
Hyperbranched Polyethylene Ionomers Containing Quaternary Ammonium Ions and Their Functionalization of Nanomaterials


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to Ionomers and Their Synthesis
2. Catalytic Synthesis of Hyperbranched Polyethylene Ionomers Containing Quaternary Ammonium Ions
3. Structure and Properties of Hyperbranched Polyethylene Ionomers
4. Functionalization of Nanomaterials with Hyperbranched Polyethylene Ionomers for Targeted Applications
4.1. Cellulose Nanocrystals
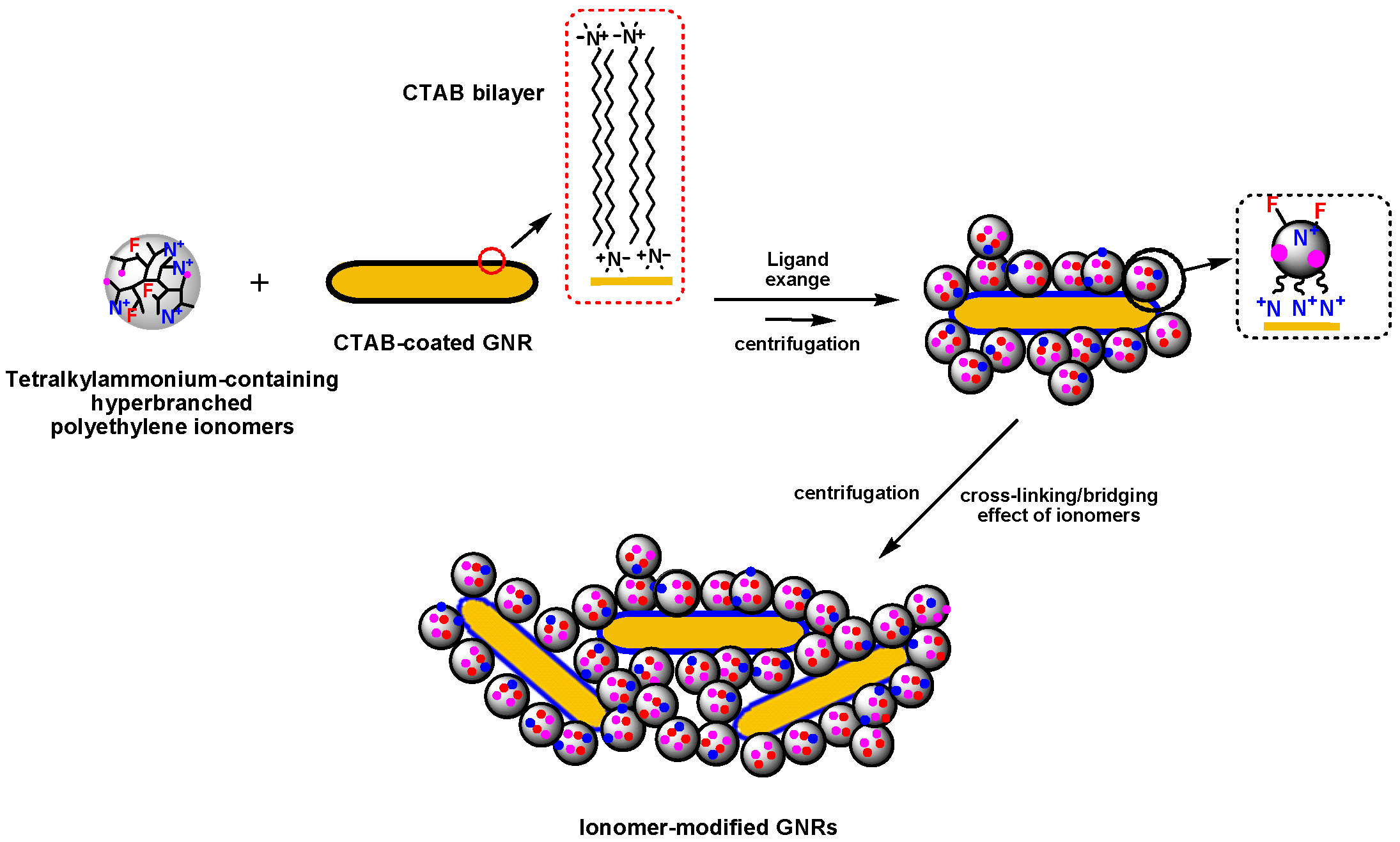
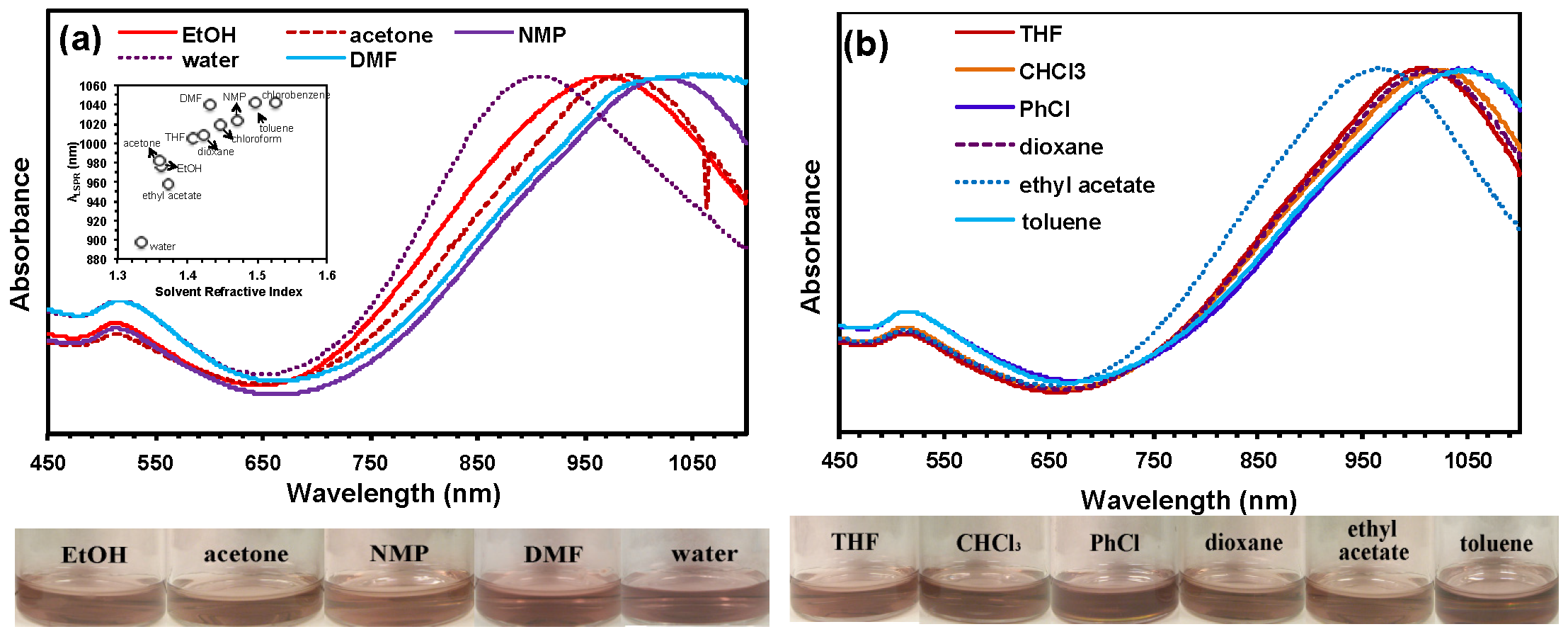
4.2. Gold Nanorods
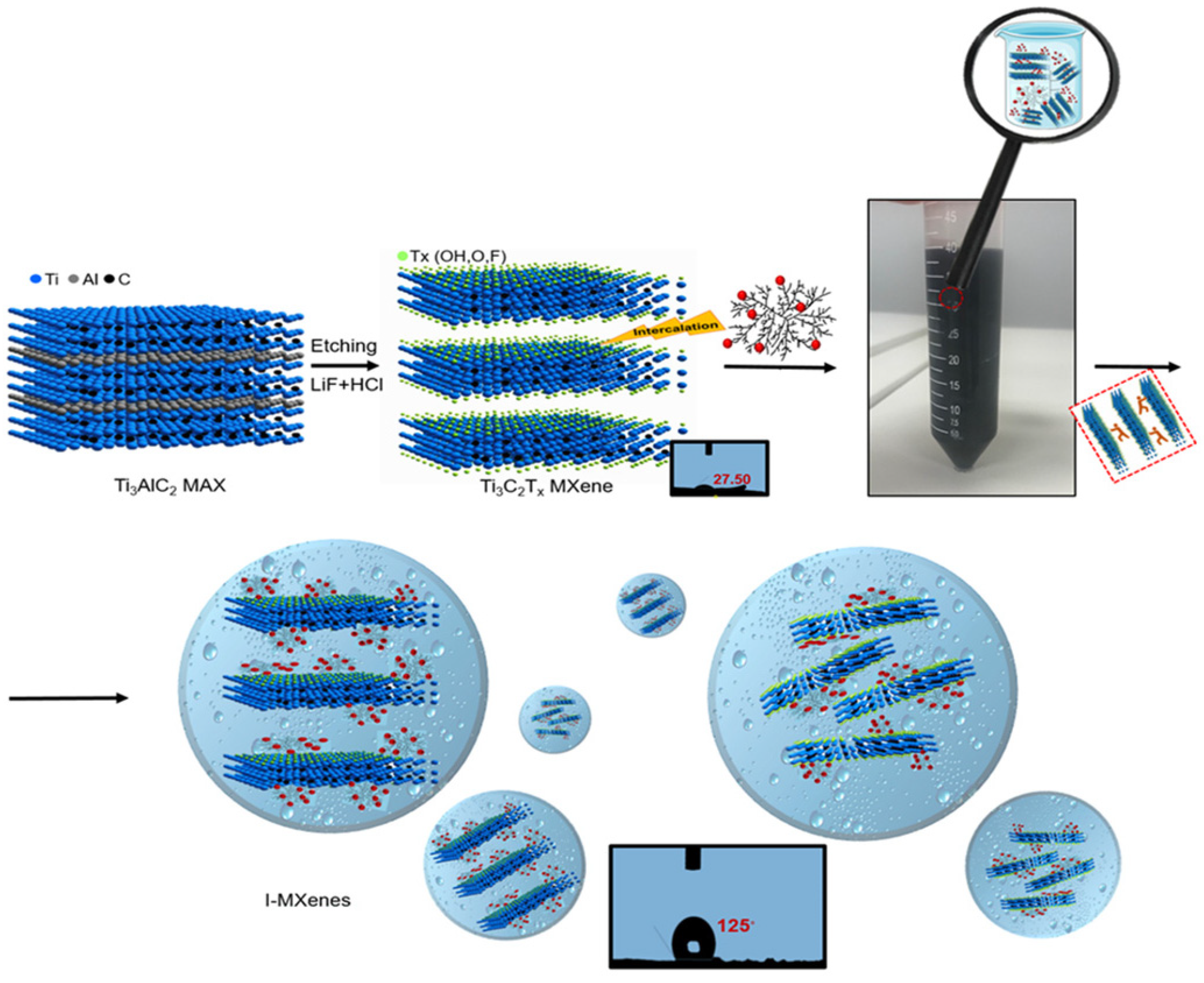
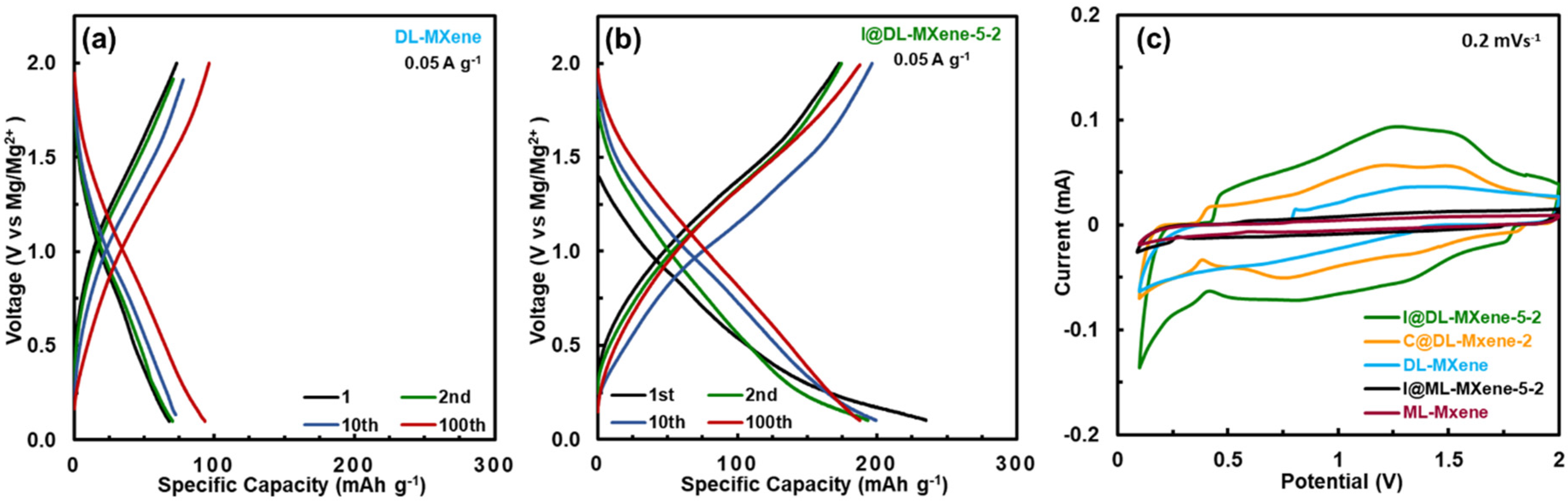
4.3. Ti3C2Tx MXene
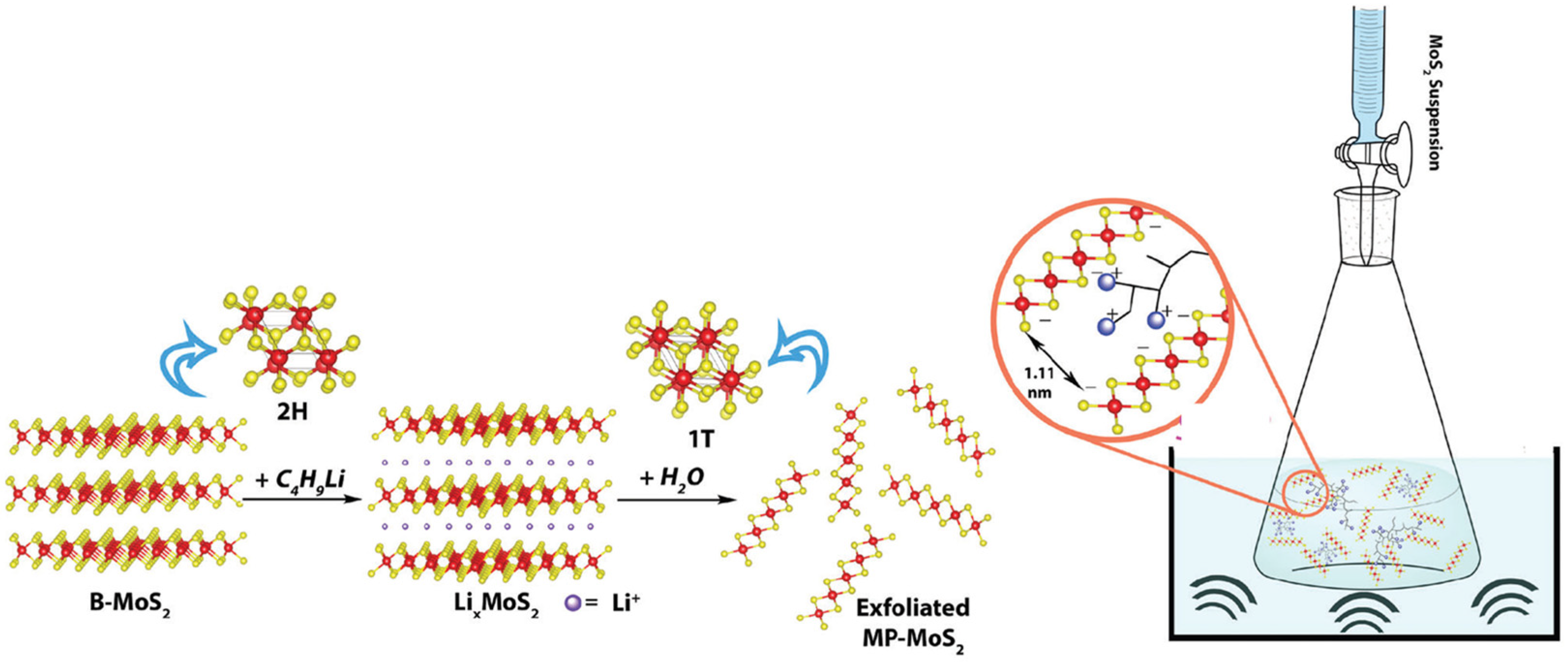
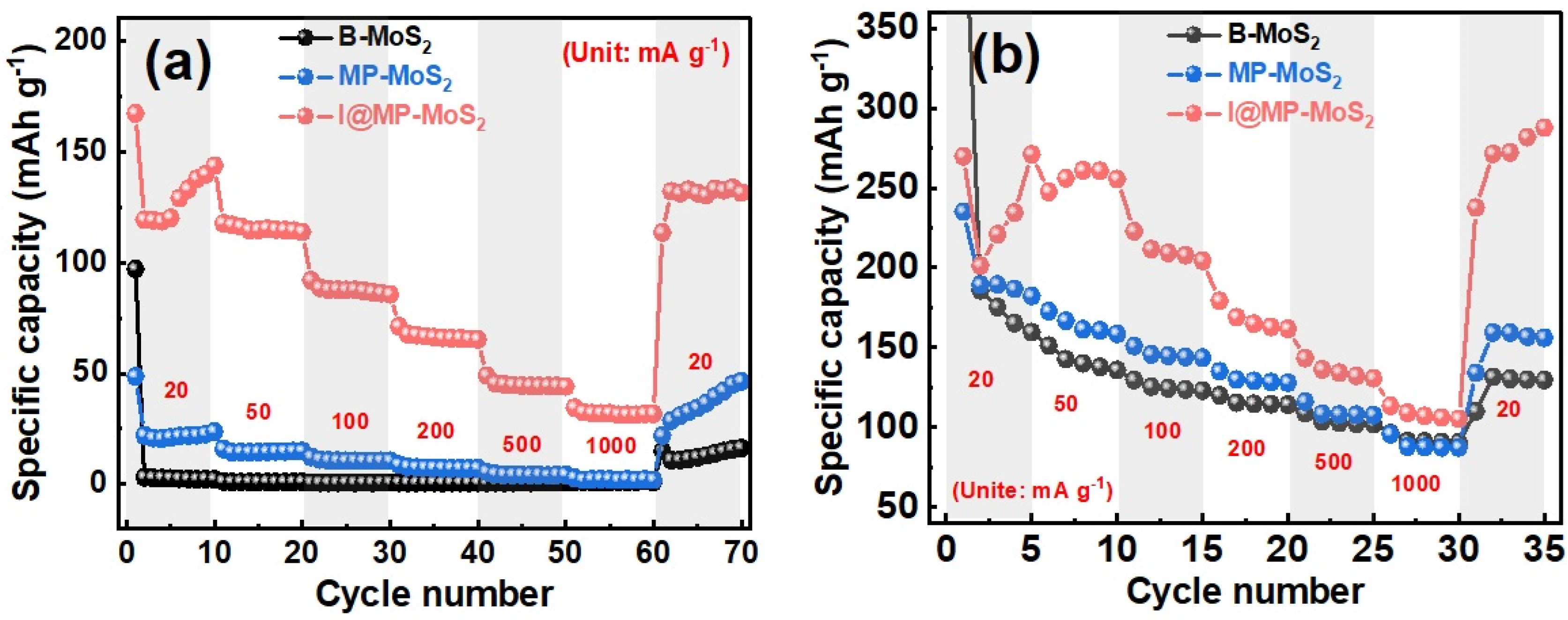
4.4. 1T/2H-MoS2
5. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eisenberg, A.; Kim, J.-S. Introduction to Ionomers; Wiley: Hoboken, NJ, USA, 1998. [Google Scholar]
- Tant, M.R.; Mauritz, K.A.; Wilkes, G.L. Ionomers: Synthesis, Structure, Properties, and Applications, 1st ed.; Blackie Academic & Professional: London, UK, 1997; ISBN 978-0-7514-0392-3. [Google Scholar]
- Grady, B.P. Review and Critical Analysis of the Morphology of Random Ionomers across Many Length Scales. Polym. Eng. Sci. 2008, 48, 1029–1051. [Google Scholar] [CrossRef]
- Xiang, P.; Ye, Z. Hyperbranched Polyethylene Ionomers Containing Cationic Tetralkylammonium Ions Synthesized by Pd–Diimine-Catalyzed Direct Ethylene Copolymerization with Ionic Liquid Comonomers. Macromolecules 2015, 48, 6096–6107. [Google Scholar] [CrossRef]
- Aitken, B.S.; Buitrago, C.F.; Heffley, J.D.; Lee, M.; Gibson, H.W.; Winey, K.I.; Wagener, K.B. Precision Ionomers: Synthesis and Thermal/Mechanical Characterization. Macromolecules 2012, 45, 681–687. [Google Scholar] [CrossRef]
