selzer, D.; Schwab, M.; Lehr, T. Data Digitizing: Accurate and Precise Data Extraction for Quantitative Systems Pharmacology and Physiologically Based Pharmacokinetic Modeling. CPT Pharmacomet. Syst. Pharmacol. 2020. accepted for publication. [Google Scholar] [CrossRef]
- Härtter, S.; Sennewald, R.; Nehmiz, G.; Reilly, P. Oral bioavailability of dabigatran etexilate (Pradaxa®) after co-medication with verapamil in healthy subjects. Br. J. Clin. Pharmacol. 2013, 75, 1053–1062. [Google Scholar] [CrossRef]
- Boehringer Ingelheim Pharma GmbH; Co. KG. The Effect of Potent Inhibitors of Drug Transporters (Verapamil, Rifampin, Cimetidine, Probenecid) on Pharmacokinetics of A Transporter Probe Drug Cocktail Consisting of Digoxin, Furosemide, Metformin and Rosuvastatin. EudraCT 2017-001549-29. Available online: https://clinicaltrials.gov/ct2/show/record/NCT03307252 (accessed on 25 March 2020).
- Open Systems Pharmacology Suite Community PK-Sim Ontog-eny Database Documentation, Version 7.3. Available online: https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/PK-SimOntogenyDatabaseVersion7.3.pdf (accessed on 25 March 2020).
- Sandström, R.; Karlsson, A.; Knutson, L.; Lennernäs, H. Jejunal absorption and metabolism of R/S-verapamil in humans. Pharm. Res. 1998, 15, 856–862. [Google Scholar] [CrossRef]
- Luurtsema, G.; Molthoff, C.F.M.; Windhorst, A.D.; Smit, J.W.; Keizer, H.; Boellaard, R.; Lammertsma, A.A.; Franssen, E.J.F. (R)- and (S)-[11C]verapamil as PET-tracers for measuring P-glycoprotein function: In vitro and in vivo evaluation. Nucl. Med. Biol. 2003, 30, 747–751. [Google Scholar] [CrossRef]
- Engman, H.; Tannergren, C.; Artursson, P.; Lennernäs, H. Enantioselective transport and CYP3A4-mediated metabolism of R/S-verapamil in Caco-2 cell monolayers. Eur. J. Pharm. Sci. 2003, 19, 57–65. [Google Scholar] [CrossRef]
- Verschraagen, M.; Koks, C.H.W.; Schellens, J.H.M.; Beijnen, J.H. P-glycoprotein system as a determinant of drug interactions: The case of digoxin-verapamil. Pharmacol. Res. 1999, 40, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, J.; Fujita, T.; Hayashi, Y.; Iwamoto, K.; Watanabe, J. pKa determination of verapamil by liquid-liquid partition. J. Pharm. Sci. 1984, 73, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Vogelpoel, H.; Welink, J.; Amidon, G.L.; Junginger, H.E.; Midha, K.K.; Möller, H.; Olling, M.; Shah, V.P.; Barends, D.M. Biowaiver monographs for immediate release solid oral dosage forms based on biopharmaceutics classification system (BCS) literature data: Verapamil hydrochloride, propranolol hydrochloride, and atenolol. J. Pharm. Sci. 2004, 93, 1945–1956. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Hoekman, D. Exploring QSAR: Hydrophobic, Electronic, and Steric Constants; American Chemical Society: Washington, DC, USA, 1995. [Google Scholar]
- Sanaee, F.; Clements, J.D.; Waugh, A.W.G.; Fedorak, R.N.; Lewanczuk, R.; Jamali, F. Drug-disease interaction: Crohn’s disease elevates verapamil plasma concentrations but reduces response to the drug proportional to disease activity. Br. J. Clin. Pharmacol. 2011, 72, 787–797. [Google Scholar] [CrossRef]
- Shirasaka, Y.; Sakane, T.; Yamashita, S. Effect of P-glycoprotein expression levels on the concentration-dependent permeability of drugs to the cell membrane. J. Pharm. Sci. 2008, 97, 553–565. [Google Scholar] [CrossRef]
- Döppenschmitt, S.; Langguth, P.; Regårdh, C.G.; Andersson, T.B.; Hilgendorf, C.; Spahn-Langguth, H. Characterization of binding properties to human P-glycoprotein: Development of a [3H]verapamil radioligand-binding assay. J. Pharmacol. Exp. Ther. 1999, 288, 348–357. [Google Scholar]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically based pharmacokinetic modeling 1: Predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modeling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef] [PubMed]
- Open Systems Pharmacology Suite Community Open Systems Pharmacology Suite Manual, Version 7.4. Available online: https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/OpenSystemsPharmacologySuite.pdf (accessed on 25 March 2020).
