Vitamin B Supplementation and Nutritional Intake of Methyl Donors in Patients with Chronic Kidney Disease: A Critical Review of the Impact on Epigenetic Machinery


Abstract
1. Introduction
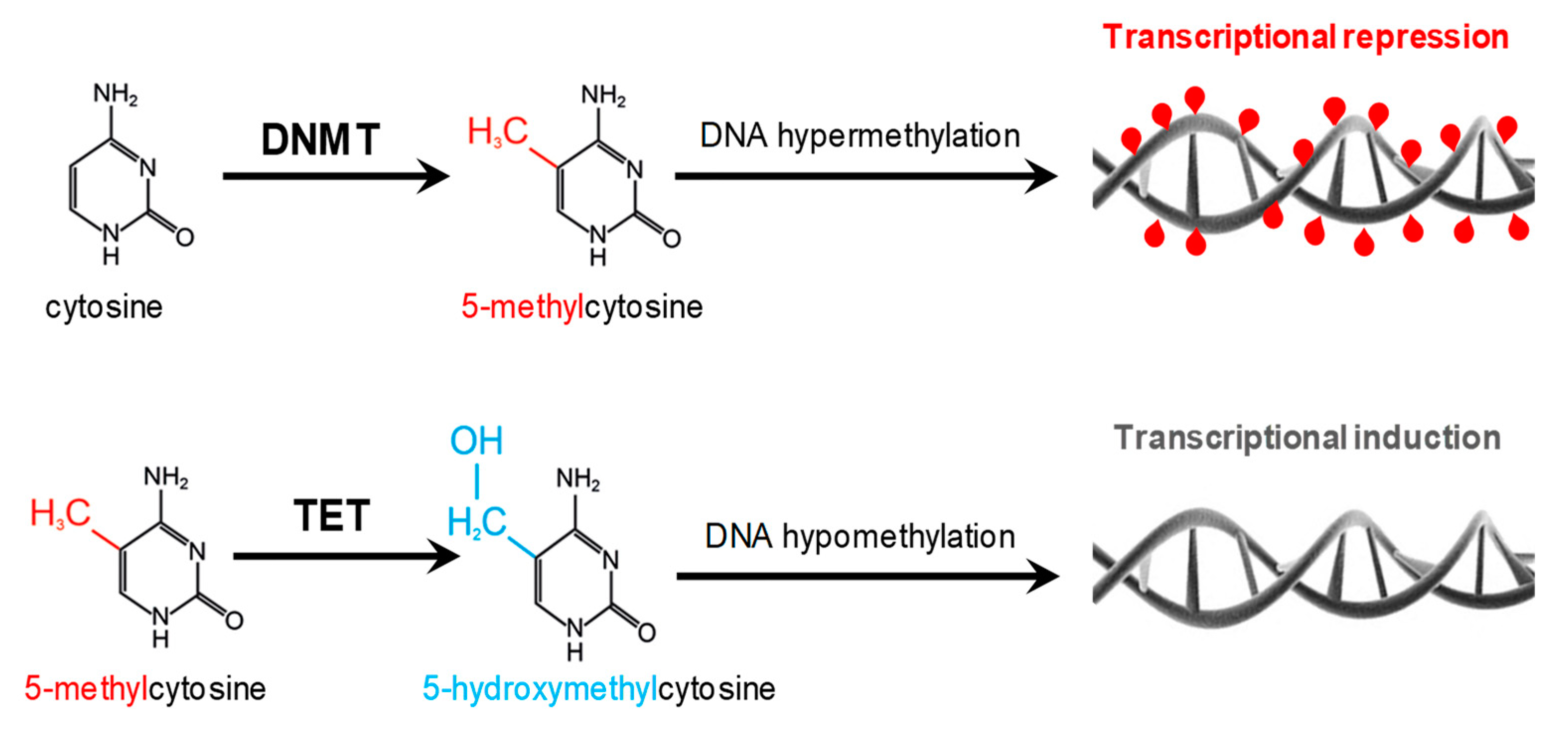
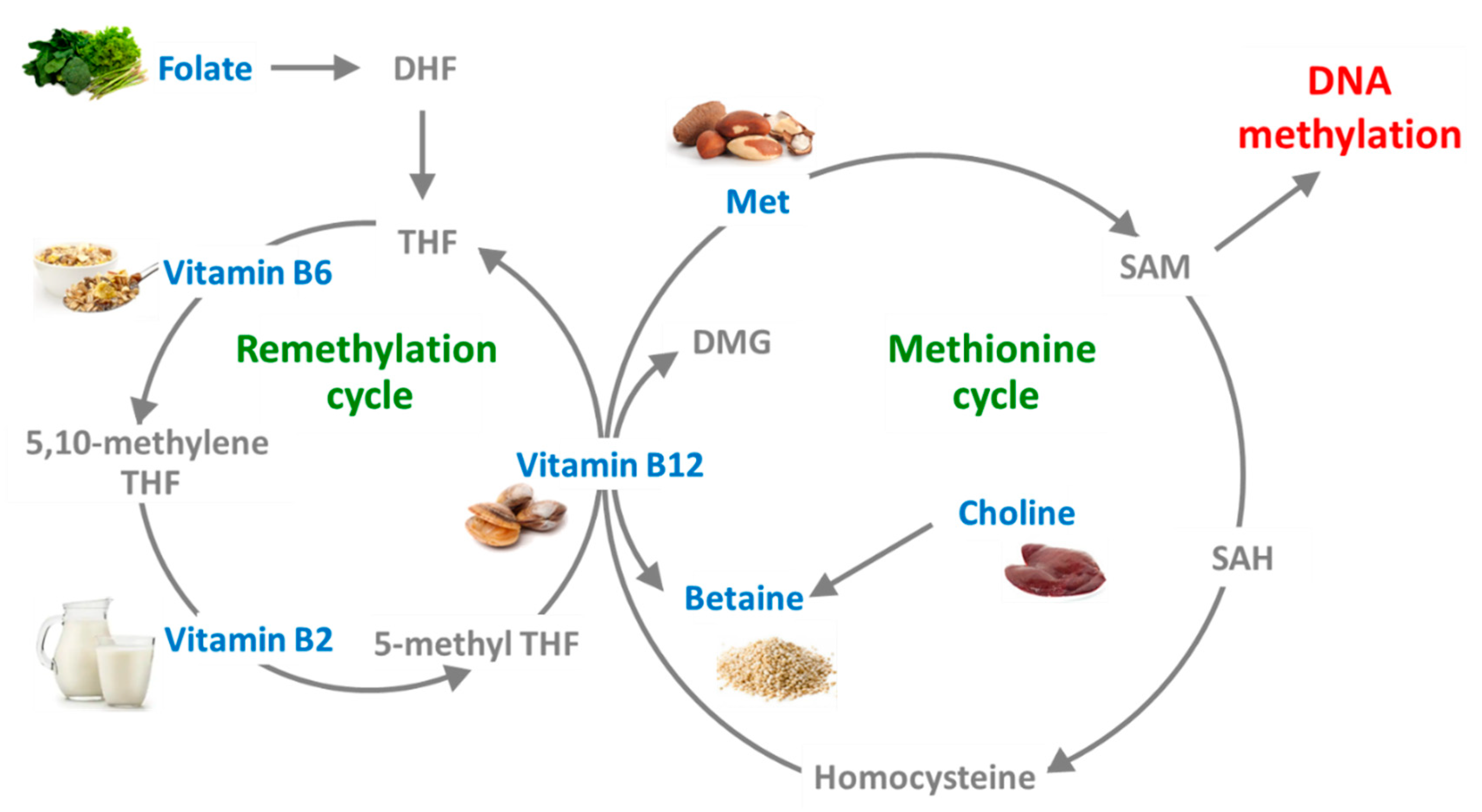
2. Methyl-Donor Mediated Epigenetic Effects in CKD
3. Effects of Supplementation and Dietary Intake of B-Vitamins and Methyl Donors on Epigenetic Changes in Renal Disease
3.1. Folate
3.2. Cobalamin (Vitamin B12)
3.3. Vitamin B6
3.4. Methionine
3.5. Choline
3.6. Betaine
3.7. Other Forms of Vitamin B: The Role of Niacin
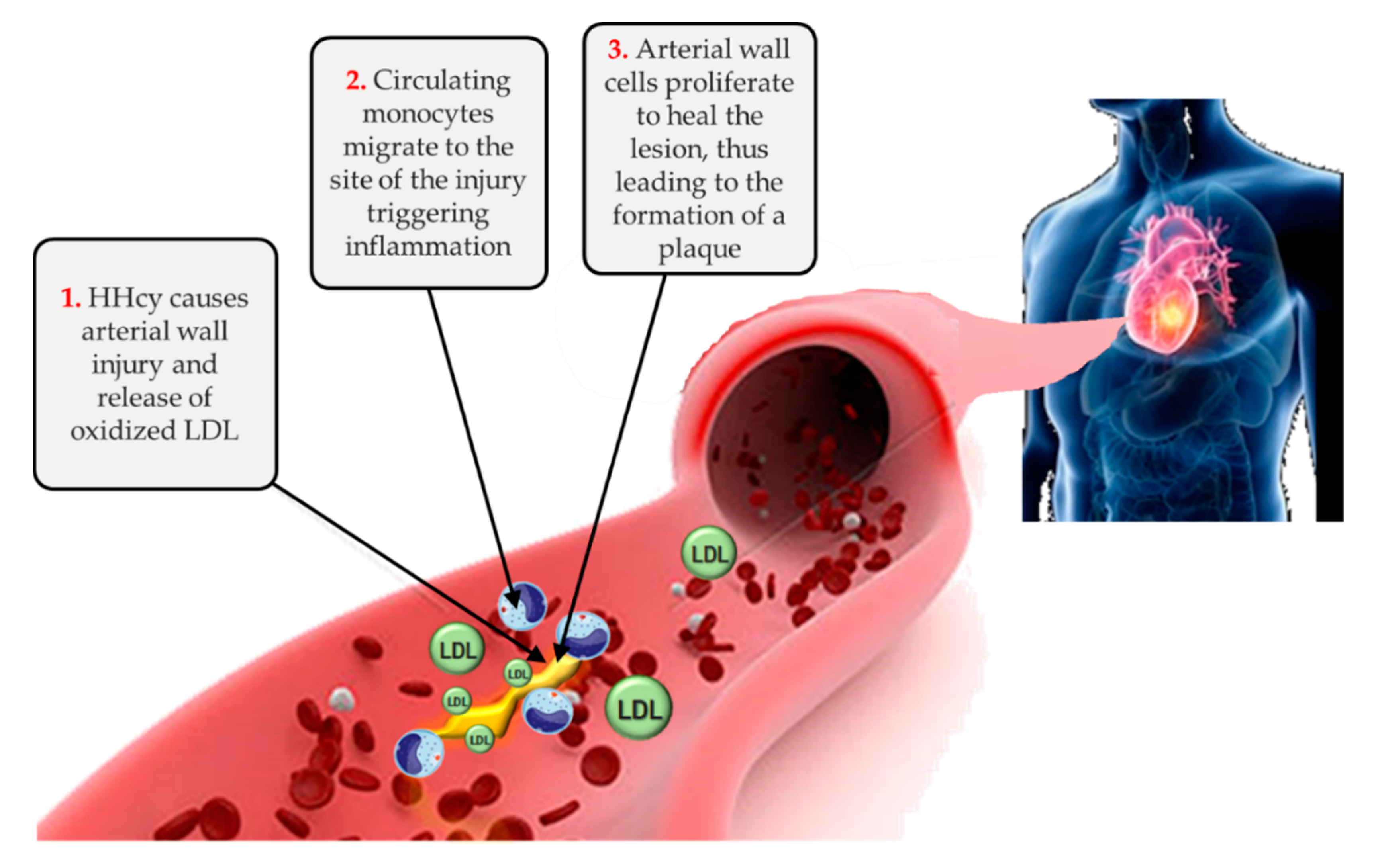
4. New Insights in the Network of Methyl Donors, Hyperhomocysteinemia, Epigenetic Landscape, Cardiovascular Risk and CKD Progression
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saran, R.; Robinson, B.; Abbott, K.C.; Bragg-Gresham, J.; Chen, X.; Gipson, D.; Gu, H.; Hirth, R.A.; Hutton, D.; Jin, Y.; et al. US Renal Data System 2019 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. 2020, 75, A6–A7. [Google Scholar] [CrossRef] [PubMed]
- USRDS. Chapter 4: Cardiovascular Disease in Patients with CKD. Am. J. Kidney Dis. 2019, 73 (Suppl. 1), S79–S98. [Google Scholar] [CrossRef]
- Cianciolo, G.; De Pascalis, A.; Di Lullo, L.; Ronco, C.; Zannini, C.; La Manna, G. Folic Acid and Homocysteine in Chronic Kidney Disease and Cardiovascular Disease Progression: Which Comes First? Cardiorenal Med. 2017, 7, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Suliman, M.E.; Lindholm, B.; Bárány, P.; Qureshi, A.R.; Stenvinkel, P. Homocysteine-lowering is not a primary target for cardiovascular disease prevention in chronic kidney disease patients. Semin. Dial. 2007, 20, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Soto, G.A.; Madrid, T. Is folic acid supplementation useful for chronic kidney disease? Medwave 2016, 16 (Suppl. 5), e6591. [Google Scholar] [CrossRef]
- Li, Y.; Spence, J.D.; Wang, X.; Huo, Y.; Xu, X.; Qin, X. Effect of Vitamin B12 Levels on the Association Between Folic Acid Treatment and CKD Progression: A Post Hoc Analysis of a Folic Acid Interventional Trial. Am. J. Kidney Dis. 2020, 75, 325–332. [Google Scholar] [CrossRef]
- Cianciolo, G.; La Manna, G.; Colì, L.; Donati, G.; D’Addio, F.; Persici, E.; Comai, G.; Wratten, M.; Dormi, A.; Mantovani, V.; et al. 5-methyltetrahydrofolate administration is associated with prolonged survival and reduced inflammation in ESRD patients. Am. J. Nephrol. 2008, 28, 941–948. [Google Scholar] [CrossRef]
- Capelli, I.; Cianciolo, G.; Gasperoni, L.; Zappulo, F.; Tondolo, F.; Cappuccilli, M.; La Manna, G. Folic Acid and Vitamin B12 Administration in CKD, Why Not? Nutrients 2019, 11, 383. [Google Scholar] [CrossRef]
- Suliman, M.; Stenvinkel, P.; Qureshi, A.R.; Kalantar-Zadeh, K.; Bárány, P.; Heimbürger, O.; Vonesh, E.F.; Lindholm, B. The reverse epidemiology of plasma total homocysteine as a mortality risk factor is related to the impact of wasting and inflammation. Nephrol. Dial. Transplant. 2007, 22, 209–217. [Google Scholar] [CrossRef]
- Chmielewski, M.; Verduijn, M.; Drechsler, C.; Lindholm, B.; Stenvinkel, P.; Rutkowski, B.; Boeschoten, E.W.; Krediet, R.T.; Dekker, F.W. Low cholesterol in dialysis patients--causal factor for mortality or an effect of confounding? Nephrol. Dial. Transplant. 2011, 26, 3325–3331. [Google Scholar] [CrossRef]