- Weiss, R.A.; Agarwal, P.K. Influence of Intermolecular Interactions on the Melt Rheology of a Propylene–Acrylic Acid Copolymer and Its Salts. J. Appl. Polym. Sci. 1981, 26, 449–462. [Google Scholar] [CrossRef]
- Landoll, L.M.; Breslow, D.S. Polypropylene Ionomers. J. Polym. Sci. Part Polym. Chem. 1989, 27, 2189–2201. [Google Scholar] [CrossRef]
- Dassaud, J.P.; Gallot, B.; Guyot, A.; Spitz, R.; Beautemps, J.P.; Williams, C.; Bourgaux, C.; Eisenberg, A. Sulfonated Polypropylene Ionomers. Polym. Adv. Technol. 1994, 5, 79–89. [Google Scholar] [CrossRef]
- Lee, J.A.; Kontopoulou, M.; Parent, J.S. Synthesis and Characterization of Polyethylene-Based Ionomer Nanocomposites. Polymer 2005, 46, 5040–5049. [Google Scholar] [CrossRef]
- Zhang, M.; Kim, H.K.; Chalkova, E.; Mark, F.; Lvov, S.N.; Chung, T.C.M. New Polyethylene Based Anion Exchange Membranes (PE–AEMs) with High Ionic Conductivity. Macromolecules 2011, 44, 5937–5946. [Google Scholar] [CrossRef]
- Zhang, M.; Yuan, X.; Wang, L.; Chung, T.C.M.; Huang, T.; deGroot, W. Synthesis and Characterization of Well-Controlled Isotactic Polypropylene Ionomers Containing Ammonium Ion Groups. Macromolecules 2014, 47, 571–581. [Google Scholar] [CrossRef]
- Clark, T.J.; Robertson, N.J.; Kostalik IV, H.A.; Lobkovsky, E.B.; Mutolo, P.F.; Abruña, H.D.; Coates, G.W. A Ring-Opening Metathesis Polymerization Route to Alkaline Anion Exchange Membranes: Development of Hydroxide-Conducting Thin Films from an Ammonium-Functionalized Monomer. J. Am. Chem. Soc. 2009, 131, 12888–12889. [Google Scholar] [CrossRef]
- Robertson, N.J.; Kostalik, H.A.I.; Clark, T.J.; Mutolo, P.F.; Abruña, H.D.; Coates, G.W. Tunable High Performance Cross-Linked Alkaline Anion Exchange Membranes for Fuel Cell Applications. J. Am. Chem. Soc. 2010, 132, 3400–3404. [Google Scholar] [CrossRef] [PubMed]
