
Neurological Sequelae of Acute Hydrogen Sulfide Poisoning: A Literature Review, Controversies, and Knowledge Gaps


Abstract
:1. Introduction
Methodology
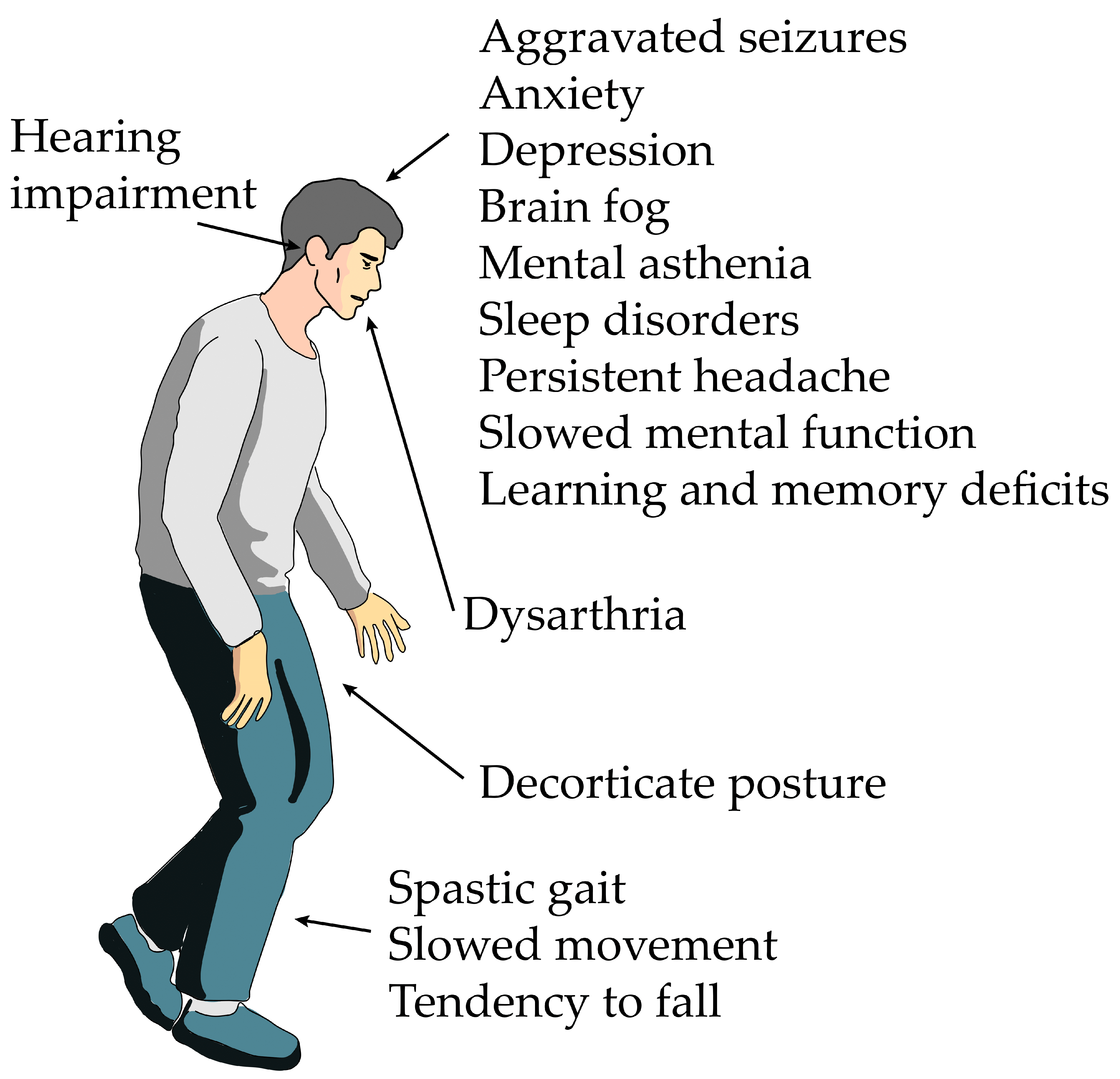
2. Results of the Literature Review on the Neurological Sequelae of Acute H2S Poisoning in Humans
3. Distribution of Delayed Neurological, Anatomical, and Functional Lesions Following Acute H2S Poisoning
4. A Review of the Literature on Animal Models
5. Results of the Literature Review on the Neurological Sequelae of Acute Cyanide Poisoning in Humans
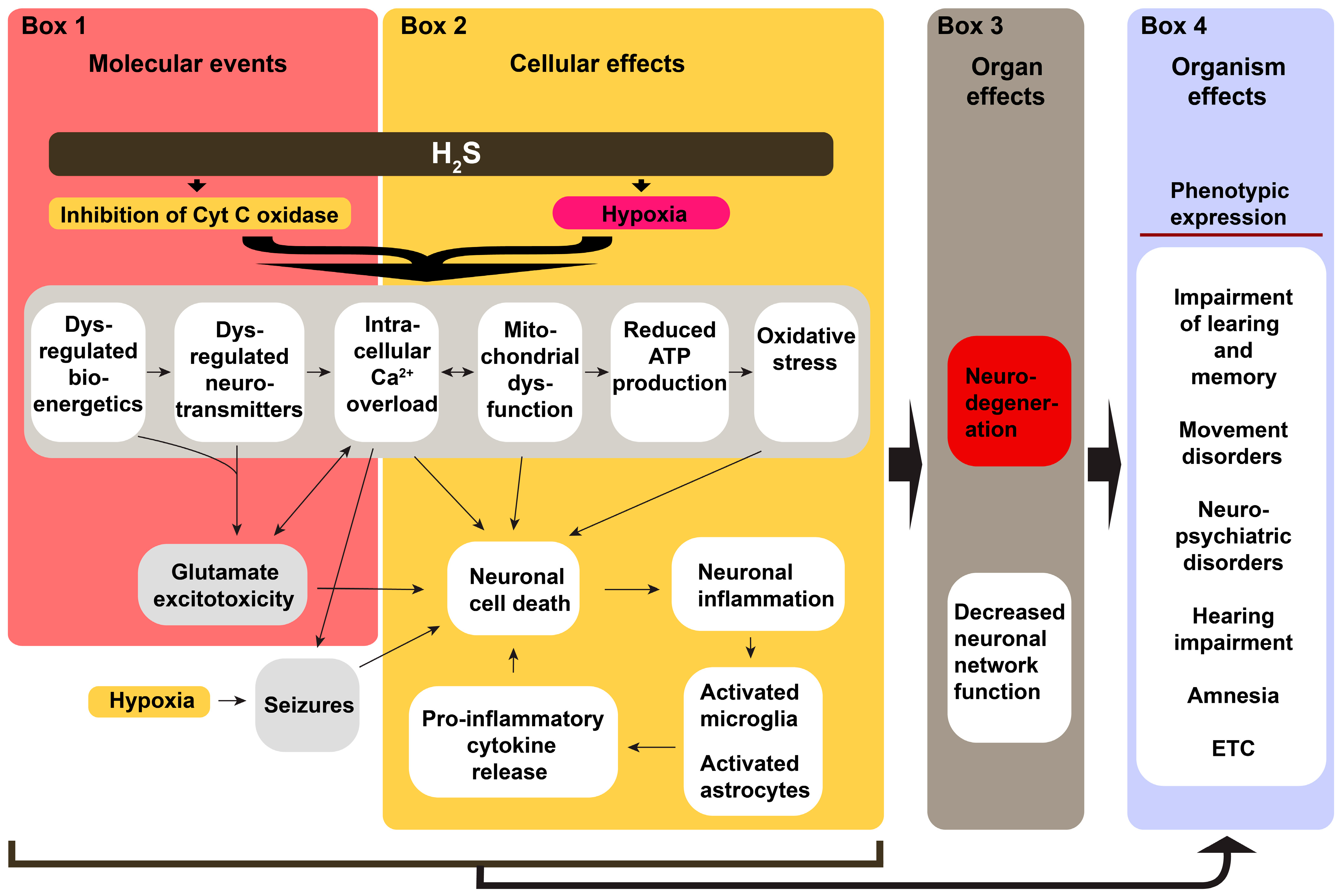
6. Potential Mechanisms Underlying H2S-Induced Neurological Sequelae
7. Discussion
8. Conclusions
9. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| H2S | hydrogen sulfide |
| ATP | adenosine triphosphate |
| CCO | cytochrome c oxidase |
| FDA | Food and Drug Administration |
| MRI | magnetic resonance imaging |
| PET | positron emission tomography |
| CT | computed tomography |
| SPECT | single-photon emission computed tomography |
| ALI | acute lung injury |
References
- Rumbeiha, W.; Whitley, E.; Anantharam, P.; Kim, D.S.; Kanthasamy, A. Acute hydrogen sulfide-induced neuropathology and neurological sequelae: Challenges for translational neuroprotective research. Ann. N. Y. Acad. Sci. 2016, 1378, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Skolnik, A.; Heise, C.W. Critical Care Toxicology; Brent, J., Burkhart, K., Dargan, P., Hatten, B., Megarbane, B., Palmer, R., White, J., Eds.; Springer Nature: Berlin/Heidelberg, Germany, 2017; pp. 1963–1971. [Google Scholar]
- Agency for Toxic Substances and Disease Registry. Toxicological profile for hydrogen sulfide and carbonyl sulfide. In Toxicological Profile for Hydrogen Sulfide and Carbonyl Sulfide; Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2016. [Google Scholar]
- Foulkes, C.H. “Gas!” The Story of the Special Brigade; Naval and Military Press, Naval & Military Press Ltd.: Uckfield, UK, 2009; p. 408. [Google Scholar]
- Binder, M.K.; Quigley, J.M.; Tinsley, H.F. Islamic State chemical weapons: A case contained by its context? CTC Sentin. 2018, 11, 27–31. [Google Scholar]
- Stuart, R.; Hall, L. Sydney Terror Plotters Tried to Blow up Etihad Plane, Unleash Poison Gas Attack. 2017. Available online: https://www.abc.net.au/news/2017-08-04/sydney-terror-raids-police-say-plane-bomb-plot-disrupted/8773752 (accessed on 1 May 2025).
- HAZMAT/SAFETY TSA Issues Security Awareness Message on Potential Use of H2S in Terrorist Attack. 2017. Available online: https://www.bulktransporter.com/hazmatsafety/tsa-issues-security-awareness-message-potential-use-h2s-terrorist-attack (accessed on 1 May 2025).
- Papadodima, S.; Papoutsis, I.; Nikolaou, P.; Spiliopoulou, C.; Stefanidou, M. “Detergent suicide” by adolescent as instructed by internet: A case report. Forensic Sci. Crimino 2017, 2, 1–2. [Google Scholar] [CrossRef]
- Anderson, A.R. Characterization of Chemical Suicides in the United States and Its Adverse Impact on Responders and Bystanders. West. J. Emerg. Med. 2016, 17, 680–683. [Google Scholar] [CrossRef]
- Ruder, J.B.; Ward, J.G.; Taylor, S.; Giles, K.; Higgins, T.; Haan, J.M. Hydrogen sulfide suicide: A new trend and threat to healthcare providers. J. Burn. Care Res. 2015, 36, e23–e25. [Google Scholar] [CrossRef]
- Sams, R.N.; Carver, H.W., 2nd; Catanese, C.; Gilson, T. Suicide with hydrogen sulfide. Am. J. Forensic Med. Pathol. 2013, 34, 81–82. [Google Scholar] [CrossRef]
- Santana Maldonado, C.M.; Kim, D.S.; Purnell, B.; Li, R.; Buchanan, G.F.; Smith, J.; Thedens, D.R.; Gauger, P.; Rumbeiha, W.K. Acute hydrogen sulfide-induced neurochemical and morphological changes in the brainstem. Toxicology 2023, 485, 153424. [Google Scholar] [CrossRef]
- Anantharam, P.; Whitley, E.M.; Mahama, B.; Kim, D.S.; Imerman, P.M.; Shao, D.; Langley, M.R.; Kanthasamy, A.; Rumbeiha, W.K. Characterizing a mouse model for evaluation of countermeasures against hydrogen sulfide-induced neurotoxicity and neurological sequelae. Ann. N. Y. Acad. Sci. 2017, 1400, 46–64. [Google Scholar] [CrossRef]
- Kim, D.S.; Santana Maldonado, C.M.; Giulivi, C.; Rumbeiha, W.K. Metabolomic Signatures of Brainstem in Mice following Acute and Subchronic Hydrogen Sulfide Exposure. Metabolites 2024, 14. [Google Scholar] [CrossRef]
- Rumbeiha, W.K.; Kim, D.S.; Min, A.; Nair, M.; Giulivi, C. Disrupted brain mitochondrial morphology after in vivo hydrogen sulfide exposure. Sci. Rep. 2023, 13, 18129. [Google Scholar] [CrossRef]