- Blume, H.; Mutschler, E. Bioäquivalenz: Qualitätsbewertung wirkstoffgleicher Fertigarzneimittel: Anleitung, Methoden, Materialien; Govi-Verlag: Eschborn, Germany, 1989. [Google Scholar]
- Sigma-Aldrich Inc. A Case Study in SPE Method Development—Understanding the Dual Interaction Properties of Discovery DSC-SCX SPE Using Verapamil (and Metabolite) from Serum as a Test Example. Available online: https://www.sigmaaldrich.com/technical-documents/articles/reporter-eu/a-case-study-in-spe.html (accessed on 25 February 2020).
- Tracy, T.S.; Korzekwa, K.R.; Gonzalez, F.J.; Wainer, I.W. Cytochrome P450 isoforms involved in metabolism of the enantiomers of verapamil and norverapamil. Br. J. Clin. Pharmacol. 1999, 47, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Streit, M.; Göggelmann, C.; Dehnert, C.; Burhenne, J.; Riedel, K.-D.; Menold, E.; Mikus, G.; Bärtsch, P.; Haefeli, W.E. Cytochrome P450 enzyme-mediated drug metabolism at exposure to acute hypoxia (corresponding to an altitude of 4,500 m). Eur. J. Clin. Pharmacol. 2005, 61, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.S.; Benyunes, M.C.; Bjornsson, T.D.; Shand, D.G.; Pritchett, E.L.C. Influence of cimetidine on verapamil kinetics and dynamics. Clin. Pharmacol. Ther. 1984, 36, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Abernethy, D.R.; Wainer, I.W.; Longstreth, J.A.; Andrawis, N.S. Stereoselective verapamil disposition and dynamics in aging during racemic verapamil administration. J. Pharmacol. Exp. Ther. 1993, 266, 904–911. [Google Scholar]
- Maeda, K.; Takano, J.; Ikeda, Y.; Fujita, T.; Oyama, Y.; Nozawa, K.; Kumagai, Y.; Sugiyama, Y. Nonlinear pharmacokinetics of oral quinidine and verapamil in healthy subjects: A clinical microdosing study. Clin. Pharmacol. Ther. 2011, 90, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, W.; Janicki, S. Pharmacokinetics of verapamil and its metabolite norverapamil from a buccal drug formulation. Int. J. Pharm. 2002, 238, 181–189. [Google Scholar] [CrossRef]
- ratiopharm GmbH Fachinformation Verapamil-ratiopharm® N 40 mg/80 mg Filmtabletten. Available online: https://nanopdf.com/download/verapamil-ratiopharm-n-40-mg-80-mg-filmtabletten_pdf (accessed on 25 February 2020).