- Baughman, T.W.; Chan, C.D.; Winey, K.I.; Wagener, K.B. Synthesis and Morphology of Well-Defined Poly(Ethylene-Co-Acrylic Acid) Copolymers. Macromolecules 2007, 40, 6564–6571. [Google Scholar] [CrossRef]
- Aitken, B.S.; Lee, M.; Hunley, M.T.; Gibson, H.W.; Wagener, K.B. Synthesis of Precision Ionic Polyolefins Derived from Ionic Liquids. Macromolecules 2010, 43, 1699–1701. [Google Scholar] [CrossRef]
- Johnson, L.K.; Killian, C.M.; Brookhart, M. New Pd(II)- and Ni(II)-Based Catalysts for Polymerization of Ethylene and Alpha-Olefins. J. Am. Chem. Soc. 1995, 117, 6414–6415. [Google Scholar] [CrossRef]
- Ittel, S.D.; Johnson, L.K.; Brookhart, M. Late-Metal Catalysts for Ethylene Homo- and Copolymerization. Chem. Rev. 2000, 100, 1169–1204. [Google Scholar] [CrossRef]
- Johnson, L.K.; Mecking, S.; Brookhart, M. Copolymerization of Ethylene and Propylene with Functionalized Vinyl Monomers by Palladium(II) Catalysts. J. Am. Chem. Soc. 1996, 118, 267–268. [Google Scholar] [CrossRef]
- Chen, G.; Ma, X.S.; Guan, Z. Synthesis of Functional Olefin Copolymers with Controllable Topologies Using a Chain-Walking Catalyst. J. Am. Chem. Soc. 2003, 125, 6697–6704. [Google Scholar] [CrossRef] [PubMed]
- Held, A.; Bauers, F.M.; Mecking, S. Coordination Polymerization of Ethylene in Water by Pd(Ii) and Ni(Ii) Catalysts. Chem. Commun. 2000, 301–302. [Google Scholar] [CrossRef]
- Guan, Z.; Cotts, P.M.; McCord, E.F.; McLain, S.J. Chain Walking: A New Strategy to Control Polymer Topology. Science 1999, 283, 2059–2062. [Google Scholar] [CrossRef]
- Ye, Z.; Zhu, S. Newtonian Flow Behavior of Hyperbranched High-Molecular-Weight Polyethylenes Produced with a Pd−Diimine Catalyst and Its Dependence on Chain Topology. Macromolecules 2003, 36, 2194–2197. [Google Scholar] [CrossRef]
- Xiang, P.; Ye, Z.; Morgan, S.; Xia, X.; Liu, W. Tuning Polyethylene Chain Topology via Ring Incorporation in Chain Walking Ethylene Polymerization. Macromolecules 2009, 42, 4946–4949. [Google Scholar] [CrossRef]