- Kim, D.S.; Pessah, I.N.; Santana, C.M.; Purnell, B.; Li, R.; Buchanan, G.F.; Rumbeiha, W.K. Investigations into hydrogen sulfide-induced suppression of neuronal activity in vivo and calcium dysregulation in vitro. Toxicol. Sci. 2023, 192, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Anantharam, P.; Padhi, P.; Thedens, D.R.; Li, G.; Gilbreath, E.; Rumbeiha, W.K. Transcriptomic profile analysis of brain inferior colliculus following acute hydrogen sulfide exposure. Toxicology 2020, 430, 152345. [Google Scholar] [CrossRef] [PubMed]
- Anantharam, P.; Kim, D.S.; Whitley, E.M.; Mahama, B.; Imerman, P.; Padhi, P.; Rumbeiha, W.K. Midazolam Efficacy Against Acute Hydrogen Sulfide-Induced Mortality and Neurotoxicity. J. Med. Toxicol. 2018, 14, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Anantharam, P.; Whitley, E.M.; Mahama, B.; Kim, D.S.; Sarkar, S.; Santana, C.; Chan, A.; Kanthasamy, A.G.; Kanthasamy, A.; Boss, G.R.; et al. Cobinamide is effective for treatment of hydrogen sulfide-induced neurological sequelae in a mouse model. Ann. N. Y. Acad. Sci. 2017, 1408, 61–78. [Google Scholar] [CrossRef]
- Jiang, J.; Chan, A.; Ali, S.; Saha, A.; Haushalter, K.J.; Lam, W.L.; Glasheen, M.; Parker, J.; Brenner, M.; Mahon, S.B.; et al. Hydrogen Sulfide-Mechanisms of Toxicity and Development of an Antidote. Sci. Rep. 2016, 6, 20831. [Google Scholar] [CrossRef]
- Haouzi, P.; Sonobe, T.; Judenherc-Haouzi, A. Hydrogen sulfide intoxication induced brain injury and methylene blue. Neurobiol. Dis. 2020, 133, 104474. [Google Scholar] [CrossRef]
- Beauchamp, R.O., Jr.; Bus, J.S.; Popp, J.A.; Boreiko, C.J.; Andjelkovich, D.A. A critical review of the literature on hydrogen sulfide toxicity. Crit. Rev. Toxicol. 1984, 13, 25–97. [Google Scholar] [CrossRef]
- Guidotti, T.L. Handbook of Clinical Neurology; Lotti, M., Bleecker, M.L., Eds.; Elservier: Amsterdam, The Netherlands, 2015; Volume 131. [Google Scholar]
- World Health Organization. Hydrogen Sulfide; World Health Organization: Geneva, Switzerland, 1981. [Google Scholar]
- Wasch, H.H.; Estrin, W.J.; Yip, P.; Bowler, R.; Cone, J.E. Prolongation of the P-300 latency associated with hydrogen sulfide exposure. Arch. Neurol. 1989, 46, 902–904. [Google Scholar] [CrossRef]
- Tvedt, B.; Edland, A.; Skyberg, K.; Forberg, O. Delayed neuropsychiatric sequelae after acute hydrogen sulfide poisoning: Affection of motor function, memory, vision and hearing. Acta Neurol. Scand. 1991, 84, 348–351. [Google Scholar] [CrossRef]
- Nam, B.; Kim, H.; Choi, Y.; Lee, H.; Hong, E.S.; Park, J.K.; Lee, K.M.; Kim, Y. Neurologic sequela of hydrogen sulfide poisoning. Ind. Health 2004, 42, 83–87. [Google Scholar] [CrossRef]
- Hoidal, C.R.; Hall, A.H.; Robinson, M.D.; Kulig, K.; Rumack, B.H. Hydrogen sulfide poisoning from toxic inhalations of roofing asphalt fumes. Ann. Emerg. Med. 1986, 15, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, A.R. Hydrogen sulfide exposure without loss of consciousness: Chronic effects in four cases. Toxicol. Ind. Health 2002, 18, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Gallen, P.; Nogue, S.; Palomar, M.; Rodriguez, M.; Marti, M.J.; Munne, P. [Acute poisoning caused by hydrogen sulphide: Clinical features of 3 cases]. An. Med. Interna 1994, 11, 392–394. [Google Scholar]
- Hurwitz, L.J.; Taylor, G.I. Poisoning by sewer gas with unusual sequelae. Lancet 1954, 266, 1110–1112. [Google Scholar] [CrossRef]
- Shivanthan, M.C.; Perera, H.; Jayasinghe, S.; Karunanayake, P.; Chang, T.; Ruwanpathirana, S.; Jayasinghe, N.; De Silva, Y.; Jayaweerabandara, D. Hydrogen sulphide inhalational toxicity at a petroleum refinery in Sri Lanka: A case series of seven survivors following an industrial accident and a brief review of medical literature. J. Occup. Med. Toxicol. 2013, 8, 9. [Google Scholar] [CrossRef]
- Fenga, C.; Cacciola, A.; Micali, E. [Cognitive sequelae of acute hydrogen sulphide poisoning. A case report]. La Med. Del Lav. 2002, 93, 322–328. [Google Scholar]
- Kilburn, K.H. Case report: Profound neurobehavioral deficits in an oil field worker overcome by hydrogen sulfide. Am. J. Med. Sci. 1993, 306, 301–305. [Google Scholar] [CrossRef]
- Matsuo, F.; Cummins, J.W.; Anderson, R.E. Neurological sequelae of massive hydrogen sulfide inhalation. Arch. Neurol. 1979, 36, 451–452. [Google Scholar] [CrossRef]
- Schneider, J.S.; Tobe, E.H.; Mozley, P.D., Jr.; Barniskis, L.; Lidsky, T.I. Persistent cognitive and motor deficits following acute hydrogen sulphide poisoning. Occup. Med. 1998, 48, 255–260. [Google Scholar] [CrossRef]
- Burnett, W.W.; King, E.G.; Grace, M.; Hall, W.F. Hydrogen sulfide poisoning: Review of 5 years’ experience. Can. Med. Assoc. J. 1977, 117, 1277–1280. [Google Scholar]
- Arnold, I.M.; Dufresne, R.M.; Alleyne, B.C.; Stuart, P.J. Health implication of occupational exposures to hydrogen sulfide. J. Occup. Med. 1985, 27, 373–376. [Google Scholar] [CrossRef] [PubMed]
