
Urolithins: Diet-Derived Bioavailable Metabolites to Tackle Diabetes

 , and
, and 
Abstract
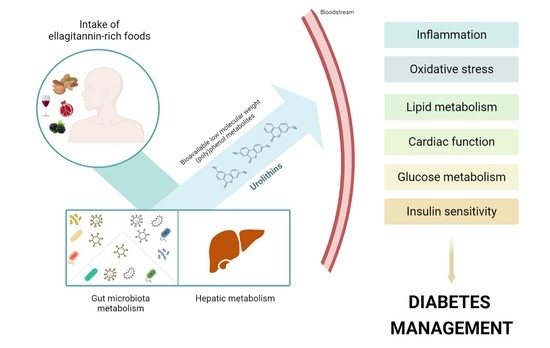
{kind=link}
{kind=link}
{kind=link}
1. Diabetes Mellitus
1.1. β-Cell Dysfunction and Insulin Resistance
1.2. Chronic Inflammation and Diabetes
1.3. Islet Amyloid Pathology
1.4. Role of Autophagy in Β-Cell Function and Survival
1.5. Dysbiosis in Metabolic Dysfunction
2. (Poly)Phenols in Diabetes
2.1. Ellagitannins and Ellagic Acid
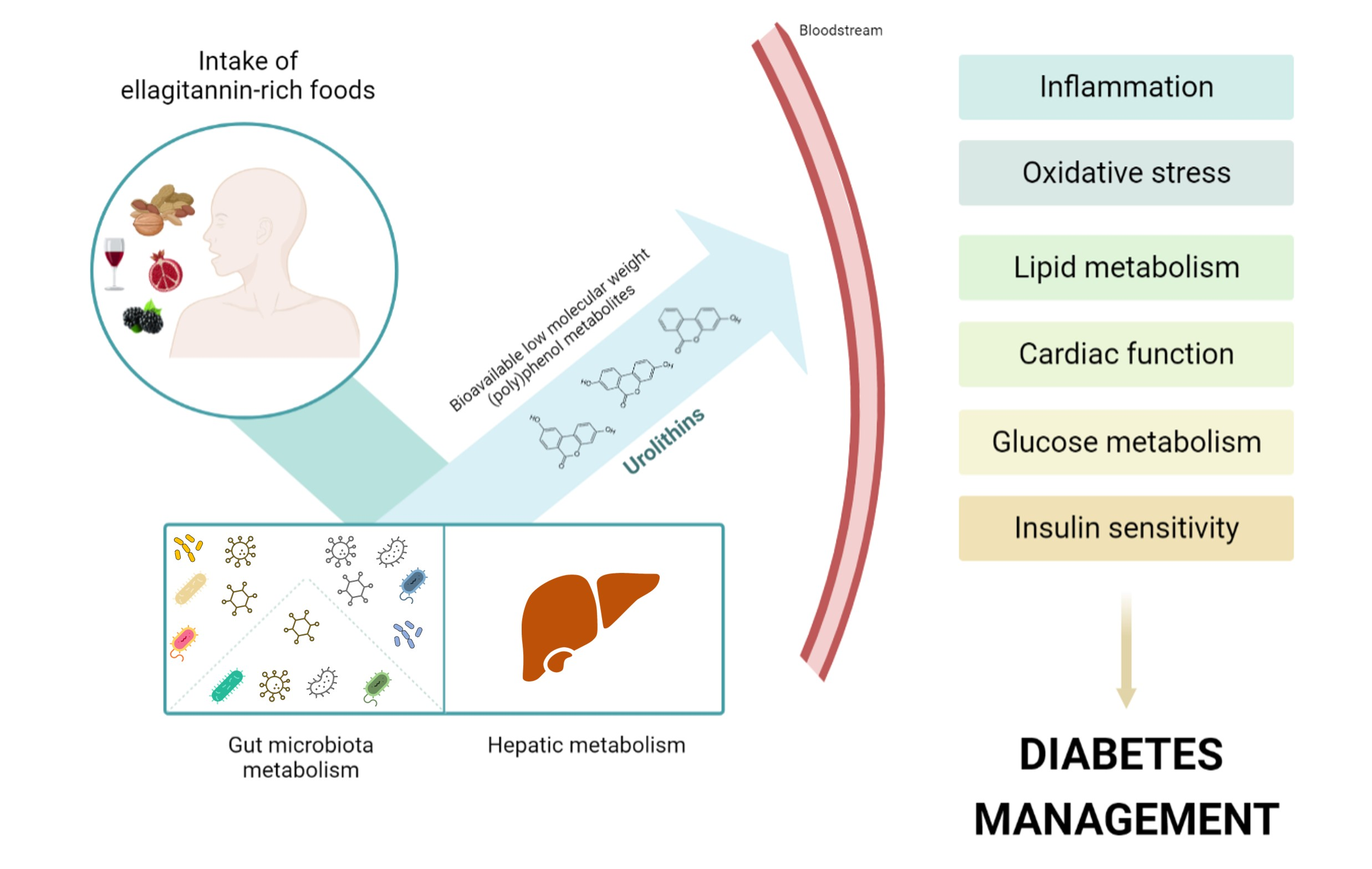
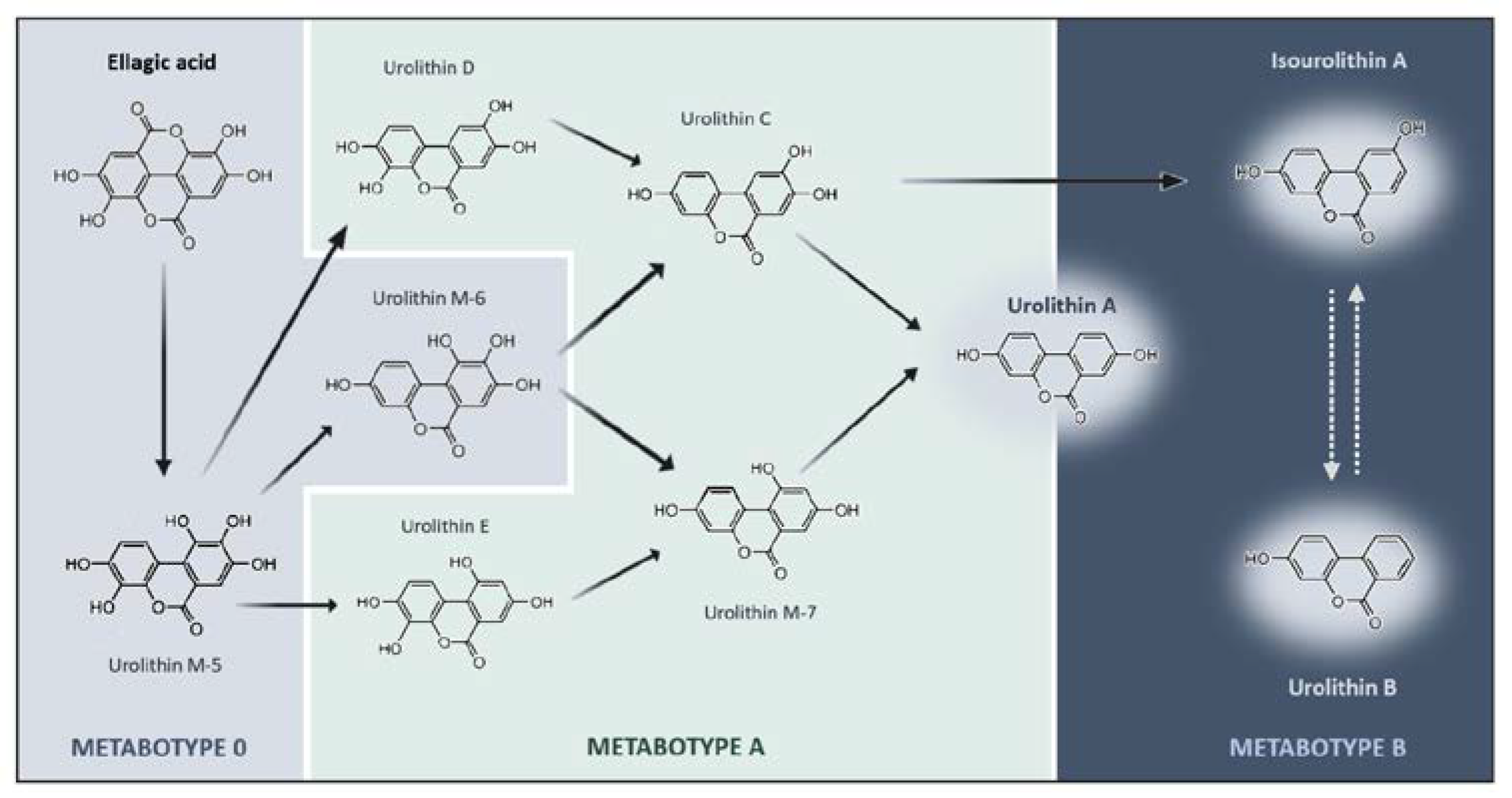
2.2. Urolithins and Metabotypes
2.2.1. Phase-II Metabolism
2.2.2. Bioavailability
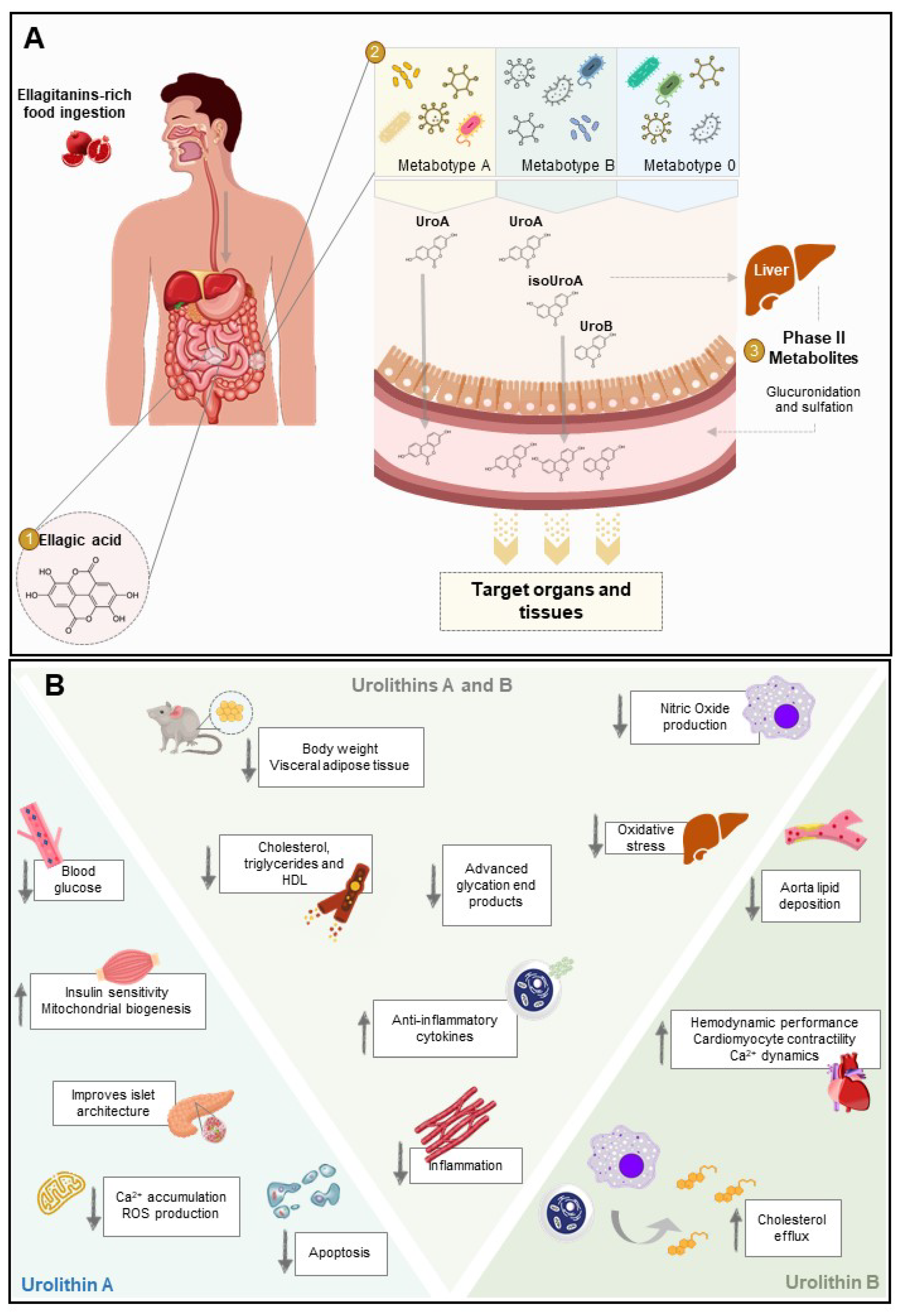
3. Biological Activity against the Pathological Processes of Diabetes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Classification of Diabetes Mellitus; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef]
- Stumvoll, M.; Goldstein, B.J.; van Haeften, T.W. Type 2 diabetes: Principles of pathogenesis and therapy. Lancet (London, England) 2005, 365, 1333–1346. [Google Scholar] [CrossRef]
- Tan, S.Y.; Mei Wong, J.L.; Sim, Y.J.; Wong, S.S.; Elhassan, S.A.M.; Tan, S.H.; Lim, G.P.L.; Tay, N.W.R.; Annan, N.C.; Bhattamisra, S.K.; et al. Type 1 and 2 diabetes mellitus: A review on current treatment approach and gene therapy as potential intervention. Diabetes Syndr. Clin. Res. Rev. 2019, 13, 364–372. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. IDF Diabetes Atlas, 8th ed.; International Diabetes Federation: Brussels, Belgium, 2017; Available online: https://diabetesatlas.org/atlas/ninth-edition/ (accessed on 6 August 2021).