- Gottfried, A.C.; Brookhart, M. Living Polymerization of Ethylene Using Pd(II) α-Diimine Catalysts. Macromolecules 2001, 34, 1140–1142. [Google Scholar] [CrossRef]
- Gottfried, A.C.; Brookhart, M. Living and Block Copolymerization of Ethylene and α-Olefins Using Palladium(II)−α-Diimine Catalysts. Macromolecules 2003, 36, 3085–3100. [Google Scholar] [CrossRef]
- Ye, Z.; Xu, L.; Dong, Z.; Xiang, P. Designing Polyethylenes of Complex Chain Architectures via Pd–Diimine-Catalyzed “Living” Ethylene Polymerization. Chem. Commun. 2013, 49, 6235. [Google Scholar] [CrossRef]
- Zhang, K.; Ye, Z.; Subramanian, R. Synthesis of Block Copolymers of Ethylene with Styrene and n -Butyl Acrylate via a Tandem Strategy Combining Ethylene “Living” Polymerization Catalyzed by a Functionalized Pd−Diimine Catalyst with Atom Transfer Radical Polymerization. Macromolecules 2008, 41, 640–649. [Google Scholar] [CrossRef]
- Xu, Y.; Xiang, P.; Ye, Z.; Wang, W.-J. Hyperbranched−Linear Polyethylene Block Polymers Constructed with Chain Blocks of Hybrid Chain Topologies via One-Pot Stagewise Chain Walking Ethylene “Living” Polymerization. Macromolecules 2010, 43, 8026–8038. [Google Scholar] [CrossRef]
- Zhang, K.; Ye, Z.; Subramanian, R. A Trinuclear Pd−Diimine Catalyst for “Core-First” Synthesis of Three-Arm Star Polyethylenes via Ethylene “Living” Polymerization. Macromolecules 2009, 42, 2313–2316. [Google Scholar] [CrossRef]
- Zhang, Z.; Ye, Z. A Ligand Exchange Strategy for One-Pot Sequential Synthesis of (Hyperbranched Polyethylene)-b-(Linear Polyketone) Block Polymers. Chem. Commun. 2012, 48, 7940–7942. [Google Scholar] [CrossRef]
- Xu, L.; Ye, Z. A Pd–Diimine Catalytic Inimer for Synthesis of Polyethylenes of Hyperbranched-on-Hyperbranched and Star Architectures. Chem. Commun. 2013, 49, 8800–8802. [Google Scholar] [CrossRef]