- Mikus, G.; Eichelbaum, M.; Fischer, C.; Gumulka, S.; Klotz, U.; Kroemer, H.K. Interaction of verapamil and cimetidine: Stereochemical aspects of drug metabolism, drug disposition and drug action. J. Pharmacol. Exp. Ther. 1990, 253, 1042–1048. [Google Scholar]
- Hanke, N.; Frechen, S.; Moj, D.; Britz, H.; Eissing, T.; Wendl, T.; Lehr, T. PBPK Models for CYP3A4 and P-gp DDI Prediction: A Modeling Network of Rifampicin, Itraconazole, Clarithromycin, Midazolam, Alfentanil, and Digoxin. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 647–659. [Google Scholar] [CrossRef]
- Hanke, N.; Türk, D.; Selzer, D.; Ishiguro, N.; Ebner, T.; Wiebe, S.; Müller, F.; Stopfer, P.; Nock, V.; Lehr, T. A Comprehensive Whole-Body Physiologically Based Pharmacokinetic Drug-Drug-Gene Interaction Model of Metformin and Cimetidine in Healthy Adults and Renally Impaired Individuals. Clin. Pharmacokinet. 2020. [Google Scholar] [CrossRef]
- Pedersen, K.E.; Dorph-Pedersen, A.; Hvidt, S.; Klitgaard, N.A.; Pedersen, K.K. The long-term effect of verapamil on plasma digoxin concentration and renal digoxin clearance in healthy subjects. Eur. J. Clin. Pharmacol. 1982, 22, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Türk, D.; Hanke, N.; Wolf, S.; Frechen, S.; Eissing, T.; Wendl, T.; Schwab, M.; Lehr, T. Physiologically Based Pharmacokinetic Models for Prediction of Complex CYP2C8 and OATP1B1 (SLCO1B1) Drug-Drug-Gene Interactions: A Modeling Network of Gemfibrozil, Repaglinide, Pioglitazone, Rifampicin, Clarithromycin and Itraconazole. Clin. Pharmacokinet. 2019, 58, 1595–1607. [Google Scholar] [CrossRef]
- Wrighton, S.A.; Ring, B.J. Inhibition of human CYP3A catalyzed 1′-hydroxy midazolam formation by ketoconazole, nifedipine, erythromycin, cimetidine, and nizatidine. Pharm. Res. 1994, 11, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Barbarash, R.A.; Bauman, J.L.; Fischer, J.H.; Kondos, G.T.; Batenhorst, R.L. Near-total reduction in verapamil bioavailability by rifampin. Electrocardiographic correlates. Chest 1988, 94, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Guest, E.J.; Aarons, L.; Houston, J.B.; Rostami-Hodjegan, A.; Galetin, A. Critique of the two-fold measure of prediction success for ratios: Application for the assessment of drug-drug interactions. Drug Metab. Dispos. 2011, 39, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Eichelbaum, M.; Mikus, G.; Vogelgesang, B. Pharmacokinetics of (+)-, (-)- and (+/−)-verapamil after intravenous administration. Br. J. Clin. Pharmacol. 1984, 17, 453–458. [Google Scholar] [CrossRef]
- von Richter, O.; Burk, O.; Fromm, M.F.; Thon, K.P.; Eichelbaum, M.; Kivistö, K.T. Cytochrome P450 3A4 and P-glycoprotein expression in human small intestinal enterocytes and hepatocytes: A comparative analysis in paired tissue specimens. Clin. Pharmacol. Ther. 2004, 75, 172–183. [Google Scholar] [CrossRef]