- McCabe, L.C.; Clayton, G.D. Air pollution by hydrogen sulfide in Poza Rica, Mexico; an evaluation of the incident of Nov. 24, 1950. AMA Arch. Ind. Hyg. Occup. Med. 1952, 6, 199–213. [Google Scholar] [PubMed]
- Gerasimon, G.; Bennett, S.; Musser, J.; Rinard, J. Acute hydrogen sulfide poisoning in a dairy farmer. Clin. Toxicol. 2007, 45, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Vos, T. Global Burden of Neurological Disorders: From Global Burden of Disease Estimates to Actions. Neuroepidemiology 2019, 52, 1–2. [Google Scholar] [CrossRef]
- Feigin, V.L.; Vos, T.; Nichols, E.; Owolabi, M.O.; Carroll, W.M.; Dichgans, M.; Deuschl, G.; Parmar, P.; Brainin, M.; Murray, C. The global burden of neurological disorders: Translating evidence into policy. Lancet Neurol. 2020, 19, 255–265. [Google Scholar] [CrossRef]
- Wang, D.X. [A review of 152 cases of acute poisoning of hydrogen sulfide]. Zhonghua Yu Fang Yi Xue Za Zhi 1989, 23, 330–332. [Google Scholar]
- Tvedt, B.; Skyberg, K.; Aaserud, O.; Hobbesland, A.; Mathiesen, T. Brain damage caused by hydrogen sulfide: A follow-up study of six patients. Am. J. Ind. Med. 1991, 20, 91–101. [Google Scholar] [CrossRef]
- Snyder, J.W.; Safir, E.F.; Summerville, G.P.; Middleberg, R.A. Occupational fatality and persistent neurological sequelae after mass exposure to hydrogen sulfide. Am. J. Emerg. Med. 1995, 13, 199–203. [Google Scholar] [CrossRef]
- Tang, D.; Tian, N.; Cai, J.; Ma, J.; Wang, T.; Zhang, H.; Sheng, F. Analysis of CT and MR imaging features of the brain in patients with hydrogen sulfide poisoning based on clinical symptom grading. BMC Neurol. 2022, 22, 413. [Google Scholar] [CrossRef]
- Grandas, F.; Artieda, J.; Obeso, J.A. Clinical and CT scan findings in a case of cyanide intoxication. Mov. Disord. 1989, 4, 188–193. [Google Scholar] [CrossRef]
- Messing, B. Extrapyramidal disturbances after cyanide poisoning (first MRT-investigation of the brain). J. Neural Transm. Suppl. 1991, 33, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Kasamo, K.; Okuhata, Y.; Satoh, R.; Ikeda, M.; Takahashi, S.; Kamata, R.; Nogami, Y.; Kojima, T. Chronological changes of MRI findings on striatal damage after acute cyanide intoxication: Pathogenesis of the damage and its selectivity, and prevention for neurological sequelae: A case report. Eur. Arch. Psychiatry Clin. Neurosci. 1993, 243, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Rosenow, F.; Herholz, K.; Lanfermann, H.; Weuthen, G.; Ebner, R.; Kessler, J.; Ghaemi, M.; Heiss, W.D. Neurological sequelae of cyanide intoxication—The patterns of clinical, magnetic resonance imaging, and positron emission tomography findings. Ann. Neurol. 1995, 38, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Pentore, R.; Venneri, A.; Nichelli, P. Accidental choke-cherry poisoning: Early symptoms and neurological sequelae of an unusual case of cyanide intoxication. Ital. J. Neurol. Sci. 1996, 17, 233–235. [Google Scholar] [CrossRef]
- Rachinger, J.; Fellner, F.A.; Stieglbauer, K.; Trenkler, J. MR changes after acute cyanide intoxication. AJNR Am. J. Neuroradiol. 2002, 23, 1398–1401. [Google Scholar]
- Mohan, A.; Lee, T.; Sachdev, P. Surviving acute cyanide poisoning: A longitudinal neuropsychological investigation with interval MRI. BMJ Case Rep. 2014, 2014, bcr2013203025. [Google Scholar] [CrossRef]
- Alqahtani, R.M.; Alyousef, M.Y.; AlWatban, Z.H.; Ghandour, M.K. Long-Term Neuropsychiatric Sequelae in a Survivor of Cyanide Toxicity Patient With Arterialization. Cureus 2020, 12, e8430. [Google Scholar] [CrossRef]
- Yang, D.; Chen, G.; Zhang, R. Estimated Public Health Exposure to H2S Emissions from a Sour Gas Well Blowout in Kaixian County, China. Aerosol Air Qual. Res. 2006, 6, 430–443. [Google Scholar] [CrossRef]
- Mooyaart, E.A.Q.; Gelderman, E.L.G.; Nijsten, M.W.; de Vos, R.; Hirner, J.M.; de Lange, D.W.; Leuvenink, H.D.G.; van den Bergh, W.M. Outcome after hydrogen sulphide intoxication. Resuscitation 2016, 103, 1–6. [Google Scholar] [CrossRef]
- Gaitonde, U.B.; Sellar, R.J.; O’Hare, A.E. Long term exposure to hydrogen sulphide producing subacute encephalopathy in a child. Br. Med. J. 1987, 294, 614. [Google Scholar] [CrossRef]
- Warenycia, M.W.; Goodwin, L.R.; Benishin, c.G.; Reiffenstein, R.J.; Francom, D.M.; Taylor, J.D.; Dieken, F.P. Acute Hydrogen sulfide poisoning. Biochem. Pharmacol. 1989, 38, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Leavesley, H.B.; Li, L.; Prabhakaran, K.; Borowitz, J.L.; Isom, G.E. Interaction of cyanide and nitric oxide with cytochrome c oxidase: Implications for acute cyanide toxicity. Toxicol. Sci. 2008, 101, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.C.; Mlady, G.W.; Kwon, Y.H.; Rose, G.M. Chronic in vivo sodium azide infusion induces selective and stable inhibition of cytochrome c oxidase. J. Neurochem. 1996, 66, 2606–2611. [Google Scholar] [CrossRef] [PubMed]