- Tsirpanlis, G.; Boufidou, F.; Zoga, M.; Triantafyllis, G.; Fatourou, A.; Stamatelou, K.; Bleta, A.; Petrihou, C.; Chatzipanagiotou, S.; Nicolaou, C. Low cholesterol along with inflammation predicts morbidity and mortality in hemodialysis patients. Hemodial. Int. 2009, 13, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Cianciolo, G.; La Manna, G.; Donati, G.; Persici, E.; Dormi, A.; Cappuccilli, M.L.; Corsini, S.; Fattori, R.; Russo, V.; Nastasi, V.; et al. Coronary calcifications in end-stage renal disease patients: A new link between osteoprotegerin, diabetes and body mass index? Blood Purif. 2010, 29, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Akgul, A.; Bilgic, A.; Sezer, S.; Arat, Z.; Ozdemir, F.N.; Haberal, M. Low total plasma homocysteine level in relation to malnutrition, inflammation, and outcome in hemodialysis patients. J. Ren. Nutr. 2008, 18, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Block, G.; Humphreys, M.H.; McAllister, C.J.; Kopple, J.D. A low, rather than a high, total plasma homocysteine is an indicator of poor outcome in hemodialysis patients. J. Am. Soc. Nephrol. 2004, 15, 442–453. [Google Scholar] [CrossRef]
- Ingrosso, D.; Cimmino, A.; Perna, A.F.; Masella, L.; De Santo, N.G.; De Bonis, M.L.; Vacca, M.; D’Esposito, M.; D’Urso, M.; Galletti, P.; et al. Folate treatment and unbalanced methylation and changes of allelic expression induced by hyperhomocysteinaemia in patients with uraemia. Lancet 2003, 361, 1693–1699. [Google Scholar] [CrossRef]
- Kanbay, M.; Yerlikaya, A.; Sag, A.A.; Ortiz, A.; Kuwabara, M.; Covic, A.; Wiecek, A.; Stenvinkel, P.; Afsar, B. A journey from microenvironment to macroenvironment: The role of metaflammation and epigenetic changes in cardiorenal disease. Clin. Kidney J. 2019, 12, 861–870. [Google Scholar] [CrossRef]
- La Manna, G.; Cappuccilli, M.L.; Cianciolo, G.; Conte, D.; Comai, G.; Carretta, E.; Scolari, M.P.; Stefoni, S. Cardiovascular disease in kidney transplant recipients: The prognostic value of inflammatory cytokine genotypes. Transplantation 2010, 89, 1001–1008. [Google Scholar] [CrossRef]
- Pushpakumar, S.; Kundu, S.; Narayanan, N.; Sen, U. DNA hypermethylation in hyperhomocysteinemia contributes to abnormal extracellular matrix metabolism in the kidney. FASEB J. 2015, 29, 4713–4725. [Google Scholar] [CrossRef]
- Smyth, L.J.; McKay, G.J.; Maxwell, A.P.; McKnight, A.J. DNA hypermethylation and DNA hypomethylation is present at different loci in chronic kidney disease. Epigenetics 2014, 9, 366–376. [Google Scholar] [CrossRef]
- Yang, J.; Fang, P.; Yu, D.; Zhang, L.; Zhang, D.; Jiang, X.; Yang, W.Y.; Bottiglieri, T.; Kunapuli, S.P.; Yu, J.; et al. Chronic Kidney Disease Induces Inflammatory CD40+ Monocyte Differentiation via Homocysteine Elevation and DNA Hypomethylation. Circ. Res. 2016, 119, 1226–1241. [Google Scholar] [CrossRef]
- Khalil, H.; Tazi, M.; Caution, K.; Ahmed, A.; Kanneganti, A.; Assani, K.; Kopp, B.; Marsh, C.; Dakhlallah, D.; Amer, A.O. Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics 2016, 11, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Pascual, J.L.; Marchant, V.; Rodrigues-Diez, R.; Dolade, N.; Suarez-Alvarez, B.; Kerr, B.; Valdivielso, J.M.; Ruiz-Ortega, M.; Rayego-Mateos, S. Epigenetic Modification Mechanisms Involved in Inflammation and Fibrosis in Renal Pathology. Mediators Inflamm. 2018, 2018, 2931049. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.; Rakyan, V.K. The methylome: Approaches for global DNA methylation profiling. Trends Genet. 2008, 24, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S. Choline, Other Methyl-Donors and Epigenetics. Nutrients 2017, 9, 445. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; van Vliet, P.P.; Andelfinger, G.; Puceat, M. Role of Epigenetics in Cardiac Development and Congenital Diseases. Physiol. Rev. 2018, 98, 2453–2475. [Google Scholar] [CrossRef]
- Aleksandrova, K.; Romero-Mosquera, B.; Hernandez, V. Diet, Gut Microbiome and Epigenetics: Emerging Links with Inflammatory Bowel Diseases and Prospects for Management and Prevention. Nutrients 2017, 9, 962. [Google Scholar] [CrossRef]
- Sharp, G.C.; Relton, C.L. Epigenetics and noncommunicable diseases. Epigenomics 2017, 9, 789–791. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Ingrosso, D.; Perna, A.F. Epigenetics in hyperhomocysteinemic states. A special focus on uremia. Biochim. Biophys. Acta 2009, 1790, 892–899. [Google Scholar] [CrossRef]