- Rautio, J.; Humphreys, J.E.; Webster, L.O.; Balakrishnan, A.; Keogh, J.P.; Kunta, J.R.; Serabjit-Singh, C.J.; Polli, J.W. In vitro p-glycoprotein inhibition assays for assessment of clinical drug interaction potential of new drug candidates: A recommendation for probe substrates. Drug Metab. Dispos. 2006, 34, 786–792. [Google Scholar] [CrossRef]
- Varma, M.V.S.; Ashokraj, Y.; Dey, C.S.; Panchagnula, R. P-glycoprotein inhibitors and their screening: A perspective from bioavailability enhancement. Pharmacol. Res. 2003, 48, 347–359. [Google Scholar] [CrossRef]
- Neuhoff, S.; Yeo, K.R.; Barter, Z.; Jamei, M.; Turner, D.B.; Rostami-Hodjegan, A. Application of permeability-limited physiologically-based pharmacokinetic models: Part II—Prediction of P-glycoprotein mediated drug-drug interactions with digoxin. J. Pharm. Sci. 2013, 102, 3161–3173. [Google Scholar] [CrossRef]
- Kimoto, E.; Chupka, J.; Xiao, Y.; Bi, Y.-A.; Duignan, D.B. Characterization of digoxin uptake in sandwich-cultured human hepatocytes. Drug Metab. Dispos. 2011, 39, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Lumen, A.A.; Li, L.; Li, J.; Ahmed, Z.; Meng, Z.; Owen, A.; Ellens, H.; Hidalgo, I.J.; Bentz, J. Transport inhibition of digoxin using several common P-gp expressing cell lines is not necessarily reporting only on inhibitor binding to P-gp. PLoS ONE 2013, 8, e69394. [Google Scholar] [CrossRef] [PubMed]
- Taub, M.E.; Mease, K.; Sane, R.S.; Watson, C.A.; Chen, L.; Ellens, H.; Hirakawa, B.; Reyner, E.L.; Jani, M.; Lee, C.A. Digoxin is not a substrate for organic anion-transporting polypeptide transporters OATP1A2, OATP1B1, OATP1B3, and OATP2B1 but is a substrate for a sodium-dependent transporter expressed in HEK293 cells. Drug Metab. Dispos. 2011, 39, 2093–2102. [Google Scholar] [CrossRef] [PubMed]
- Wing, L.M.; Miners, J.O.; Lillywhite, K.J. Verapamil disposition--effects of sulphinpyrazone and cimetidine. Br. J. Clin. Pharmacol. 1985, 19, 385–391. [Google Scholar] [CrossRef]
- Abernethy, D.R.; Schwartz, J.B.; Todd, E.L. Lack of interaction between verapamil and cimetidine. Clin. Pharmacol. Ther. 1985, 38, 342–349. [Google Scholar] [CrossRef]
- Perdaems, N.; Blasco, H.; Vinson, C.; Chenel, M.; Whalley, S.; Cazade, F.; Bouzom, F. Predictions of metabolic drug-drug interactions using physiologically based modelling: Two cytochrome P450 3A4 substrates coadministered with ketoconazole or verapamil. Clin. Pharmacokinet. 2010, 49, 239–258. [Google Scholar] [CrossRef]
- Lippert, J.; Burghaus, R.; Edginton, A.; Frechen, S.; Karlsson, M.; Kovar, A.; Lehr, T.; Milligan, P.; Nock, V.; Ramusovic, S.; et al. Open Systems Pharmacology Community-An Open Access, Open Source, Open Science Approach to Modeling and Simulation in Pharmaceutical Sciences. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 878–882. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Unit | Source | Literature | Reference | Value | Unit | Source | Literature | Reference | Description |
|---|---|---|---|---|---|---|---|---|---|---|---|
| R-Verapamil | S-Verapamil | ||||||||||
| MW | 454.611 | g/mol | Lit. | 454.611 | [29] | 454.611 | g/mol | Lit. | 454.611 | [29] | Molecular weight |