- Wieloch, T. Neurochemical correlates to selective neuronal vulnerability. Prog. Brain Res. 1985, 63, 69–85. [Google Scholar] [CrossRef]
- Milby, T.H.; Baselt, R.C. Hydrogen sulfide poisoning: Clarification of some controversial issues. Am. J. Ind. Med. 1999, 35, 192–195. [Google Scholar] [CrossRef]
- Lund, O.E.; Wieland, H. [Pathologic-anatomic findings in experimental hydrogen sulfide poisoning (H2S). A study on rhesus monkeys]. Int. Arch. Fur Arbeitsmedizin 1966, 22, 46–54. [Google Scholar]
- Baldelli, R.J.; Green, F.H.; Auer, R.N. Sulfide toxicity: Mechanical ventilation and hypotension determine survival rate and brain necrosis. J. Appl. Physiol. 1993, 75, 1348–1353. [Google Scholar] [CrossRef]
- Sonobe, T.; Chenuel, B.; Cooper, T.K.; Haouzi, P. Immediate and Long-Term Outcome of Acute H2S Intoxication Induced Coma in Unanesthetized Rats: Effects of Methylene Blue. Mir Oxidative Stress 2015, 10, e0131340. [Google Scholar] [CrossRef]
- Kim, D.S.; Anantharam, P.; Hoffmann, A.; Meade, M.L.; Grobe, N.; Gearhart, J.M.; Whitley, E.M.; Mahama, B.; Rumbeiha, W.K. Broad spectrum proteomics analysis of the inferior colliculus following acute hydrogen sulfide exposure. Toxicol. Appl. Pharmacol. 2018, 355, 28–42. [Google Scholar] [CrossRef]
- Judenherc-Haouz, I.A.; Sonobe, T.; Bebarta, V.S.; Haouzi, P. On the Efficacy of Cardio-Pulmonary Resuscitation and Epinephrine Following Cyanide- and H2S Intoxication-Induced Cardiac Asystole. Cardiovasc. Toxicol. 2018, 18, 436–449. [Google Scholar] [CrossRef]
- Sonobe, T.; Haouzi, P. H2S induced coma and cardiogenic shock in the rat: Effects of phenothiazinium chromophores. Clin. Toxicol. 2015, 53, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Haouzi, P.; Sonobe, T.; Chenuel, B. Oxygen-related chemoreceptor drive to breathe during H(2)S infusion. Respir. Physiol. Neurobiol. 2014, 201, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Chenuel, B.; Sonobe, T.; Haouzi, P. Effects of infusion of human methemoglobin solution following hydrogen sulfide poisoning. Clin. Toxicol. 2015, 53, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Haouzi, P.; Tubbs, N.; Cheung, J.; Judenherc-Haouzi, A. Methylene Blue Administration During and After Life-Threatening Intoxication by Hydrogen Sulfide: Efficacy Studies in Adult Sheep and Mechanisms of Action. Toxicol. Sci. 2019, 168, 443–459. [Google Scholar] [CrossRef]
- Cronican, A.A.; Frawley, K.L.; Ahmed, H.; Pearce, L.L.; Peterson, J. Antagonism of Acute Sulfide Poisoning in Mice by Nitrite Anion without Methemoglobinemia. Chem. Res. Toxicol. 2015, 28, 1398–1408. [Google Scholar] [CrossRef]
- Ding, Y.; Li, X.; Chen, C.; Ling, J.; Li, W.; Guo, Y.; Yan, J.; Zha, L.; Cai, J. A rapid evaluation of acute hydrogen sulfide poisoning in blood based on DNA-Cu/Ag nanocluster fluorescence probe. Sci. Rep. 2017, 7, 9638. [Google Scholar] [CrossRef]
- Lopez, A.; Prior, M.G.; Reiffenstein, R.J.; Goodwin, L.R. Peracute toxic effects of inhaled hydrogen sulfide and injected sodium hydrosulfide on the lungs of rats. Fundam. Appl. Toxicol. 1989, 12, 367–373. [Google Scholar] [CrossRef]
- Shen, Y.; Zhao, G.; Lin, J.; Wang, J.; Luo, B.; Liu, J.; Zhang, Y.; Huang, J. Case series and clinical analysis of acute hydrogen sulfide poisoning: Experience from 10 cases at a hospital in Zhoushan. Toxicol. Ind. Health 2025, 41, 151–162. [Google Scholar] [CrossRef]
- Zhang, D.; Lee, B.; Nutter, A.; Song, P.; Dolatabadi, N.; Parker, J.; Sanz-Blasco, S.; Newmeyer, T.; Ambasudhan, R.; McKercher, S.R.; et al. Protection from cyanide-induced brain injury by the Nrf2 transcriptional activator carnosic acid. J. Neurochem. 2015, 133, 898–908. [Google Scholar] [CrossRef]
- Carella, F.; Grassi, M.P.; Savoiardo, M.; Contri, P.; Rapuzzi, B.; Mangoni, A. Dystonic-Parkinsonian syndrome after cyanide poisoning: Clinical and MRI findings. J. Neurol. Neurosurg. Psychiatry 1988, 51, 1345–1348. [Google Scholar] [CrossRef]
- Borgohain, R.; Singh, A.K.; Radhakrishna, H.; Rao, V.C.; Mohandas, S. Delayed onset generalised dystonia after cyanide poisoning. Clin. Neurol. Neurosurg. 1995, 97, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Finelli, P.F. Case report. Changes in the basal ganglia following cyanide poisoning. J. Comput. Assist. Tomogr. 1981, 5, 755–756. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.L.; Myers, J.A.; Martin, W.R. Cyanide-induced parkinsonism: Clinical, MRI, and 6-fluorodopa PET studies. Neurology 1989, 39, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, R.; Court, J.; Godoy, J. Delayed cyanide induced dystonia. J. Neurol. Neurosurg. Psychiatry 1992, 55, 198–199. [Google Scholar] [CrossRef]