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019; Available online: https://diabetesatlas.org/atlas/eighth-edition/ (accessed on 6 August 2021).
- Yang, J.S.; Lu, C.C.; Kuo, S.C.; Hsu, Y.M.; Tsai, S.C.; Chen, S.Y.; Chen, Y.T.; Lin, Y.J.; Huang, Y.C.; Chen, C.J.; et al. Autophagy and its link to type II diabetes mellitus. BioMedicine 2017, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Tabák, A.G.; Jokela, M.; Akbaraly, T.N.; Brunner, E.J.; Kivimäki, M.; Witte, D.R. Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: An analysis from the Whitehall II study. Lancet 2009, 373, 2215–2221. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Brüning, J.C.; Winnay, J.N.; Postic, C.; Magnuson, M.A.; Kahn, C.R. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 1999, 96, 329–339. [Google Scholar] [CrossRef]
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front. Endocrinol. 2014, 5, 161. [Google Scholar] [CrossRef]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Maezono, K.; Osman, A.; Pendergrass, M.; Patti, M.E.; Pratipanawatr, T.; DeFronzo, R.A.; Kahn, C.R.; Mandarino, L.J. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J. Clin. Investig. 2000, 105, 311–320. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- Fosam, A.; Sikder, S.; Abel, B.S.; Tella, S.H.; Walter, M.F.; Mari, A.; Muniyappa, R. Reduced Insulin Clearance and Insulin-Degrading Enzyme Activity Contribute to Hyperinsulinemia in African Americans. J. Clin. Endocrinol. Metab. 2020, 105, E1835–E1846. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Imaizumi, T. Diabetic Vascular Complications: Pathophysiology, Biochemical Basis and Potential Therapeutic Strategy. Curr. Pharm. Des. 2005, 11, 2279–2299. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.M. Lipids and lipoproteins in patients with type 2 diabetes. Diabetes Care 2004, 27, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Erion, D.M.; Park, Hy.; Lee, H. The role of lipids in the pathogenesis and treatment of type 2 diabetes and associated co-morbidities. BMB Rep. 2016, 49, 139–148. [Google Scholar] [CrossRef]
- Chaurasia, B.; Summers, S.A. Ceramides—Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol. Metab. TEM 2015, 26, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Adiels, M.; Borén, J.; Caslake, M.J.; Stewart, P.; Soro, A.; Westerbacka, J.; Wennberg, B.; Olofsson, Sv.; Packard, C.; Taskinen, Ma. Overproduction of VLDL1 driven by hyperglycemia is a dominant feature of diabetic dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1697–1703. [Google Scholar] [CrossRef]
- Hirano, T.; Naito, H.; Kurokawa, M.; Ebara, T.; Nagano, S.; Adachi, M.; Yoshino, G. High prevalence of small LDL particles in non-insulin-dependent diabetic patients with nephropathy. Atherosclerosis 1996, 123, 57–72. [Google Scholar] [CrossRef]
- Sarwar, N.; Gao, P.; Kondapally Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; Stampfer, M.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [CrossRef] [PubMed]
- Brunham, L.R.; Kruit, J.K.; Hayden, M.R.; Verchere, C.B. Cholesterol in β-cell Dysfunction: The Emerging Connection Between HDL Cholesterol and Type 2 Diabetes. Curr. Diabetes Rep. 2010, 10, 55–60. [Google Scholar] [CrossRef]
- Ishikawa, M.; Iwasaki, Y.; Yatoh, S.; Kato, T.; Kumadaki, S.; Inoue, N.; Yamamoto, T.; Matsuzaka, T.; Nakagawa, Y.; Yahagi, N.; et al. Cholesterol accumulation and diabetes in pancreatic β-cell-specific SREBP-2 transgenic mice: A new model for lipotoxicity. J. Lipid Res. 2008, 49, 2524–2534. [Google Scholar] [CrossRef]
- Poitout, V.; Robertson, R.P. Minireview: Secondary β-Cell Failure in Type 2 Diabetes—A Convergence of Glucotoxicity and Lipotoxicity. Endocrinology 2002, 143, 339–342. [Google Scholar] [CrossRef]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C.; Mattock, M.B.; Chusney, G.D.; Burt, D. NIDDM as a disease of the innate immune system: Association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 1997, 40, 1286–1292. [Google Scholar] [CrossRef]
- Spranger, J.; Kroke, A.; Möhlig, M.; Hoffmann, K.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F.H. Inflammatory cytokines and the risk to develop type 2 diabetes: Results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes 2003, 52, 812–817. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef]
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur. Cardiol. 2019, 14, 50–59. [Google Scholar] [CrossRef]
- Sauter, N.S.; Schulthess, F.T.; Galasso, R.; Castellani, L.W.; Maedler, K. The antiinflammatory cytokine interleukin-1 receptor antagonist protects from high-fat diet-induced hyperglycemia. Endocrinology 2008, 149, 2208–2218. [Google Scholar] [CrossRef]
- Böni-Schnetzler, M.; Thorne, J.; Parnaud, G.; Marselli, L.; Ehses, J.A.; Kerr-Conte, J.; Pattou, F.; Halban, P.A.; Weir, G.C.; Donath, M.Y. Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta -cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J. Clin. Endocrinol. Metab. 2008, 93, 4065–4074. [Google Scholar] [CrossRef]
- Goldfine, A.B.; Silver, R.; Aldhahi, W.; Cai, D.; Tatro, E.; Lee, J.; Shoelson, S.E. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin. Transl. Sci. 2008, 1, 36–43. [Google Scholar] [CrossRef]
- Ruan, H.; Miles, P.D.G.; Ladd, C.M.; Ross, K.; Golub, T.R.; Olefsky, J.M.; Lodish, H.F. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-alpha: Implications for insulin resistance. Diabetes 2002, 51, 3176–3188. [Google Scholar] [CrossRef] [PubMed]