- Dai, S.; Chen, C. Palladium-Catalyzed Direct Synthesis of Various Branched, Carboxylic Acid-Functionalized Polyolefins: Characterization, Derivatization, and Properties. Macromolecules 2018, 51, 6818–6824. [Google Scholar] [CrossRef]
- Jian, Z.; Leicht, H.; Mecking, S. Direct Synthesis of Imidazolium-Functional Polyethylene by Insertion Copolymerization. Macromol. Rapid Commun. 2016, 37, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Ye, Z.; Cui, Q.; Gu, Z. Noncovalent Nonspecific Functionalization and Solubilization of Multi-Walled Carbon Nanotubes at High Concentrations with a Hyperbranched Polyethylene. Macromol. Chem. Phys. 2009, 210, 2194–2202. [Google Scholar] [CrossRef]
- Chen, M.; Jiang, B.; Wang, J.; Yang, Y.; Ye, Z. Effects of Counter Anion Type on Melt Rheological Properties of Hyperbranched Polyethylene Ionomers Containing Covalently Bound Quaternary Ammonium Cations. Macromol. React. Eng. 2022, 16, 2200021. [Google Scholar] [CrossRef]
- Register, R.A.; Prud’homme, R.K. Melt Rheology. In Ionomers: Synthesis, Structure, Properties and Applications; Tant, M.R., Mauritz, K.A., Wilkes, G.L., Eds.; Springer: Dordrecht, The Netherlands, 1997; pp. 208–260. ISBN 978-94-009-1461-2. [Google Scholar]
- Weiss, R.A.; Zhao, H. Rheological Behavior of Oligomeric Ionomers. J. Rheol. 2009, 53, 191–213. [Google Scholar] [CrossRef]
- Ling, G.H.; Wang, Y.; Weiss, R.A. Linear Viscoelastic and Uniaxial Extensional Rheology of Alkali Metal Neutralized Sulfonated Oligostyrene Ionomer Melts. Macromolecules 2012, 45, 481–490. [Google Scholar] [CrossRef]
- Huang, L.; Ye, Z.; Berry, R. Modification of Cellulose Nanocrystals with Quaternary Ammonium-Containing Hyperbranched Polyethylene Ionomers by Ionic Assembly. ACS Sustain. Chem. Eng. 2016, 4, 4937–4950. [Google Scholar] [CrossRef]