- Cianciolo, G.; Cappuccilli, M.; La Manna, G. The Hydrogen Sulfide-Vitamin B12-Folic Acid Axis: An Intriguing Issue in Chronic Kidney Disease. A Comment on Toohey JI: “Possible Involvement of Hydrosulfide in B12-Dependent Methyl Group Transfer”. Molecules 2017, 22, 1216. [Google Scholar] [CrossRef] [PubMed]
- Trimmer, E.E. Methylenetetrahydrofolate reductase: Biochemical characterization and medical significance. Curr. Pharm. Des. 2013, 19, 2574–2593. [Google Scholar] [CrossRef] [PubMed]
- Födinger, M.; Hörl, W.H.; Sunder-Plassmann, G. Molecular biology of 5,10-methylenetetrahydrofolate reductase. J. Nephrol. 2000, 13, 20–33. [Google Scholar] [PubMed]
- Ghattas, M.; El-Shaarawy, F.; Mesbah, N.; Abo-Elmatty, D. DNA methylation status of the methylenetetrahydrofolate reductase gene promoter in peripheral blood of end-stage renal disease patients. Mol. Biol. Rep. 2014, 41, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.Y.; Sun, C.Y.; Lee, C.C.; Wu, I.W.; Hsu, H.J.; Wu, M.S. Global DNA methylation not increased in chronic hemodialysis patients: A case-control study. Ren. Fail. 2012, 34, 1195–1199. [Google Scholar] [CrossRef]
- Ghigolea, A.B.; Moldovan, R.A.; Gherman-Caprioara, M. DNA methylation: Hemodialysis versus hemodiafiltration. Ther. Apher. Dial. 2015, 19, 119–124. [Google Scholar] [CrossRef]
- Mafra, D.; Esgalhado, M.; Borges, N.A.; Cardozo, L.F.M.F.; Stockler-Pinto, M.B.; Craven, H.; Buchanan, S.J.; Lindholm, B.; Stenvinkel, P.; Shiels, P.G. Methyl Donor Nutrients in Chronic Kidney Disease: Impact on the Epigenetic Landscape. J. Nutr. 2019, 149, 372–380. [Google Scholar] [CrossRef]
- Ebbing, M.; Bleie, Ø.; Ueland, P.M.; Nordrehaug, J.E.; Nilsen, D.W.; Vollset, S.E.; Refsum, H.; Pedersen, E.K.; Nygård, O. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: A randomized controlled trial. JAMA 2008, 300, 795–804. [Google Scholar] [CrossRef]
- Perna, A.F.; De Santo, N.G.; Ingrosso, D. Adverse effects of hyperhomocysteinemia and their management by folic acid. Miner. Electrolyte Metab. 1997, 23, 174–178. [Google Scholar]
- Doshi, S.N.; McDowell, I.F.; Moat, S.J.; Payne, N.; Durrant, H.J.; Lewis, M.J.; Goodfellow, J. Folic acid improves endothelial function in coronary artery disease via mechanisms largely independent of homocysteine lowering. Circulation 2002, 105, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Miguelsanz, J.; Vallecillo, N.; Garrido, F.; Reytor, E.; Pérez-Sala, D.; Pajares, M.A. Betaine homocysteine S-methyltransferase emerges as a new player of the nuclear methionine cycle. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1165–1182. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Lanza, D.; Sepe, I.; Conzo, G.; Altucci, L.; Ingrosso, D. Altered folate receptor 2 expression in uraemic patients on haemodialysis: Implications for folate resistance. Nephrol. Dial. Transplant. 2013, 28, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; He, G.W. Imbalance of Homocysteine and H2S: Significance, Mechanisms, and Therapeutic Promise in Vascular Injury. Oxid. Med. Cell Longev. 2019, 2019, 7629673. [Google Scholar] [CrossRef] [PubMed]
- Jamison, R.L.; Hartigan, P.; Kaufman, J.S.; Goldfarb, D.S.; Warren, S.R.; Guarino, P.D.; Gaziano, J.M.; Veterans Affairs Site Investigators. Effect of homocysteine lowering on mortality and vascular disease in advanced chronic kidney disease and end-stage renal disease: A randomized controlled trial. JAMA 2007, 298, 1163–1170. [Google Scholar] [CrossRef]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef]
- Qin, X.; Huo, Y.; Langman, C.B.; Hou, F.; Chen, Y.; Matossian, D.; Xu, X.; Wang, X. Folic acid therapy and cardiovascular disease in ESRD or advanced chronic kidney disease: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2011, 6, 482–488. [Google Scholar] [CrossRef]