- Kamata, K.; Mizukami, H.; Inaba, W.; Tsuboi, K.; Tateishi, Y.; Yoshida, T.; Yagihashi, S. Islet amyloid with macrophage migration correlates with augmented β-cell deficits in type 2 diabetic patients. Amyloid Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis 2014, 21, 191–201. [Google Scholar] [CrossRef]
- Horii, T.; Fujita, Y.; Ishibashi, C.; Fukui, K.; Eguchi, H.; Kozawa, J.; Shimomura, I. Islet inflammation is associated with pancreatic fatty infiltration and hyperglycemia in type 2 diabetes. BMJ Open Diabetes Res. & Care 2020, 8, e001508. [Google Scholar] [CrossRef]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef]
- Bertola, A.; Ciucci, T.; Rousseau, D.; Bourlier, V.; Duffaut, C.; Bonnafous, S.; Blin-Wakkach, C.; Anty, R.; Iannelli, A.; Gugenheim, J.; et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 2012, 61, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Meier, D.T.; Morcos, M.; Samarasekera, T.; Zraika, S.; Hull, R.L.; Kahn, S.E. Islet amyloid formation is an important determinant for inducing islet inflammation in high-fat-fed human IAPP transgenic mice. Diabetologia 2014, 57, 1884–1888. [Google Scholar] [CrossRef]
- Abedini, A.; Cao, P.; Plesner, A.; Zhang, J.; He, M.; Derk, J.; Patil, S.A.; Rosario, R.; Lonier, J.; Song, F.; et al. RAGE binds preamyloid IAPP intermediates and mediates pancreatic β cell proteotoxicity. J. Clin. Investig. 2018, 128, 682–698. [Google Scholar] [CrossRef]
- Park, Y.J.; Lee, S.; Kieffer, T.J.; Warnock, G.L.; Safikhan, N.; Speck, M.; Hao, Z.; Woo, M.; Marzban, L. Deletion of Fas protects islet beta cells from cytotoxic effects of human islet amyloid polypeptide. Diabetologia 2012. [Google Scholar] [CrossRef]
- Westermark, P.; Grimelius, L. The Pancreatic Islet Cells in Insular Amyloidosis in Human Diabetic and Non-Diabetic Adults. Acta Pathol. Microbiol. Scand. Sect. A Pathol. 1973, 81, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.H.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J. Diabetes Res. 2016, 2016, 2798269. [Google Scholar] [CrossRef] [PubMed]
- Yonemoto, I.T.; Kroon, G.J.A.; Dyson, H.J.; Balch, W.E.; Kelly, J.W. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry 2008, 47, 9900–9910. [Google Scholar] [CrossRef]
- Mulder, H.; Ahrén, B.; Sundler, F. Islet amyloid polypeptide and insulin gene expression are regulated in parallel by glucose in vivo in rats. Am. J. Physiol. Endocrinol. Metab. 1996, 271, E1008–E1014. [Google Scholar] [CrossRef]
- Kahn, S.E.; Fujimoto, W.Y.; D’Alessio, D.A.; Ensninick, J.W.; Porte, D. Glucose stimulates and potentiates islet amyloid polypeptide secretion by the B-cell. Horm. Metab. Res. 1991, 23, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.F.; Westermark, G.T. Aberrant processing of human proislet amyloid polypeptide results in increased amyloid formation. Diabetes 2005, 54, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.F.; Andersson, A.; Westermark, P.; Westermark, G.T. Intracellular amyloid-like deposits contain unprocessed pro-islet amyloid polypeptide (proIAPP) in beta cells of transgenic mice overexpressing the gene for human IAPP and transplanted human islets. Diabetologia 2006, 49, 1237–1246. [Google Scholar] [CrossRef]
- Westermark, G.T.; Steiner, D.F.; Gebre-Medhin, S.; Engström, U.; Westermark, P. Pro islet amyloid polypeptide (ProIAPP) immunoreactivity in the islets of langerhans. Upsala J. Med Sci. 2000, 105, 97–106. [Google Scholar] [CrossRef]
- Clark, A.; Wells, C.A.; Buley, I.D.; Cruickshank, J.K.; Vanhegan, R.I.; Matthews, D.R.; Cooper, G.J.; Holman, R.R.; Turner, R.C. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: Quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. (Edinb. Scotl.) 1988, 9, 151–159. [Google Scholar]
- Asthana, S.; Mallick, B.; Alexandrescu, A.T.; Jha, S. IAPP in type II diabetes: Basic research on structure, molecular interactions, and disease mechanisms suggests potential intervention strategies. Biochim. Et Biophys. Acta (BBA) Biomembr. 2018, 1860, 1765–1782. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, A.; Razzaboni, B.; Weir, G.C.; Yankner, B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994, 368, 756–760. [Google Scholar] [CrossRef]
- Montané, J.; Novials, A. The Role of Human IAPP in Stress and Inflammatory Processes in Type 2 Diabetes. In Exploring New Findings on Amyloidosis; IntechOpen: London, UK, 2016. [Google Scholar] [CrossRef]
- Anguiano, M.; Nowak, R.J.; Lansbury, P.T.J. Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry 2002, 41, 11338–11343. [Google Scholar] [CrossRef]
- Abedini, A.; Schmidt, A.M. Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett. 2013, 587, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Kapurniotu, A.; Bernhagen, J.; Greenfield, N.; Al-Abed, Y.; Teichberg, S.; Frank, R.W.; Voelter, W.; Bucala, R. Contribution of advanced glycosylation to the amyloidogenicity of islet amyloid polypeptide. Eur. J. Biochem. 1998, 251, 208–216. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Mukhopadhyay, M.; Bhattacharyya, M.; Karmakar, P. Is autophagy associated with diabetes mellitus and its complications? A review. EXCLI J. 2018, 17, 709–720. [Google Scholar] [CrossRef]