- Park, H.; Lee, C.H. The Impact of Pulmonary Disorders on Neurological Health (Lung-Brain Axis). Immune Netw. 2024, 24, e20. [Google Scholar] [CrossRef]
- Bajinka, O.; Simbilyabo, L.; Tan, Y.; Jabang, J.; Saleem, S.A. Lung-brain axis. Crit. Rev. Microbiol. 2022, 48, 257–269. [Google Scholar] [CrossRef]
- Cheung, N.S.; Peng, Z.F.; Chen, M.J.; Moore, P.K.; Whiteman, M. Hydrogen sulfide induced neuronal death occurs via glutamate receptor and is associated with calpain activation and lysosomal rupture in mouse primary cortical neurons. Neuropharmacology 2007, 53, 505–514. [Google Scholar] [CrossRef]
- Jett, D.A.; Spriggs, S.M. Translational research on chemical nerve agents. Neurobiol. Dis. 2020, 133, 104335. [Google Scholar] [CrossRef]
- Mart, M.F.; Ware, L.B. The long-lasting effects of the acute respiratory distress syndrome. Expert. Rev. Respir. Med. 2020, 14, 577–586. [Google Scholar] [CrossRef]
- Kapfhammer, H.P.; Rothenhausler, H.B.; Krauseneck, T.; Stoll, C.; Schelling, G. Posttraumatic stress disorder and health-related quality of life in long-term survivors of acute respiratory distress syndrome. Am. J. Psychiatry 2004, 161, 45–52. [Google Scholar] [CrossRef]
- Hopkins, R.O.; Weaver, L.K.; Pope, D.; Orme, J.F.; Bigler, E.D.; Larson, L.V. Neuropsychological sequelae and impaired health status in survivors of severe acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1999, 160, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Herridge, M.S.; Tansey, C.M.; Matte, A.; Tomlinson, G.; Diaz-Granados, N.; Cooper, A.; Guest, C.B.; Mazer, C.D.; Mehta, S.; Stewart, T.E.; et al. Functional disability 5 years after acute respiratory distress syndrome. N. Engl. J. Med. 2011, 364, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, M.E.; Shull, W.H.; Biester, R.C.; Taichman, D.B.; Lynch, S.; Demissie, E.; Hansen-Flaschen, J.; Christie, J.D. Cognitive, mood and quality of life impairments in a select population of ARDS survivors. Respirology 2009, 14, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, M.E.; Brummel, N.E.; Archer, K.; Ely, E.W.; Jackson, J.C.; Hopkins, R.O. Cognitive dysfunction in ICU patients: Risk factors, predictors, and rehabilitation interventions. Crit. Care Med. 2013, 41, S81–S98. [Google Scholar] [CrossRef]
- Herridge, M.S.; Moss, M.; Hough, C.L.; Hopkins, R.O.; Rice, T.W.; Bienvenu, O.J.; Azoulay, E. Recovery and outcomes after the acute respiratory distress syndrome (ARDS) in patients and their family caregivers. Intensive Care Med. 2016, 42, 725–738. [Google Scholar] [CrossRef]
- Smith, R.P.; Gosselin, R.E. On the mechanism of sulfide inactivation by methemoglobin. Toxicol. Appl. Pharmacol. 1966, 8, 159–172. [Google Scholar] [CrossRef]
- Truong, D.H.; Mihajlovic, A.; Gunness, P.; Hindmarsh, W.; O’Brien, P.J. Prevention of hydrogen sulfide (H2S)-induced mouse lethality and cytotoxicity by hydroxocobalamin (vitamin B(12a)). Toxicology 2007, 242, 16–22. [Google Scholar] [CrossRef]
- Brenner, M.; Benavides, S.; Mahon, S.B.; Lee, J.; Yoon, D.; Mukai, D.; Viseroi, M.; Chan, A.; Jiang, J.; Narula, N.; et al. The vitamin B12 analog cobinamide is an effective hydrogen sulfide antidote in a lethal rabbit model. Clin. Toxicol. 2014, 52, 490–497. [Google Scholar] [CrossRef]
- Judenherc-Haouzi, A.; Zhang, X.Q.; Sonobe, T.; Song, J.; Rannals, M.D.; Wang, J.; Tubbs, N.; Cheung, J.Y.; Haouzi, P. Methylene blue counteracts H2S toxicity-induced cardiac depression by restoring L-type Ca channel activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R1030–R1044. [Google Scholar] [CrossRef]
- Bitterman, N.; Talmi, Y.; Lerman, A.; Melamed, Y.; Taitelman, U. The effect of hyperbaric oxygen on acute experimental sulfide poisoning in the rat. Toxicol. Appl. Pharmacol. 1986, 84, 325–328. [Google Scholar] [CrossRef]
- Fang, M.; Hu, J.; Weiss, J.; Knopman, D.S.; Albert, M.; Windham, B.G.; Walker, K.A.; Sharrett, A.R.; Gottesman, R.F.; Lutsey, P.L.; et al. Lifetime risk and projected burden of dementia. Nat. Med. 2025, 31, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.; Irshad, K.; Kamal, S.; Imran, I.; Akash, M.S.H. Exposure of Environmental Contaminants and Development of Neurological Disorders. Crit. Rev. Eukaryot. Gene Expr. 2021, 31, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Buoli, M.; Grassi, S.; Caldiroli, A.; Carnevali, G.S.; Mucci, F.; Iodice, S.; Cantone, L.; Pergoli, L.; Bollati, V. Is there a link between air pollution and mental disorders? Environ. Int. 2018, 118, 154–168. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Reference Title | Authors | Age | Exposure Duration | Comments |
|---|---|---|---|---|