| pKa (base) | 8.75 | - | Lit. | 8.75 | [30] | 8.75 | - | Lit. | 8.75 | [30] | Acid dissociation constant |
| Solubility (pH 6.54) | 46.0 | g/L | Lit. | 46.0 | [31] | 46.0 | g/L | Lit. | 46.0 | [31] | Solubility |
| logP | 2.84 * | - | Optim. | 3.79 | [32] | 2.84 * | - | Optim. | 3.79 | [32] | Lipophilicity |
| fu | 5.1 | % | Lit. | 5.1 | [33] | 11.0 | % | Lit. | 11.0 | [33] | Fraction unbound |
| CYP3A4 Km → Norv | 19.59 | µmol/L | Lit. | 19.59 ‡ | [9] | 9.72 | µmol/L | Lit. | 9.72 ‡ | [9] | Michaelis–Menten constant |
| CYP3A4 kcat → Norv | 34.94 | 1/min | Optim. | - | - | 26.17 | 1/min | Optim. | - | - | Catalytic rate constant |
| CYP3A4 Km → D617 | 35.34 | µmol/L | Lit. | 35.34 ‡ | [9] | 23.64 | µmol/L | Lit. | 23.64 ‡ | [9] | Michaelis–Menten constant |
| CYP3A4 kcat → D617 | 43.98 | 1/min | Optim. | - | - | 56.42 | 1/min | Optim. | - | - | Catalytic rate constant |
| Pgp Km | 1.01 | µmol/L | Lit. | 1.01 | [34] | 1.01 | µmol/L | Lit. | 1.01 | [34] | Michaelis–Menten constant |
| Pgp kcat | 12.60 ° | 1/min | Optim. | - | - | 12.60 ° | 1/min | Optim. | - | - | Transport rate constant |
| GFR fraction | 1.00 | - | Ass. | - | - | 1.00 | - | Ass. | - | - | Filtered drug in the urine |
| EHC cont. fraction | 1.00 | - | Ass. | - | - | 1.00 | - | Ass. | - | - | Bile fraction cont. released |
| CYP3A4 MBI KI | 27.63 | µmol/L | Lit. | 27.63 ‡ | [9] | 3.85 | µmol/L | Lit. | 3.85 ‡ | [9] | Conc. for 50% inactivation |
| CYP3A4 MBI kinact | 0.038 | 1/min | Lit. | 0.038 | [9] | 0.034 | 1/min | Lit. | 0.034 | [9] | Maximum inactivation rate |
| Pgp non-competitive Ki | 0.038 * | µmol/L | Optim. | 0.31 | [35] | 0.038 * | µmol/L | Optim. | 0.31 | [35] | Conc. for 50% inhibition |
| Partition coefficients | Diverse | - | Calc. | R&R | [36,37] | Diverse | - | Calc. | R&R | [36,37] | Cell to plasma partitioning |
| Cellular permeability | 9.94 × 10−2 * | cm/min | Optim. | PK-Sim | [38] | 9.94 × 10−2 * | cm/min | Optim. | PK-Sim | [38] | Perm. into the cellular space |
| Intestinal permeability | 3.54 × 10−6 * | cm/min | Optim. | 1.21 × 10−5 | Calc. | 3.54 × 10−6 * | cm/min | Optim. | 1.21 × 10−5 | Calc. | Transcellular intestinal perm. |
| SR tablet Weibull time | 155.24 | min | Optim. | - | [39] | 155.24 | min | Optim. | - | [39] | Dissolution time (50%) |
| SR tablet Weibull shape | 2.37 | - | Optim. | - | [39] | 2.37 | - | Optim. | - | [39] | Dissolution profile shape |
| Parameter | Value | Unit | Source | Literature | Reference | Value | Unit | Source | Literature | Reference | Description |
|---|---|---|---|---|---|---|---|---|---|---|---|
| R-Norverapamil | S-Norverapamil | ||||||||||
| MW | 440.584 | g/mol | Lit. | 440.584 | [29] | 440.584 | g/mol | Lit. | 440.584 | [29] | Molecular weight |
| pKa (base) | 8.75 | - | Lit. | 8.6–8.9 | [40] | 8.75 | - | Lit. | 8.6–8.9 | [40] | Acid dissociation constant |
| logP | 2.84 * | - | Optim. | - | - | 2.84 * | - | Optim. | - | - | Lipophilicity |