- Qin, X.; Huo, Y.; Xie, D.; Hou, F.; Xu, X.; Wang, X. Homocysteine-lowering therapy with folic acid is effective in cardiovascular disease prevention in patients with kidney disease: A meta-analysis of randomized controlled trials. Clin. Nutr. 2013, 32, 722–727. [Google Scholar] [CrossRef]
- Perna, A.F.; Ingrosso, D.; Violetti, E.; Luciano, M.G.; Sepe, I.; Lanza, D.; Capasso, R.; Ascione, E.; Raiola, I.; Lombardi, C.; et al. Hyperhomocysteinemia in uremia—A red flag in a disrupted circuit. Semin. Dial. 2009, 22, 351–356. [Google Scholar] [CrossRef]
- Mahajan, A.; Sapehia, D.; Thakur, S.; Mohanraj, P.S.; Bagga, R.; Kaur, J. Effect of imbalance in folate and vitamin B12 in maternal/parental diet on global methylation and regulatory miRNAs. Sci. Rep. 2019, 9, 17602. [Google Scholar] [CrossRef]
- Warren, M.J.; Raux, E.; Schubert, H.L.; Escalante-Semerena, J.C. The biosynthesis of adenosylcobalamin (vitamin B12). Nat. Prod. Rep. 2002, 19, 390–412. [Google Scholar] [CrossRef]
- Farquharson, J.; Adams, J.F. The forms of vitamin B 12 in foods. Br. J. Nutr. 1976, 36, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Greibe, E.; Mahalle, N.; Bhide, V.; Heegaard, C.W.; Naik, S.; Nexo, E. Increase in circulating holotranscobalamin after oral administration of cyanocobalamin or hydroxocobalamin in healthy adults with low and normal cobalamin status. Eur. J. Nutr. 2018, 57, 2847–2855. [Google Scholar] [CrossRef] [PubMed]
- Randaccio, L.; Geremia, S.; Demitri, N.; Wuerges, J. Vitamin B12: Unique metalorganic compounds and the most complex vitamins. Molecules 2010, 15, 3228–3259. [Google Scholar] [CrossRef] [PubMed]
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B12, folate, and the methionine remethylation cycle—Biochemistry, pathways, and regulation. J. Inherit. Metab. Dis. 2019, 42, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Buccianti, G.; Bamonti Catena, F.; Patrosso, C.; Corghi, E.; Novembrino, C.; Baragetti, I.; Lando, G.; De Franceschi, M.; Maiolo, A.T. Reduction of the homocysteine plasma concentration by intravenously administered folinic acid and vitamin B(12) in uraemic patients on maintenance haemodialysis. Am. J. Nephrol. 2001, 21, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Stabler, S.P. Vitamin B12 deficiency. N. Engl. J. Med. 2013, 368, 149–160. [Google Scholar] [CrossRef]
- Green, R.; Allen, L.H.; Bjørke-Monsen, A.L.; Brito, A.; Guéant, J.L.; Miller, J.W.; Molloy, A.M.; Nexo, E.; Stabler, S.; Toh, B.H.; et al. Vitamin B12 deficiency. Nat. Rev. Dis. Primers 2017, 3, 17040. [Google Scholar] [CrossRef]
- Langan, R.C.; Goodbred, A.J.; Luke, S.; Residency, M. Vitamin B12 Deficiency: Recognition and Management. Am. Fam. Physician 2017, 96, 384–389. [Google Scholar]
- McMahon, G.M.; Hwang, S.J.; Tanner, R.M.; Jacques, P.F.; Selhub, J.; Muntner, P.; Fox, C.S. The association between vitamin B12, albuminuria and reduced kidney function: An observational cohort study. BMC Nephrol. 2015, 16, 7. [Google Scholar] [CrossRef]
- Soohoo, M.; Ahmadi, S.F.; Qader, H.; Streja, E.; Obi, Y.; Moradi, H.; Rhee, C.M.; Kim, T.H.; Kovesdy, C.P.; Kalantar-Zadeh, K. Association of serum vitamin B12 and folate with mortality in incident hemodialysis patients. Nephrol. Dial. Transplant. 2017, 32, 1024–1032. [Google Scholar] [CrossRef]
- Wickramasinghe, S.N.; Fida, S. Correlations between holo-transcobalamin II, holo-haptocorrin, and total B12 in serum samples from healthy subjects and patients. J. Clin. Pathol. 1993, 46, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Hannibal, L.; Lysne, V.; Bjørke-Monsen, A.L.; Behringer, S.; Grünert, S.C.; Spiekerkoetter, U.; Jacobsen, D.W.; Blom, H.J. Biomarkers and Algorithms for the Diagnosis of Vitamin B12 Deficiency. Front. Mol. Biosci. 2016, 3, 27, published correction appears in Front. Mol. Biosci. 2017, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Devalia, V.; Hamilton, M.S.; Molloy, A.M. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br. J. Haematol. 2014, 166, 496–513. [Google Scholar] [CrossRef] [PubMed]