| Air pollution by H2S in Poza Rica, Mexico; an evaluation of the incident of 24 November 1950 | McCabe and Clayton 1952 [39] | Up to 20 min | Four survivors (age not given) developed neurological sequelae; two survivors developed neuritis of the acoustic nerve; one survivor developed dysarthria; and the fourth survivor developed aggravated epilepsy. | |
| Poisoning by sewer gas with unusual sequelae | Hurwitz 1954 [31] | 46 | 30 min | A 30 min exposure in a 46 year old male manifested as neurological sequelae 3 months after exposure, with exaggerated reflexes and tremors. |
| Hydrogen sulfide poisoning: a review of 5 years’ experience | Burnett WW, et al., 1977 [37] | 19–31 | N/A | A review of 221 cases. The average age was 31 year old. All cases involved workers, with 43% between 21 and 30 year old. A low prevalence of sequelae was reported, but no specialized neurological or neuropsychiatric examinations were performed. |
| Neurological sequelae of massive hydrogen sulfide inhalation | Matsuo F et al., 1979 [35] | 45 | N/A | A 45 year old male was rendered unconscious and entered a chronic vegetative state. A CT scan showed bilateral cerebral hemispheres and lentiform nucleus lesions. He died 5 weeks after exposure despite treatment. |
| Health implications of occupational exposures to hydrogen sulfide | Arnold IMF, et al., 1985 [38] | 19–30 | N/A | A 5 year retrospective study was conducted using Compensation Board records of 250 workers: 54.6% were 21–30 year old; 98% were male. Compensation was given to 58 cases (23.2%) due to lost time from work of <1 mo duration. Completeness of the recovery of individuals could not be determined. No specialized neurological or neuropsychiatric examinations were performed. |
| Hydrogen sulfide poisoning from toxic inhalations of roofing asphalt fumes | Hoidal CR, et al., 1986 [28] | 35 | 20 | A 35 year old male exposed for 20 min entered a chronic vegetative state despite 100% oxygen therapy. |
| A review of 152 cases of acute poisoning of hydrogen sulfide | Wang DX, 1989 [43] | 18–57 | Not given | A large population study of 120 males and 32 females, with an age range of 18–57 year old. A total of 95 patients were followed for 1–10 years; 39 of them (41%) showed neuropsychiatric sequelae. |
| Brain damage caused by hydrogen sulfide: A follow up study of six patients | Tvedt B. 1991 [44] | 30–59 | 5–20 min | All males, 30–59 year old, who were rendered unconscious for 5–20 min showed persistent neurological impairment at neurological and neuropsychological re-examination 5–10 year after the accident. The authors stressed importance of long-term follow-up in order to identify neurological sequelae. |
| Delayed neuropsychiatric sequelae after acute H2S poisoning: affection of motor, memory, vision and hearing | Tvedt B, et al., 1991 [26] | 31 | 15–20 min | The 5-year follow-up of a 31 year old male exposed for 15–20 min showed cerebral atrophy and widening of the lateral ventricle (MRI and CT). Motor, memory, vision, and hearing impairment were also noted. The authors concluded that sequelae may be more common than previously reported |
| Case report: Profound neurobehavioral deficits in an oil field worker overcome by hydrogen sulfide | Kilburn KH, 1993 [34] | 24 | 10 min | A 24 year old male rendered unconscious after a 10 min exposure was treated with oxygen and released 30 min later. The subject manifested profound cognitive, memory, and neuropsychological deficits 49 months postexposure. |
| Acute poisoning caused by hydrogen sulphide: clinical features of 3 cases | Sanz-Gallen et al., 1994 [30] | 23–28 | 50–60 min | Of three 23–28 year old males exposed for 50–60 min, one entered a vegetative state, and the second developed neurologic sequelae. |
| Occupational Fatality and persistent neurological sequelae after mass exposure to hydrogen sulfide | Snyder JW et al., 1995 [45] | 24–50 | Seconds to unknown time | A case report of acute H2S poisoning, including 37 people affected (age range 24–50 year), 6 admitted, and 1 death. At least one patient who underwent hyperbaric oxygen treatment developed neurological sequelae. The authors recommended that victims of acute H2S poisoning presenting coma or evidence of neurotoxicity should undergo baseline and annual neurological and neuropsychological testing for at least 5 year because patients with long-term neurological sequelae continue to be reported. This is necessary in order to detect permanent alterations in the nervous system following acute H2S exposure. |
| Persistent cognitive and motor deficits following acute hydrogen sulfide poisoning | Schneider JS et al., 1998 [36] | 27 | N/A | A 27 year male was unconscious and treated with hyperbaric oxygen for several days. At 3 year after the accident, PET showed abnormal metabolism in basal ganglia, thalamus, and temporal and inferior parietal lobes. Neurobehavioral, neuropsychological, and neurofunctional impairment. |