- Dong, Z.; Xiang, P.; Huang, L.; Ye, Z. Efficient, Robust Surface Functionalization and Stabilization of Gold Nanorods with Quaternary Ammonium-Containing Ionomers as Multidentate Macromolecular Ligands. RSC Adv. 2016, 6, 43574–43590. [Google Scholar] [CrossRef]
- Raisi, B.; Huang, L.; Ye, Z. Modification of Ti3C2Tx MXene with Hyperbranched Polyethylene Ionomers: Stable Dispersions in Nonpolar/Low-Polarity Organic Solvents, Oxidation Protection, and Potential Application in Supercapacitors. J. Mater. Chem. A 2023, 11, 17167–17187. [Google Scholar] [CrossRef]
- Raisi, B.; Liu, X.; Rahmatinejad, J.; Ye, Z. Pillar-Structured Ti3C2Tx MXene with Engineered Interlayer Spacing for High-Performance Magnesium Batteries. Small Methods 2024, 8, 2400004. [Google Scholar] [CrossRef]
- Rahmatinejad, J.; Raisi, B.; Liu, X.; Zhang, X.; Sadeghi Chevinli, A.; Yang, L.; Ye, Z. 1T-2H Mixed-Phase MoS2 Stabilized with a Hyperbranched Polyethylene Ionomer for Mg2+/Li+ Co-Intercalation Toward High-Capacity Dual-Salt Batteries. Small 2024, 20, 2304878. [Google Scholar] [CrossRef]
- Moon, R.J.; Martini, A.; Nairn, J.; Simonsen, J.; Youngblood, J. Cellulose Nanomaterials Review: Structure, Properties and Nanocomposites. Chem. Soc. Rev. 2011, 40, 3941–3994. [Google Scholar] [CrossRef] [PubMed]
- Šturcová, A.; Davies, G.R.; Eichhorn, S.J. Elastic Modulus and Stress-Transfer Properties of Tunicate Cellulose Whiskers. Biomacromolecules 2005, 6, 1055–1061. [Google Scholar] [CrossRef]
- Dreaden, E.C.; Alkilany, A.M.; Huang, X.; Murphy, C.J.; El-Sayed, M.A. The Golden Age: Gold Nanoparticles for Biomedicine. Chem. Soc. Rev. 2012, 41, 2740–2779. [Google Scholar] [CrossRef]
- Jana, N.R.; Gearheart, L.; Murphy, C.J. Wet Chemical Synthesis of High Aspect Ratio Cylindrical Gold Nanorods. J. Phys. Chem. B 2001, 105, 4065–4067. [Google Scholar] [CrossRef]