- Heil, S.G.; de Jonge, R.; de Rotte, M.C.; van Wijnen, M.; Heiner-Fokkema, R.M.; Kobold, A.C.; Pekelharing, J.M.; Adriaansen, H.J.; Sanders, E.; Trienekens, P.H.; et al. Screening for metabolic vitamin B12 deficiency by holotranscobalamin in patients suspected of vitamin B12 deficiency: A multicentre study. Ann. Clin. Biochem. 2012, 49, 184–189. [Google Scholar] [CrossRef]
- Green, R. Vitamin B12 deficiency from the perspective of a practicing hematologist. Blood 2017, 129, 2603–2611. [Google Scholar] [CrossRef]
- Saifan, C.; Samarneh, M.; Shtaynberg, N.; Nasr, R.; El-Charabaty, E.; El-Sayegh, S. Treatment of confirmed B12 deficiency in hemodialysis patients improves Epogen® requirements. Int. J. Nephrol. Renovasc. Dis. 2013, 6, 89–93. [Google Scholar] [CrossRef]
- Delpre, G.; Stark, P.; Niv, Y. Sublingual therapy for cobalamin deficiency as an alternative to oral and parenteral cobalamin supplementation. Lancet 1999, 354, 740–741. [Google Scholar] [CrossRef]
- Strong, A.P.; Haeusler, S.; Weatherall, M.; Krebs, J. Sublingual vitamin B12 compared to intramuscular injection in patients with type 2 diabetes treated with metformin: A randomised trial. N. Z. Med. J. 2016, 129, 67–75. [Google Scholar]
- Bensky, M.J.; Ayalon-Dangur, I.; Ayalon-Dangur, R.; Naamany, E.; Gafter-Gvili, A.; Koren, G.; Shiber, S. Comparison of sublingual vs. intramuscular administration of vitamin B12 for the treatment of patients with vitamin B12 deficiency. Drug Deliv. Transl. Res. 2019, 9, 625–630. [Google Scholar] [CrossRef]
- Ink, S.L.; Henderson, L.M. Vitamin B6 metabolism. Annu. Rev. Nutr. 1984, 4, 455–470. [Google Scholar] [CrossRef]
- Johansson, S.; Lindstedt, S.; Tiselius, H.G. Metabolic interconversions of different forms of vitamin B6. J. Biol. Chem. 1974, 249, 6040–6046. [Google Scholar] [PubMed]
- Riccio, F.; Mennella, C.; Fogliano, V. Effect of cooking on the concentration of Vitamins B in fortified meat products. J. Pharm. Biomed. Anal. 2006, 41, 1592–1595. [Google Scholar] [CrossRef] [PubMed]
- House, A.A.; Eliasziw, M.; Cattran, D.C.; Churchill, D.N.; Oliver, M.J.; Fine, A.; Dresser, G.K.; Spence, J.D. Effect of B-vitamin therapy on progression of diabetic nephropathy: A randomized controlled trial. JAMA 2010, 303, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, Y.; Schöttker, B.; Brenner, H. Vitamin D status and epigenetic-based mortality risk score: Strong independent and joint prediction of all-cause mortality in a population-based cohort study. Clin. Epigenetics 2018, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Morrone, L.F.; Bolasco, P.; Camerini, C.; Cianciolo, G.; Cupisti, A.; Galassi, A.; Mazzaferro, S.; Russo, D.; Russo, L.; Cozzolino, M. Vitamin D in patients with chronic kidney disease: A position statement of the Working Group "Trace Elements and Mineral Metabolism" of the Italian Society of Nephrology. J. Nephrol. 2016, 29, 305–328. [Google Scholar] [CrossRef]
- Obeid, R.; Hübner, U.; Bodis, M.; Graeber, S.; Geisel, J. Effect of adding B-vitamins to vitamin D and calcium supplementation on CpG methylation of epigenetic aging markers. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 411–417. [Google Scholar] [CrossRef]
- Herrmann, W.; Obeid, R. Hyperhomocysteinemia and response of methionine cycle intermediates to vitamin treatment in renal patients. Clin. Chem. Lab. Med. 2005, 43, 1039–1047. [Google Scholar] [CrossRef]
- Zhang, N. Role of methionine on epigenetic modification of DNA methylation and gene expression in animals. Anim. Nutr. 2018, 4, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Mladenović, D.; Radosavljević, T.; Hrnčić, D.; Rasic-Markovic, A.; Stanojlović, O. The effects of dietary methionine restriction on the function and metabolic reprogramming in the liver and brain—Implications for longevity. Rev. Neurosci. 2019, 30, 581–593. [Google Scholar] [CrossRef]