| Cognitive sequelae three months after hydrogen sulfide poisoning | Fenga C et al., 2002 [33] | 36 | A few mins | A 36 year old male was exposed to 500 ppm for a few minutes. Three months postexposure, he manifested reduced cognition, depression, and personality changes even though a neurological exam and neuroimaging were unremarkable. |
| Hydrogen sulfide exposure without loss of consciousness: chronic effects in four cases | Hirsch AR 2002 [29] | N/A | 2.5 h | Four workers (sex and age not stated) had persistent neuropsychiatric disorders and abnormal P300 evoked responses 1 year after exposure. |
| Neurological sequela of hydrogen sulfide poisoning | Nam B et al., 2004 [27] | 25 | 10 min | A 25 year old male exposed for 10 min was hospitalized in a coma. Necrosis of basal ganglia and motor cortex were identified by MRI 30 days postexposure. Neurological cognitive deficits were identified up to 5 mo postexposure. |
| Acute hydrogen sulfide poisoning in a dairy farmer | Gerasimon G et al., 2007 [40] | 36 | 5 min | A 36 year old male exposed for 5 min showed MRI lesions in superior the cerebral hemispheres, basal ganglia, and thalamus. Problems with balance, dysarthria, and difficulty eating for several months improved with intense neuro-rehabilitation. |
| Hydrogen sulfide inhalation toxicity at a petroleum refinery in Sri Lanka | Shivanthan M, et al., 2013 [32] | 36 | 10 min | Dysarthria, status epilepticus and retrograde amnesia were identified in one 36 year old male survivor after a 10 min exposure. |
| Analysis of CT and MR imaging features of the brain in patients with hydrogen sulfide poisoning based on clinical symptom grading | Tang D et al., 2022 [46] | 18–54 | N/A | A retrospective analysis of CT and MRI data from 40 patients (35 males, 5 females, age 18–54 year, median age 37.5 year) with acute poisoning clinically graded according to central nervous system score (minor n = 10, moderate n = 17, severe n = 13). Generalized brain edema was found in most severe cases and in one moderate case. Symmetrical abnormal intensities in basal ganglia and around lateral ventricles were seen in most of the severe cases. Subarachnoid or intracerebral hemorrhage and cerebral tonsillar herniation were present is a few severe cases that also had a poor prognosis. Brain lesions were still present in some patients 5.5 months after exposure. One victim who was exposed for 50 min developed severe neurological sequelae. |
| Reference Title | Authors | Age | Exposure Dose | Comments |
|---|---|---|---|---|
| Clinical and CT scan findings in a case of cyanide intoxication | Grandas 1983 [47] | 39 | Unknown | A 39-year-old man developed brain lesions in basal ganglia and the front cortex 1 month after exposure and putamen and external globus pallidus 2 to 3 years after exposure. The patient manifested delayed neurological sequelae, including signs of Parkinsonism (bradykinesia, resting tremor, and postural instability). His neurological sequelae were permanent. |
| Extrapyramidal disturbances after cyanide poisoning (first MRT investigation of the brain) | Messing 1991 [48] | 29 | 500 mg of potassium cyanide | A 29-year-old man developed delayed dysarthria and signs of Parkinsonism, including bradykinesia and monosynaptic reflexes. CT scans and MRI imaging revealed lesions in the basal ganglia 5 months after attempting suicide. |
| Chronological changes of MRI findings on striatal damage after acute cyanide intoxication: pathogenesis of the damage and its selectivity, and prevention for neurological sequelae: a case report | Kasamo 1993 [49] | 31 | 20–40 g of potassium cyanide | A patient developed delayed neurological sequelae characterized by lesions restricted to the caudate nuclei and putamina using MRI. The lesions in putamina were still present at 9 months after exposure to cyanide. |
| Neurological sequelae of cyanide intoxication—the patterns of clinical, magnetic resonance imaging, and positron emission tomography findings | Rosenow 1995 [50] | 22 and 43 | 1.3 g of potassium cyanide for a 22-year-old man Unknown amount for a 43-year-old man | Both patients had delayed extrapyramidal motor and cerebral symptoms, including bradykinesia, months after exposure to cyanide. MRI imaging showed lesions in the globus pallidus, putamen, substantia nigra, subthalamic nucleus, and cerebellum. A 22-year-old patient showed reduced glucose metabolism and dopamine uptake in the putamen and caudate. The 22-year-old patient recovered 3 years after exposure to cyanide, whereas the 43-year-old man still had brain lesions in the pallidum, posterior putamen, and substantia nigra 5 years after exposure to cyanide. |