| fu | 5.1 a | % | Ass. | - | - | 11.0 b | % | Ass. | - | - | Fraction unbound |
| CYP3A4 Km → D620 | 144.0 | µmol/L | Lit. | 144.0 | [41] | 36.0 | µmol/L | Lit. | 36.0 | [41] | Michaelis–Menten constant |
| CYP3A4 kcat → D620 | 145.64 | 1/min | Optim. | - | - | 41.10 | 1/min | Optim. | - | - | Catalytic rate constant |
| Pgp Km | 1.01 * | µmol/L | Ass. | - | - | 1.01 * | µmol/L | Ass. | - | - | Michaelis–Menten constant |
| Pgp kcat | 3.39 ° | 1/min | Optim. | - | - | 3.39 ° | 1/min | Optim. | - | - | Transport rate constant |
| GFR fraction | 1.00 | - | Ass. | - | - | 1.00 | - | Ass. | - | - | Filtered drug in the urine |
| EHC cont. fraction | 1.00 | - | Ass. | - | - | 1.00 | - | Ass. | - | - | Bile fraction cont. released |
| CYP3A4 MBI KI | 6.10 | µmol/L | Lit. | 6.10 ‡ | [9] | 2.90 | µmol/L | Lit. | 2.90 ‡ | [9] | Conc. for 50% inactivation |
| CYP3A4 MBI kinact | 0.048 | 1/min | Lit. | 0.048 | [9] | 0.080 | 1/min | Lit. | 0.080 | [9] | Maximum inactivation rate |
| Pgp non-competitive Ki | 0.038 * | µmol/L | Optim. | 0.30 c | [11] | 0.038 * | µmol/L | Optim. | 0.30 c | [11] | Conc. for 50% inhibition |
| Partition coefficients | Diverse | - | Calc. | R&R | [36,37] | Diverse | - | Calc. | R&R | [36,37] | Cell to plasma partitioning |
| Cellular permeability | 9.94 × 10−2 * | cm/min | Optim. | PK-Sim | [38] | 9.94 × 10−2 * | cm/min | Optim. | PK-Sim | [38] | Perm. into the cellular space |
| Intestinal permeability | 3.54 × 10−6 * | cm/min | Optim. | 1.40 × 10−5 | Calc. | 3.54 × 10−6 * | cm/min | Optim. | 1.40 × 10−5 | Calc. | Transcellular intestinal perm. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanke, N.; Türk, D.; Selzer, D.; Wiebe, S.; Fernandez, É.; Stopfer, P.; Nock, V.; Lehr, T. A Mechanistic, Enantioselective, Physiologically Based Pharmacokinetic Model of Verapamil and Norverapamil, Built and Evaluated for Drug–Drug Interaction Studies. Pharmaceutics 2020, 12, 556. https://doi.org/10.3390/pharmaceutics12060556
Hanke N, Türk D, Selzer D, Wiebe S, Fernandez É, Stopfer P, Nock V, Lehr T. A Mechanistic, Enantioselective, Physiologically Based Pharmacokinetic Model of Verapamil and Norverapamil, Built and Evaluated for Drug–Drug Interaction Studies. Pharmaceutics. 2020; 12(6):556. https://doi.org/10.3390/pharmaceutics12060556
Chicago/Turabian StyleHanke, Nina, Denise Türk, Dominik Selzer, Sabrina Wiebe, Éric Fernandez, Peter Stopfer, Valerie Nock, and Thorsten Lehr. 2020. "A Mechanistic, Enantioselective, Physiologically Based Pharmacokinetic Model of Verapamil and Norverapamil, Built and Evaluated for Drug–Drug Interaction Studies" Pharmaceutics 12, no. 6: 556. https://doi.org/10.3390/pharmaceutics12060556
APA StyleHanke, N., Türk, D., Selzer, D., Wiebe, S., Fernandez, É., Stopfer, P., Nock, V., & Lehr, T. (2020). A Mechanistic, Enantioselective, Physiologically Based Pharmacokinetic Model of Verapamil and Norverapamil, Built and Evaluated for Drug–Drug Interaction Studies. Pharmaceutics, 12(6), 556. https://doi.org/10.3390/pharmaceutics12060556