- Nikoobakht, B.; El-Sayed, M.A. Evidence for Bilayer Assembly of Cationic Surfactants on the Surface of Gold Nanorods. Langmuir 2001, 17, 6368–6374. [Google Scholar] [CrossRef]
- Takahashi, H.; Niidome, Y.; Niidome, T.; Kaneko, K.; Kawasaki, H.; Yamada, S. Modification of Gold Nanorods Using Phosphatidylcholine to Reduce Cytotoxicity. Langmuir 2006, 22, 2–5. [Google Scholar] [CrossRef]
- Naguib, M.; Mashtalir, O.; Carle, J.; Presser, V.; Lu, J.; Hultman, L.; Gogotsi, Y.; Barsoum, M.W. Two-Dimensional Transition Metal Carbides. ACS Nano 2012, 6, 1322–1331. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Anasori, B. The Rise of MXenes. ACS Nano 2019, 13, 8491–8494. [Google Scholar] [CrossRef]
- Maleski, K.; Mochalin, V.N.; Gogotsi, Y. Dispersions of Two-Dimensional Titanium Carbide MXene in Organic Solvents. Chem. Mater. 2017, 29, 1632–1640. [Google Scholar] [CrossRef]
- Naguib, M.; Mashtalir, O.; Lukatskaya, M.R.; Dyatkin, B.; Zhang, C.; Presser, V.; Gogotsi, Y.; Barsoum, M.W. One-Step Synthesis of Nanocrystalline Transition Metal Oxides on Thin Sheets of Disordered Graphitic Carbon by Oxidation of MXenes. Chem. Commun. 2014, 50, 7420–7423. [Google Scholar] [CrossRef]
- Attias, R.; Salama, M.; Hirsch, B.; Goffer, Y.; Aurbach, D. Anode-Electrolyte Interfaces in Secondary Magnesium Batteries. Joule 2019, 3, 27–52. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, Y.; Miao, Y.; Yang, M.; Zhao, X.; Shen, X. High-Energy Interlayer-Expanded Copper Sulfide Cathode Material in Non-Corrosive Electrolyte for Rechargeable Magnesium Batteries. Adv. Mater. 2020, 32, 1905524. [Google Scholar] [CrossRef]
- Hadler-Jacobsen, J.; Schnell, S.K. The Importance of Stacking and Coordination for Li, Na, and Mg Diffusion and Intercalation in Ti3C2T2 MXene. Adv. Mater. Interfaces 2022, 9, 2200014. [Google Scholar] [CrossRef]