- Plaisance, E.P.; Greenway, F.L.; Boudreau, A.; Hill, K.L.; Johnson, W.D.; Krajcik, R.A.; Perrone, C.E.; Orentreich, N.; Cefalu, W.T.; Gettys, T.W. Dietary methionine restriction increases fat oxidation in obese adults with metabolic syndrome. J. Clin. Endocrinol. Metab. 2011, 96, E836–E840. [Google Scholar] [CrossRef]
- Bárcena, C.; Quirós, P.M.; Durand, S.; Mayoral, P.; Rodríguez, F.; Caravia, X.M.; Mariño, G.; Garabaya, C.; Fernández-García, M.T.; Kroemer, G.; et al. Methionine Restriction Extends Lifespan in Progeroid Mice and Alters Lipid and Bile Acid Metabolism. Cell Rep. 2018, 24, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- McIsaac, R.S.; Lewis, K.N.; Gibney, P.A.; Buffenstein, R. From yeast to human: Exploring the comparative biology of methionine restriction in extending eukaryotic life span. Ann. N. Y. Acad. Sci. 2016, 1363, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Pradas, I.; Jové, M.; Cabré, R.; Ayala, V.; Mota-Martorell, N.; Pamplona, R. Effects of Aging and Methionine Restriction on Rat Kidney Metabolome. Metabolites 2019, 9, 280. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Wang, W.J.; Liu, J.Q.; Song, Y.H.; Li, P.; Sun, X.; Cai, G.Y.; Chen, X. Methionine restriction delays senescence and suppresses the senescence-associated secretory phenotype in the kidney through endogenous hydrogen sulfide. Cell Cycle 2019, 18, 1573–1587. [Google Scholar] [CrossRef]
- Cooke, D.; Ouattara, A.; Ables, G.P. Dietary methionine restriction modulates renal response and attenuates kidney injury in mice. FASEB J. 2018, 32, 693–702. [Google Scholar] [CrossRef]
- Grant, L.; Lees, E.K.; Forney, L.A.; Mody, N.; Gettys, T.; Brown, P.A.; Wilson, H.M.; Delibegovic, M. Methionine restriction improves renal insulin signalling in aged kidneys. Mech. Ageing Dev. 2016, 157, 35–43. [Google Scholar] [CrossRef]
- IoM (US). Choline. In Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline; National Academies Press: Washington, DC, USA, 1998. [Google Scholar]
- Mun, J.G.; Legette, L.L.; Ikonte, C.J.; Mitmesser, S.H. Choline and DHA in Maternal and Infant Nutrition: Synergistic Implications in Brain and Eye Health. Nutrients 2019, 11, 1125. [Google Scholar] [CrossRef]
- Rennick, B.; Acara, M.; Hysert, P.; Mookerjee, B. Choline loss during hemodialysis: Homeostatic control of plasma choline concentrations. Kidney Int. 1976, 10, 329–335. [Google Scholar] [CrossRef]
- Ilcol, Y.O.; Dilek, K.; Yurtkuran, M.; Ulus, I.H. Changes of plasma free choline and choline-containing compounds’ concentrations and choline loss during hemodialysis in ESRD patients. Clin. Biochem. 2002, 35, 233–239. [Google Scholar] [CrossRef]
- Shafi, T.; Powe, N.R.; Meyer, T.W.; Hwang, S.; Hai, X.; Melamed, M.L.; Banerjee, T.; Coresh, J.; Hostetter, T.H. Trimethylamine N-Oxide and Cardiovascular Events in Hemodialysis Patients. J. Am. Soc. Nephrol. 2017, 28, 321–331. [Google Scholar] [CrossRef]
- Romano, K.A.; Martinez-Del Campo, A.; Kasahara, K.; Chittim, C.L.; Vivas, E.I.; Amador-Noguez, D.; Balskus, E.P.; Rey, F.E. Metabolic, Epigenetic, and Transgenerational Effects of Gut Bacterial Choline Consumption. Cell Host Microbe 2017, 22, 279–290.e7. [Google Scholar] [CrossRef]
- Pummer, S.; Dantzler, W.H.; Lien, Y.H.; Moeckel, G.W.; Völker, K.; Silbernagl, S. Reabsorption of betaine in Henle’s loops of rat kidney in vivo. Am. J. Physiol. Renal Physiol. 2000, 278, F434–F439. [Google Scholar] [CrossRef] [PubMed]
- Kempson, S.A.; Vovor-Dassu, K.; Day, C. Betaine transport in kidney and liver: Use of betaine in liver injury. Cell Physiol. Biochem. 2013, 32, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Kempson, S.A. Differential activation of system A and betaine/GABA transport in MDCK cell membranes by hypertonic stress. Biochim. Biophys. Acta 1998, 1372, 117–123. [Google Scholar] [CrossRef][Green Version]