- Marasco, M.R.; Linnemann, A.K. β-Cell Autophagy in Diabetes Pathogenesis. Endocrinology 2018, 159, 2127–2141. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy Is Important in Islet Homeostasis and Compensatory Increase of Beta Cell Mass in Response to High-Fat Diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, A.; Kimura-Koyanagi, M.; Asahara, S.I.; Guillén, C.; Inoue, H.; Teruyama, K.; Shimizu, S.; Kanno, A.; García-Aguilar, A.; Koike, M.; et al. Pancreatic β-cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes 2014, 63, 2996–3008. [Google Scholar] [CrossRef]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional Regulation of Autophagy: Mechanisms and Diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef]
- Ji, J.; Petropavlovskaia, M.; Khatchadourian, A.; Patapas, J.; Makhlin, J.; Rosenberg, L.; Maysinger, D. Type 2 diabetes is associated with suppression of autophagy and lipid accumulation in β-cells. J. Cell. Mol. Med. 2019, 23, 2890–2900. [Google Scholar] [CrossRef] [PubMed]
- Shigihara, N.; Fukunaka, A.; Hara, A.; Komiya, K.; Honda, A.; Uchida, T.; Abe, H.; Toyofuku, Y.; Tamaki, M.; Ogihara, T.; et al. Human IAPP-induced pancreatic β cell toxicity and its regulation by autophagy. J. Clin. Investig. 2014, 124, 3634–3644. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cheon, H.; Jeong, Y.T.; Quan, W.; Kim, K.H.; Cho, J.M.; Lim, Y.M.; Oh, S.H.; Jin, S.M.; Kim, J.H.; et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient β cells induces diabetes. J. Clin. Investig. 2014, 124, 3311–3324. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, K.; Kim, M.J.; Lim, H.; Kim, K.H.; Kim, S.W.; Lee, E.S.; Kim, H.H.; Kim, S.J.; Hur, K.Y.; et al. An autophagy enhancer ameliorates diabetes of human IAPP-transgenic mice through clearance of amyloidogenic oligomer. Nat. Commun. 2021, 12, 183. [Google Scholar] [CrossRef]
- Meijnikman, A.S.; Gerdes, V.E.; Nieuwdorp, M.; Herrema, H. Evaluating Causality of Gut Microbiota in Obesity and Diabetes in Humans. Endocr. Rev. 2018, 39, 133–153. [Google Scholar] [CrossRef]
- Clooney, A.G.; Fouhy, F.; Sleator, R.D.; O’Driscoll, A.; Stanton, C.; Cotter, P.D.; Claesson, M.J. Comparing apples and oranges?: Next generation sequencing and its impact on microbiome analysis. PLoS ONE 2016, 11, e0148028. [Google Scholar] [CrossRef]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef] [PubMed]
- Aw, W.; Fukuda, S. Understanding the role of the gut ecosystem in diabetes mellitus. J. Diabetes Investig. 2018, 9, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Goodarzi, M.O. Metabolites Linking the Gut Microbiome with Risk for Type 2 Diabetes. Curr. Nutr. Rep. 2020, 9, 83–93. [Google Scholar] [CrossRef]
- Sikalidis, A.K.; Maykish, A. The Gut Microbiome and Type 2 Diabetes Mellitus: Discussing A Complex Relationship. Biomedicines 2020, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Mandaliya, D.K.; Seshadri, S. Short Chain Fatty Acids, pancreatic dysfunction and type 2 diabetes. Pancreatology 2019, 19, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Todesco, T.; Rao A v Bosello, O.; Jenkins, D.J.A. Propionate lowers blood glucose and alters lipid metabolism in healthy subjects. Am. J. Clin. Nutr. 1991, 54, 860–865. [Google Scholar] [CrossRef] [PubMed]
- Zaky, A.; Glastras, S.J.; Wong, M.Y.W.; Pollock, C.A.; Saad, S. The Role of the Gut Microbiome in Diabetes and Obesity-Related Kidney Disease. Int. J. Mol. Sci. 2021, 22, 9641. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef]
- Kovatcheva-Datchary, P.; Nilsson, A.; Akrami, R.; Lee, Y.S.; De Vadder, F.; Arora, T.; Hallen, A.; Martens, E.; Björck, I.; Bäckhed, F. Dietary Fiber-Induced Improvement in Glucose Metabolism Is Associated with Increased Abundance of Prevotella. Cell Metab. 2015, 22, 971–982. [Google Scholar] [CrossRef] [PubMed]
- le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, J.L.; Bäckhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef]
- Wang, J.; Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- Raimundo, A.F.; Félix, F.; Andrade, R.; García-Conesa, M.T.; González-Sarrías, A.; Gilsa-Lopes, J.; do Ó, D.; Raimundo, A.; Ribeiro, R.; Rodriguez-Mateos, A.; et al. Combined effect of interventions with pure or enriched mixtures of (poly)phenols and anti-diabetic medication in type 2 diabetes management: A meta-analysis of randomized controlled human trials. Eur. J. Nutr. 2020, 59, 1329–1343. [Google Scholar] [CrossRef] [PubMed]
- Rosado-Ramos, R.; Godinho-Pereira, J.; Figueira, I.; Jardim, C.; Garcia, G.; Menezes, R. Exploring the Benefits of Cellular Models to Uncover Bioactive Polyphenols for Neurodegeneration. Curr. Pharm. Des. 2018, 24, 2076–2106. [Google Scholar] [CrossRef]
- Zhang, J.; Li, L.; Kim, S.H.; Hagerman, A.E.; Lü, J. Anti-cancer, anti-diabetic and other pharmacologic and biological activities of penta-galloyl-glucose. Pharm. Res. 2009, 26, 2066–2080. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zheng, J.; Li, Y.; Xu, D.P.; Li, S.; Chen, Y.M.; Li, H.B. Natural polyphenols for prevention and treatment of cancer. Nutrients 2016, 8, 515. [Google Scholar] [CrossRef]
- Al-Harbi, S.A.; Abdulrahman, A.O.; Zamzami, M.A.; Khan, M.I. Urolithins: The Gut Based Polyphenol Metabolites of Ellagitannins in Cancer Prevention, a Review. Front. Nutr. 2021, 8, 258. [Google Scholar] [CrossRef] [PubMed]
- González-Sarrías, A.; Tomás-Barberán, F.A.; García-Villalba, R. Structural Diversity of Polyphenols and Distribution in Foods. In Dietary Polyphenols; John Wiley & Sons, Inc.: Hoboken, NJ, EUA, 2020; pp. 1–29. [Google Scholar] [CrossRef]