- Chhowalla, M.; Shin, H.S.; Eda, G.; Li, L.-J.; Loh, K.P.; Zhang, H. The Chemistry of Two-Dimensional Layered Transition Metal Dichalcogenide Nanosheets. Nat. Chem. 2013, 5, 263–275. [Google Scholar] [CrossRef]
- Shi, S.; Sun, Z.; Hu, Y.H. Synthesis, Stabilization and Applications of 2-Dimensional 1T Metallic MoS2. J. Mater. Chem. A 2018, 6, 23932–23977. [Google Scholar] [CrossRef]
- Voiry, D.; Mohite, A.; Chhowalla, M. Phase Engineering of Transition Metal Dichalcogenides. Chem. Soc. Rev. 2015, 44, 2702–2712. [Google Scholar] [CrossRef]
- Rahmatinejad, J.; Ye, Z. Advanced MoS2 Nanocomposites for Post-Lithium-Ion Batteries. Chem. Eng. J. 2024, 500, 156872. [Google Scholar] [CrossRef]
- Rahmatinejad, J.; Liu, X.; Raisi, B.; Ye, Z. Synergistic Cathode Design for High-Performance Dual-Salt Magnesium/Lithium-Ion Batteries Using 2D/2D 1T/2H-MoS2@Ti3C2T MXene Nanocomposite. Small 2024, 20, 2401391. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Z.; Rahmatinejad, J.; Raisi, B.; Dai, P. Hyperbranched Polyethylene Ionomers Containing Quaternary Ammonium Ions and Their Functionalization of Nanomaterials. Nanomaterials 2025, 15, 525. https://doi.org/10.3390/nano15070525
Ye Z, Rahmatinejad J, Raisi B, Dai P. Hyperbranched Polyethylene Ionomers Containing Quaternary Ammonium Ions and Their Functionalization of Nanomaterials. Nanomaterials. 2025; 15(7):525. https://doi.org/10.3390/nano15070525
Chicago/Turabian StyleYe, Zhibin, Jalal Rahmatinejad, Bahareh Raisi, and Peishuai Dai. 2025. "Hyperbranched Polyethylene Ionomers Containing Quaternary Ammonium Ions and Their Functionalization of Nanomaterials" Nanomaterials 15, no. 7: 525. https://doi.org/10.3390/nano15070525
APA StyleYe, Z., Rahmatinejad, J., Raisi, B., & Dai, P. (2025). Hyperbranched Polyethylene Ionomers Containing Quaternary Ammonium Ions and Their Functionalization of Nanomaterials. Nanomaterials, 15(7), 525. https://doi.org/10.3390/nano15070525