- Kirkland, J.B.; Meyer-Ficca, M.L. Niacin. Adv. Food Nutr. Res. 2018, 83, 83–149. [Google Scholar] [PubMed]
- Wing, M.R.; Devaney, J.M.; Joffe, M.M.; Xie, D.; Feldman, H.I.; Dominic, E.A.; Guzman, N.J.; Ramezani, A.; Susztak, K.; Herman, J.G.; et al. DNA methylation profile associated with rapid decline in kidney function: Findings from the CRIC study. Nephrol. Dial. Transplant. 2014, 29, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, W.; McGoohan, S.; Zeisberg, E.M.; Müller, G.A.; Kalbacher, H.; Salant, D.J.; Müller, C.A.; Kalluri, R.; Zeisberg, M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 2010, 16, 544–550. [Google Scholar] [CrossRef]
- Tampe, B.; Steinle, U.; Tampe, D.; Carstens, J.L.; Korsten, P.; Zeisberg, E.M.; Müller, G.A.; Kalluri, R.; Zeisberg, M. Low-dose hydralazine prevents fibrosis in a murine model of acute kidney injury-to-chronic kidney disease progression. Kidney Int. 2017, 91, 157–176. [Google Scholar] [CrossRef]
- Spence, J.D.; Urquhart, B.L.; Bang, H. Effect of renal impairment on atherosclerosis: Only partially mediated by homocysteine. Nephrol. Dial. Transplant. 2016, 31, 937–944. [Google Scholar] [CrossRef]
- Di Giuseppe, D.; Priora, R.; Coppo, L.; Ulivelli, M.; Bartalini, S.; Summa, D.; Margaritis, A.; Frosali, S.; Di Simplicio, P. The control of hyperhomocysteinemia through thiol exchange mechanisms by mesna. Amino Acids 2014, 46, 429–439. [Google Scholar] [CrossRef]
- Spence, J.D. Effects of the intestinal microbiome on constituents of red meat and egg yolks: A new window opens on nutrition and cardiovascular disease. Can. J. Cardiol. 2014, 30, 150–151. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.N.; Bostom, A.G.; Levey, A.S.; Rosenberg, I.H.; Selhub, J.; Pierratos, A. Plasma total homocysteine levels among patients undergoing nocturnal versus standard hemodialysis. J. Am. Soc. Nephrol. 2002, 13, 265–268. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Foods | Content | RDA (%) | |
|---|---|---|---|
| Folate | Asparagus | 263 μg/100 g | 66 |
| Cooked spinaches | 262 μg/100 g | 66 | |
| Cooked lentils | 179 μg/100 g | 45 | |
| Black eyed peas | 179 μg/100 g | 45 | |
| Romaine lettuce | 114 μg/100 g | 29 | |
| Great grains cereals | 114 μg/100 g | 29 | |
| Cooked broccoli | 78 μg/100 g | 20 | |
| Sunflower seeds | 76 μg/100 g | 19 | |
| Fresh orange juice | 75 μg/100 g | 19 | |
| Cooked beets | 69 μg/100 g | 17 | |
| Kidney beans | 65 μg/100 g | 16 | |
| Vitamin B12 | Clams | 98.9 μg/100 g | 4944 |
| Liver | 85.6 μg/100 g | 4280 | |
| Fortified cereals | 20.3 μg/100 g | 1017 | |
| Mackerel | 19 μg/100 g | 949 | |
| Beef | 7.5 μg/100 g | 376 | |
| Crab | 6.7 μg/100 g | 335 | |
| Swiss cheese | 3 μg/100 g | 150 | |
| Fortified tofu | 1.4 μg/100 g | 69 | |
| Eggs (whole) | 0.8 μg/100 g | 41 | |
| Skimmed milk | 0.5 μg/100 g | 26 | |
| Vitamin B6 | Cornflakes/branflakes | 1.4 mg/100 g | 107 |
| Lean pork meat | 0.72 mg/100 g | 55.3 | |
| Lean rump meat | 0.65 mg/100 g | 49.8 | |
| Chicken/turkey breast | 0.63 mg/100 g | 48.6 | |
| Lamb’s kidney | 0.56 mg/100 g | 43.1 | |
| Calf’s liver | 0.48 mg/100 g | 36.9 | |
| Lean minced beef | 0.42 mg/100 g | 32.3 | |
| Avocado | 0.36 mg/100 g | 27.7 | |
| Grilled sardines | 0.36 mg/100 g | 27.7 | |
| Mackerel/plaice | 0.27 mg/100 g | 20.9 | |
| Pomegranate | 10.26 mg/100 g | 20.1 | |
| Methionine | Brazil nuts | 1124 mg/100 g | 154 |
| Poultry | 931 mg/100 g | 128 | |
| Red meat | 905 mg/100 g | 124 | |
| Tuna | 885 mg/100 g | 122 | |
| Pork meat | 850 mg/100 g | 117 | |
| Eggs (whole) | 332 mg/100 g | 45 | |
| Ricotta cheese | 284 mg/100 g | 39 | |
| Tofu | 211 mg/100 g | 29 | |
| Large white beans | 146 mg/100 g | 20 | |
| Quinoa | 96 mg/100 g | 13 | |
| Milk | 88 mg/100 g | 12 | |
| Choline | Beef liver | 350 mg/100 g | 63 |
| Chicken liver | 330 mg/100 g | 60 | |
| Hard boiled eggs | 230 mg/100 g | 42 | |