- El-Missiry, M.A.; Amer, M.A.; Hemieda, F.A.E.; Othman, A.I.; Sakr, D.A.; Abdulhadi, H.L. Cardioameliorative effect of punicalagin against streptozotocin-induced apoptosis, redox imbalance, metabolic changes and inflammation. Egypt. J. Basic Appl. Sci. 2015, 2, 247–260. [Google Scholar] [CrossRef]
- Atrahimovich, D.; Samson, A.O.; Khattib, A.; Vaya, J.; Khatib, S. Punicalagin Decreases Serum Glucose Levels and Increases PON1 Activity and HDL Anti-Inflammatory Values in Balb/c Mice Fed a High-Fat Diet. Oxidative Med. Cell. Longev. 2018, 2018, 2673076. [Google Scholar] [CrossRef]
- Banihani, S.A.; Makahleh, S.M.; El-Akawi, Z.; Al-Fashtaki, R.A.; Khabour, O.F.; Gharibeh, M.Y.; Saadah, N.A.; Al-Hashimi, F.H.; Al-Khasieb, N.J. Fresh pomegranate juice ameliorates insulin resistance, enhances β-cell function, and decreases fasting serum glucose in type 2 diabetic patients. Nutr. Res. 2014, 34, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Banihani, S.A.; Fashtaky, R.A.; Makahleh, S.M.; El-Akawi, Z.J.; Khabour, O.F.; Saadeh, N.A. Effect of fresh pomegranate juice on the level of melatonin, insulin, and fasting serum glucose in healthy individuals and people with impaired fasting glucose. Food Sci. Nutr. 2020, 8, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Cerdá, B.; Llorach, R.; Cerón, J.J.; Espín, J.C.; Tomás-Barberán, F.A. Evaluation of the bioavailability and metabolism in the rat of punicalagin, an antioxidant polyphenol from pomegranate juice. Eur. J. Nutr. 2003, 42, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Cerdá, B.; Tomás-Barberán, F.A.; Espín, J.C. Metabolism of antioxidant and chemopreventive ellagitannins from strawberries, raspberries, walnuts, and oak-aged wine in humans: Identification of biomarkers and individual variability. J. Agric. Food Chem. 2005, 53, 227–235. [Google Scholar] [CrossRef]
- Cerdá, B.; Espín, J.C.; Parra, S.; Martínez, P.; Tomás-Barberán, F.A. The potent in vitro antioxidant ellagitannins from pomegranate juice are metabolised into bioavailable but poor antioxidant hydroxy-6H-dibenzopyran-6-one derivatives by the colonic microflora of healthy humans. Eur. J. Nutr. 2004, 43, 205–220. [Google Scholar] [CrossRef]
- Larrosa, M.; Tomás-Barberán, F.A.; Espín, J.C. The dietary hydrolysable tannin punicalagin releases ellagic acid that induces apoptosis in human colon adenocarcinoma Caco-2 cells by using the mitochondrial pathway. J. Nutr. Biochem. 2006, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- García-Villalba, R.; Giménez-Bastida, J.A.; Ávila-Gálvez, M.A.; Tomás-Barberán, F.A.; Espín, J.C.; González-Sarrías, A. Ellagitannins and Their Gut Microbiota-Derived Metabolites: Urolithins. In Dietary Polyphenols; Wiley: Hoboken, NJ, USA, 2020; pp. 319–364. [Google Scholar] [CrossRef]
- González-Sarrías, A.; García-Villalba, R.; Núñez-Sánchez, M.Á.; Tomé-Carneiro, J.; Zafrilla, P.; Mulero, J.; Tomás-Barberán, F.A.; Espín, J.C. Identifying the limits for ellagic acid bioavailability: A crossover pharmacokinetic study in healthy volunteers after consumption of pomegranate extracts. J. Funct. Foods 2015, 19, 225–235. [Google Scholar] [CrossRef]
- Selma, M.V.; González-Sarrías, A.; Salas-Salvadó, J.; Andrés-Lacueva, C.; Alasalvar, C.; Örem, A.; Tomás-Barberán, F.A.; Espín, J.C. The gut microbiota metabolism of pomegranate or walnut ellagitannins yields two urolithin-metabotypes that correlate with cardiometabolic risk biomarkers: Comparison between normoweight, overweight-obesity and metabolic syndrome. Clin. Nutr. 2018, 37, 897–905. [Google Scholar] [CrossRef] [PubMed]
- González-Sarrías, A.; García-Villalba, R.; Romo-Vaquero, M.; Alasalvar, C.; Örem, A.; Zafrilla, P.; Tomás-Barberán, F.A.; Selma, M.V.; Espín, J.C. Clustering according to urolithin metabotype explains the interindividual variability in the improvement of cardiovascular risk biomarkers in overweight-obese individuals consuming pomegranate: A randomized clinical trial. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Barberán, F.A.; García-Villalba, R.; González-Sarrías, A.; Selma, M.V.; Espín, J.C. Ellagic acid metabolism by human gut microbiota: Consistent observation of three urolithin phenotypes in intervention trials, independent of food source, age, and health status. J. Agric. Food Chem. 2014, 62, 6535–6538. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Martín, A.; García-Villalba, R.; González-Sarrías, A.; Romo-Vaquero, M.; Loria-Kohen, V.; Ramírez-de-Molina, A.; Tomás-Barberán, F.A.; Selma, M.V.; Espín, J.C. The gut microbiota urolithin metabotypes revisited: The human metabolism of ellagic acid is mainly determined by aging. Food Funct. 2018, 9, 4100–4106. [Google Scholar] [CrossRef] [PubMed]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef] [PubMed]
- González-Sarrías, A.; Giménez-Bastida, J.A.; García-Conesa, M.T.; Gómez-Sánchez, M.B.; García-Talavera, N.V.; Gil-Izquierdo, A.; Sánchez-Álvarez, C.; Fontana-Compiano, L.O.; Morga-Egea, J.P.; Pastor-Quirante, F.A.; et al. Occurrence of urolithins, gut microbiota ellagic acid metabolites and proliferation markers expression response in the human prostate gland upon consumption of walnuts and pomegranate juice. Mol. Nutr. Food Res. 2010, 54, 311–322. [Google Scholar] [CrossRef]