| Accidental choke-cherry poisoning: early symptoms and neurological sequelae of an unusual case of cyanide intoxication | Pentore 1996 [51] | 56 | Unknown | A 56 year old woman experienced breathing difficulty and entered coma. For 2 weeks, she had neurological symptoms including confusion, disorientation, and agitation. About 30 days post-admission, she developed signs of Parkinsonism and reduced bilateral visual acuity. The patient fully recovered at 15 months after admission. |
| MR changes after acute cyanide intoxication | Rachinger 2002 [52] | 35 | Unknown | A 35 year old female immediately went into a coma after cyanide poisoning and showed agitation and akinetic mutism after withdrawing sedation and removal of respiratory support in hospital. Six weeks after cyanide poisoning, MRI imaging revealed hemorrhage and hyperintense signal change in the striatum, globus pallidus, and basal ganglia. |
| Surviving acute cyanide poisoning: a longitudinal neuropsychological investigation with interval MRI | Mohan 2014 [53] | 22 | Unknown | A 22-year-old woman developed neuropsychological effects after ingestion of cyanide. Around day 5 post-ingestion, she had deficits in episodic memory. She was assessed to have impaired episodic memory and retrograde amnesia. She also had attention deficits. The patient did not have movement disorder. MRI neuroimaging revealed deficits in the basal ganglia, thalamus, and cerebral hemispheres and edema in the hippocampus. At 5 months, these lesions were still present, albeit smaller, but the hippocampus was reported to have atrophied. A neuropsychological assessment at 3 weeks post-injury revealed reduced cognitive functioning and problem-solving skills. These defects were significantly reversed at 5 months. |
| Long-term neuropsychiatric sequelae in a survivor of cyanide toxicity patient with arterialization | Alqahtani 2020 [54] | 45 | Unknown inhalation | A 45-year-old woman developed cognitive deficits, dysarthria, dystonia, and altered sleep patterns 3 months after cyanide poisoning. The authors emphasized conducting full neurological and intellectual evaluations to find neuropsychiatric sequelae in cases of acute cyanide poisoning. In this case, the CT scan and MRI were normal. |
| Drug | Mode of Action |
|---|---|
| Methemoglobin inducers | Sodium nitrite induces the formation of methemoglobin heme in vivo, which reduces the inhibition of cytochrome c oxidase [93]. It was also proposed that methemoglobin inducers may serve as a NO donor, which reverses the sulfide inhibition of cytochrome c oxidase [72]. |
| Epinephrine | Epinephrine with chest compressions counteracts sulfide-induced pulseless electrical activity (PEA) [67]. |
| Hydroxocobalamin | Hydroxocobalamin binds to sulfide in the blood to prevent free sulfide from entering tissues [94]. |
| Cobinamide | Cobinamide is an analog of hydroxocobalamin and a precursor in hydroxocobalamin synthesis. Cobinamide has more sulfide binding sites than hydroxocobolamine [95]. |
| Midazolam | Midazolam is an anticonvulsant drug and a potential antidote to counteract sulfide-induced epileptic activity. The prevention of seizure-like activity prevents mortality and reduces neuropathology in mice [18]. |
| Methylene blue | Methylene blue is potential antioxidant drug. It has shown efficacy in the treatment of methemoglobinemia. Methylene blue is reported to restore cellular redox potential, mitochondrial function, and cardiac myocyte function [71,96]. It has also shown efficacy in reducing H2S-induced neurological sequelae in a rat model. |
| Hyperbaric oxygen | Hyperbaric oxygen treatment provides supplemental oxygen to reduce the hypoxic effects of sulfide [97]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rumbeiha, W.K.; Kim, D.-S. Neurological Sequelae of Acute Hydrogen Sulfide Poisoning: A Literature Review, Controversies, and Knowledge Gaps. Neurol. Int. 2025, 17, 71. https://doi.org/10.3390/neurolint17050071
Rumbeiha WK, Kim D-S. Neurological Sequelae of Acute Hydrogen Sulfide Poisoning: A Literature Review, Controversies, and Knowledge Gaps. Neurology International. 2025; 17(5):71. https://doi.org/10.3390/neurolint17050071
Chicago/Turabian StyleRumbeiha, Wilson K., and Dong-Suk Kim. 2025. "Neurological Sequelae of Acute Hydrogen Sulfide Poisoning: A Literature Review, Controversies, and Knowledge Gaps" Neurology International 17, no. 5: 71. https://doi.org/10.3390/neurolint17050071
APA StyleRumbeiha, W. K., & Kim, D.-S. (2025). Neurological Sequelae of Acute Hydrogen Sulfide Poisoning: A Literature Review, Controversies, and Knowledge Gaps. Neurology International, 17(5), 71. https://doi.org/10.3390/neurolint17050071