| Smoked salmon | 220 mg/100 g | 40 | |
| Cooked salmon | 91 mg/100 g | 17 | |
| Soy protein powder | 86 mg/100 g | 16 | |
| Roasted chicken | 79 mg/100 g | 14 | |
| Peanut butter | 66 mg/100 g | 12 | |
| Almonds | 52 mg/100 g | 29 | |
| Cruciferous vegetables | 40 mg/100 g | 7 | |
| Betaine | Quinoa | 630 mg/100 g | 97 |
| Rye | 146 mg/100 g | 60 | |
| Beets | 129 mg/100 g | 20 | |
| White bread | 102 mg/100 g | 16 | |
| Spinaches | 89 mg/100 g | 14 | |
| Bulgur | 83 mg/100 g | 13 | |
| Sweet potato | 34.6 mg/100 g | 5.3 | |
| Veal | 33.9 mg/100 g | 5.2 | |
| Oat flour | 30.7 mg/100 g | 4.7 | |
| Tilapia | 26.3 mg/100 g | 4 |
| Study, Year | Design, Duration | Population (n) | Treatment | Homocysteine Decrease | Achievement of Endpoints |
|---|---|---|---|---|---|
| Cianciolo, 2008 [8] | Randomized prospective study, 55 months | 341 hemodialysis patients | Group A treated with 50 mg i.v. 5-MTHF vs. Group B treated with 5 mg/day oral folic acid (both groups also received i.v. vitamins B6 and B12) | About 50% within the first 6 months of treatment in both groups | Treatment with 5-MTHF reduced inflammation (lower CRP) and increased overall survival rate |
| Qin, 2011 [46] | Meta-analysis of RCTs from January 1966 to August 2010 | 3886 patients with ESRD/ACKD from 7 qualified RCTs | Among the 7 selected RCTs, 3 trials used <5 mg of folic acid daily and 4 used ≥5 mg of folic acid daily. | Homocysteine reduction was achieved in the selected RCTs | Folic acid therapy reduced cardiovascular risk in patients with ESRD/ACKD by 15% |
| Qin, 2013 [47] | Meta-analysis of RCTs from January 1966 to July 2012 | 8234 patients with kidney disease from 9 qualified RCTs | Folic acid (from 2.5 mg/d to 40 mg/d) alone or with vitamins B6and B12 | Homocysteine reduction was achieved in the selected RCTs, but did not significantly correlate with cardiovascular risk | When pooling the 9 RCTs, folic acid therapy reduced cardiovascular risk by 10% |
| Saifan, 2013 [66] | Short pilot interventional study, 4 months | 52 hemodialysis patients | 1000 mcg of intramuscular vitamin B12 weekly for the first month and then monthly for 3 consecutive months | B12, homocysteine, and MMA levels were not analyzed as markers of B12 deficiency | Vitamin B12 supplementation resulted in a reduced dose of ESA required to maintain stable hemoglobin levels |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappuccilli, M.; Bergamini, C.; Giacomelli, F.A.; Cianciolo, G.; Donati, G.; Conte, D.; Natali, T.; La Manna, G.; Capelli, I. Vitamin B Supplementation and Nutritional Intake of Methyl Donors in Patients with Chronic Kidney Disease: A Critical Review of the Impact on Epigenetic Machinery. Nutrients 2020, 12, 1234. https://doi.org/10.3390/nu12051234
Cappuccilli M, Bergamini C, Giacomelli FA, Cianciolo G, Donati G, Conte D, Natali T, La Manna G, Capelli I. Vitamin B Supplementation and Nutritional Intake of Methyl Donors in Patients with Chronic Kidney Disease: A Critical Review of the Impact on Epigenetic Machinery. Nutrients. 2020; 12(5):1234. https://doi.org/10.3390/nu12051234
Chicago/Turabian StyleCappuccilli, Maria, Camilla Bergamini, Floriana A. Giacomelli, Giuseppe Cianciolo, Gabriele Donati, Diletta Conte, Teresa Natali, Gaetano La Manna, and Irene Capelli. 2020. "Vitamin B Supplementation and Nutritional Intake of Methyl Donors in Patients with Chronic Kidney Disease: A Critical Review of the Impact on Epigenetic Machinery" Nutrients 12, no. 5: 1234. https://doi.org/10.3390/nu12051234
APA StyleCappuccilli, M., Bergamini, C., Giacomelli, F. A., Cianciolo, G., Donati, G., Conte, D., Natali, T., La Manna, G., & Capelli, I. (2020). Vitamin B Supplementation and Nutritional Intake of Methyl Donors in Patients with Chronic Kidney Disease: A Critical Review of the Impact on Epigenetic Machinery. Nutrients, 12(5), 1234. https://doi.org/10.3390/nu12051234