- González-Sarrías, A.; Giménez-Bastida, J.A.; Núñez-Sánchez, M.Á.; Larrosa, M.; García-Conesa, M.T.; Tomás-Barberán, F.A.; Espín, J.C. Phase-II metabolism limits the antiproliferative activity of urolithins in human colon cancer cells. Eur. J. Nutr. 2014, 53, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Gálvez, M.Á.; Espín, J.C.; González-Sarrías, A. Physiological Relevance of the Antiproliferative and Estrogenic Effects of Dietary Polyphenol Aglycones versus Their Phase-II Metabolites on Breast Cancer Cells: A Call of Caution. J. Agric. Food Chem. 2018, 66, 8547–8555. [Google Scholar] [CrossRef] [PubMed]
- Piwowarski, J.P.; Stanisławska, I.; Granica, S.; Stefanska, J.; Kiss, A.K. Phase II conjugates of urolithins isolated from human urine and potential role of β-glucuronidases in their disposition. Drug Metab. Dispos. 2017, 45, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Gálvez, M.A.; Giménez-Bastida, J.A.; González-Sarrías, A.; Espín, J.C. Tissue deconjugation of urolithin A glucuronide to free urolithin A in systemic inflammation. Food Funct. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Seeram, N.P.; Henning, S.M.; Zhang, Y.; Suchard, M.; Li, Z.; Heber, D. Pomegranate juice ellagitannin metabolites are present in human plasma and some persist in urine for up to 48 hours. J. Nutr. 2006, 136, 2481–2485. [Google Scholar] [CrossRef]
- Seeram, N.P.; Aronson, W.J.; Zhang, Y.; Henning, S.M.; Moro, A.; Lee, R.P.; Sartippour, M.; Harris, D.M.; Rettig, M.; Suchard, M.A.; et al. Pomegranate ellagitannin-derived metabolites inhibit prostate cancer growth and localize to the mouse prostate gland. J. Agric. Food Chem. 2007, 55, 7732–7737. [Google Scholar] [CrossRef]
- Gasperotti, M.; Passamonti, S.; Tramer, F.; Masuero, D.; Guella, G.; Mattivi, F.; Vrhovsek, U. Fate of Microbial Metabolites of Dietary Polyphenols in Rats: Is the Brain Their Target Destination? ACS Chem. Neurosci. 2015, 6, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Savi, M.; Bocchi, L.; Mena, P.; Dall’Asta, M.; Crozier, A.; Brighenti, F.; Stilli, D.; Del Rio, D. In vivo administration of urolithin A and B prevents the occurrence of cardiac dysfunction in streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2017, 16. [Google Scholar] [CrossRef]
- Freedland, S.J.; Carducci, M.; Kroeger, N.; Partin, A.; Rao, J.Y.; Jin, Y.; Kerkoutian, S.; Wu, H.; Li, Y.; Creel, P.; et al. A double-blind, randomized, neoadjuvant study of the tissue effects of POMx pills in men with prostate cancer before radical prostatectomy. Cancer Prev. Res. 2013, 6, 1120–1127. [Google Scholar] [CrossRef]
- Nuñez-Sánchez, M.A.; García-Villalba, R.; Monedero-Saiz, T.; Garcıa-Talavera, N.V.; Gomez-Sanchez, M.B.; Sanchez-Alvarez, C.; Garcia-Albert, A.M.; Rodriguez-Gil, F.J.; Ruiz-Marin, M.; Pastor-Quirante, F.A.; et al. Targeted metabolic profiling of pomegranate polyphenols and urolithins in plasma, urine and colon tissues from colorectal cancer patients. Mol. Nutr. Food Res. 2014, 58, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Shi, X.C.; Xie, B.C.; Zhu, M.Q.; Chen, Y.; Chu, X.Y.; Cai, G.H.; Liu, M.; Yang, S.Z.; Mitchell, G.A.; et al. Urolithin A exerts antiobesity effects through enhancing adipose tissue thermogenesis in mice. PLoS Biol. 2020, 18, e3000688. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Y.; Henning, S.M.; Chan, B.; Long, J.; Zhong, J.; Acin-Perez, R.; Petcherski, A.; Shirihai, O.; Heber, D.; et al. Ellagic Acid and Its Microbial Metabolite Urolithin A Alleviate Diet-Induced Insulin Resistance in Mice. Mol. Nutr. Food Res. 2020, 64, 2000091. [Google Scholar] [CrossRef] [PubMed]
- Verzelloni, E.; Pellacani, C.; Tagliazucchi, D.; Tagliaferri, S.; Calani, L.; Costa, L.G.; Brighenti, F.; Borges, G.; Crozier, A.; Conte, A.; et al. Antiglycative and neuroprotective activity of colon-derived polyphenol catabolites. Mol. Nutr. Food Res. 2011, 55 (Suppl. 1), S35–S43. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ma, H.; Frost, L.; Yuan, T.; Dain, J.A.; Seeram, N.P. Pomegranate phenolics inhibit formation of advanced glycation endproducts by scavenging reactive carbonyl species. Food Funct. 2014, 5, 2996–3004. [Google Scholar] [CrossRef] [PubMed]
- Puupponen-Pimiä, R.; Seppänen-Laakso, T.; Kankainen, M.; Maukonen, J.; Törrönen, R.; Kolehmainen, M.; Leppänen, T.; Moilanen, E.; Nohynek, L.; Aura, A.M.; et al. Effects of ellagitannin-rich berries on blood lipids, gut microbiota, and urolithin production in human subjects with symptoms of metabolic syndrome. Mol. Nutr. Food Res. 2013, 57, 2258–2263. [Google Scholar] [CrossRef] [PubMed]
- Esmaillzadeh, A.; Tahbaz, F.; Gaieni, I.; Alavi-Majd, H.; Azadbakht, L. Concentrated pomegranate juice improves lipid profiles in diabetic patients with hyperlipidemia. J. Med. Food 2004, 7, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, L.; Haller, V.; Ritsch, A. A Novel Candidate for Prevention and Treatment of Atherosclerosis: Urolithin B Decreases Lipid Plaque Deposition in apoE −/− Mice and Increases Early Stages of Reverse Cholesterol Transport in ox-LDL Treated Macrophages Cells. Mol. Nutr. Food Res. 2019, 63, e1800887. [Google Scholar] [CrossRef]
- Kang, I.; Kim, Y.; Tomás-Barberán, F.A.; Espín, J.C.; Chung, S. Urolithin A, C, and D, but not iso-urolithin A and urolithin B, attenuate triglyceride accumulation in human cultures of adipocytes and hepatocytes. Mol. Nutr. Food Res. 2016, 60, 1129–1138. [Google Scholar] [CrossRef]
- Kang, I.; Tomás-Barberán, F.; Carlos Espín, J.; Chung, S. Urolithin C, a Gut Microbiota Metabolite Derived from Ellagic Acid, Attenuates Triglyceride Accumulation in Human Adipocytes and Hepatoma Huh7 Cells. FASEB J. 2015. [Google Scholar] [CrossRef]
- Manigandan, S.; Won Yun, J. Urolithin A Induces Brown-like Phenotype in 3T3-L1 White Adipocytes via β3-adrenergic Receptor-p38 MAPK Signaling Pathway. Biotechnol. Bioprocess Eng. 2020, 25, 345–355. [Google Scholar] [CrossRef]
- Les, F.; Arbonés-Mainar, J.M.; Valero, M.S.; López, V. Pomegranate polyphenols and urolithin A inhibit α-glucosidase, dipeptidyl peptidase-4, lipase, triglyceride accumulation and adipogenesis related genes in 3T3-L1 adipocyte-like cells. J. Ethnopharmacol. 2018, 220, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Abdulrahman, A.O.; Kuerban, A.; Alshehri, Z.A.; Abdulaal, W.H.; Khan, J.A.; Khan, M.I. Urolithins attenuate multiple symptoms of obesity in rats fed on a high-fat diet. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 3337–3348. [Google Scholar] [CrossRef]
- Sala, R.; Mena, P.; Savi, M.; Brighenti, F.; Crozier, A.; Miragoli, M.; Stilli, D.; Del Rio, D. Urolithins at physiological concentrations affect the levels of pro-inflammatory cytokines and growth factor in cultured cardiac cells in hyperglucidic conditions. J. Funct. Foods 2015, 15, 97–105. [Google Scholar] [CrossRef]
- Bobowska, A.; Granica, S.; Filipek, A.; Melzig, M.F.; Moeslinger, T.; Zentek, J.; Kruk, A.; Piwowarski, J.P. Comparative studies of urolithins and their phase II metabolites on macrophage and neutrophil functions. Eur. J. Nutr. 2020, 60, 1957–1972. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.F.; Gurlo, T.; Daval, M.; Huang, C.J.; Matveyenko, A.V.; Butler, P.C.; Costes, S. Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic Β-cells: Protective role of p62-positive cytoplasmic inclusions. Cell Death Differ. 2011, 18, 415–426. [Google Scholar] [CrossRef]
- Rivera, J.F.; Costes, S.; Gurlo, T.; Glabe, C.G.; Butler, P.C. Autophagy defends pancreatic β cells from Human islet amyloid polypeptide-induced toxicity. J. Clin. Investig. 2014, 124, 3489–3500. [Google Scholar] [CrossRef] [PubMed]
- Araújo, A.R.; Reis, R.L.; Pires, R.A. Natural Polyphenols as Modulators of the Fibrillization of Islet Amyloid Polypeptide. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2020; Volume 1250, pp. 159–176. [Google Scholar] [CrossRef]
- Pithadia, A.; Brender, J.R.; Fierke, C.A.; Ramamoorthy, A. Inhibition of IAPP Aggregation and Toxicity by Natural Products and Derivatives. J. Diabetes Res. 2016, 2016, 2046327. [Google Scholar] [CrossRef]
- Bruno, E.; Pereira, C.; Roman, K.P.; Takiguchi, M.; Kao, P.Y.; Nogaj, L.A.; Moffet, D.A. IAPP aggregation and cellular toxicity are inhibited by 1,2,3,4,6-penta-O-galloyl-β-d-glucose. Amyloid 2013, 20, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Huang, J.; Xu, B.; Ou, Z.; Zhang, L.; Lin, X.; Ye, X.; Kong, X.; Long, D.; Sun, X.; et al. Urolithin A attenuates memory impairment and neuroinflammation in APP/PS1 mice. J. Neuroinflammation 2019, 16, 62. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Chae, C.W.; Lim, J.R.; Kim, S.Y.; Yoon, J.H.; Cho, J.H.; Lee, S.J.; et al. Urolithin A suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death Differ. 2021, 28, 184–202. [Google Scholar] [CrossRef] [PubMed]
- Tuohetaerbaike, B.; Zhang, Y.; Tian, Y.; Zhang, N.N.; Kang, J.; Mao, X.; Zhang, Y.; Li, X. Pancreas protective effects of Urolithin A on type 2 diabetic mice induced by high fat and streptozotocin via regulating autophagy and AKT/mTOR signaling pathway. J. Ethnopharmacol. 2020, 250, 112479. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Tian, L.; Dong, H.; Halemahebai, G.; Aisker, G. Urolithin A, a pomegranate metabolite, protects pancreatic β Cells from apoptosis by activating autophagy. J. Ethnopharmacol. 2020, 272, 113628. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-dit-Félix, A.A.; Williams, E.G.; Jha, P.; Sasso, G.L.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Iglesias-Aguirre, C.E.; Cortés-Martín, A.; Ávila-Gálvez, M.Á.; Giménez-Bastida, J.A.; Selma, M.V.; González-Sarrías, A.; Espín, J.C. Main drivers of (poly)phenol effects on human health: Metabolite production and/or gut microbiota-associated metabotypes? Food Funct. 2021, 12, 10324–10355. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raimundo, A.F.; Ferreira, S.; Tomás-Barberán, F.A.; Santos, C.N.; Menezes, R. Urolithins: Diet-Derived Bioavailable Metabolites to Tackle Diabetes. Nutrients 2021, 13, 4285. https://doi.org/10.3390/nu13124285
Raimundo AF, Ferreira S, Tomás-Barberán FA, Santos CN, Menezes R. Urolithins: Diet-Derived Bioavailable Metabolites to Tackle Diabetes. Nutrients. 2021; 13(12):4285. https://doi.org/10.3390/nu13124285
Chicago/Turabian StyleRaimundo, Ana F., Sofia Ferreira, Francisco A. Tomás-Barberán, Claudia N. Santos, and Regina Menezes. 2021. "Urolithins: Diet-Derived Bioavailable Metabolites to Tackle Diabetes" Nutrients 13, no. 12: 4285. https://doi.org/10.3390/nu13124285
APA StyleRaimundo, A. F., Ferreira, S., Tomás-Barberán, F. A., Santos, C. N., & Menezes, R. (2021). Urolithins: Diet-Derived Bioavailable Metabolites to Tackle Diabetes. Nutrients, 13(12), 4285. https://doi.org/10.3390/nu13124285