
Influence of Electron Transfer Mediators in the Pd(II)-Catalyzed Oxidative Carbonylation of Aniline


Abstract

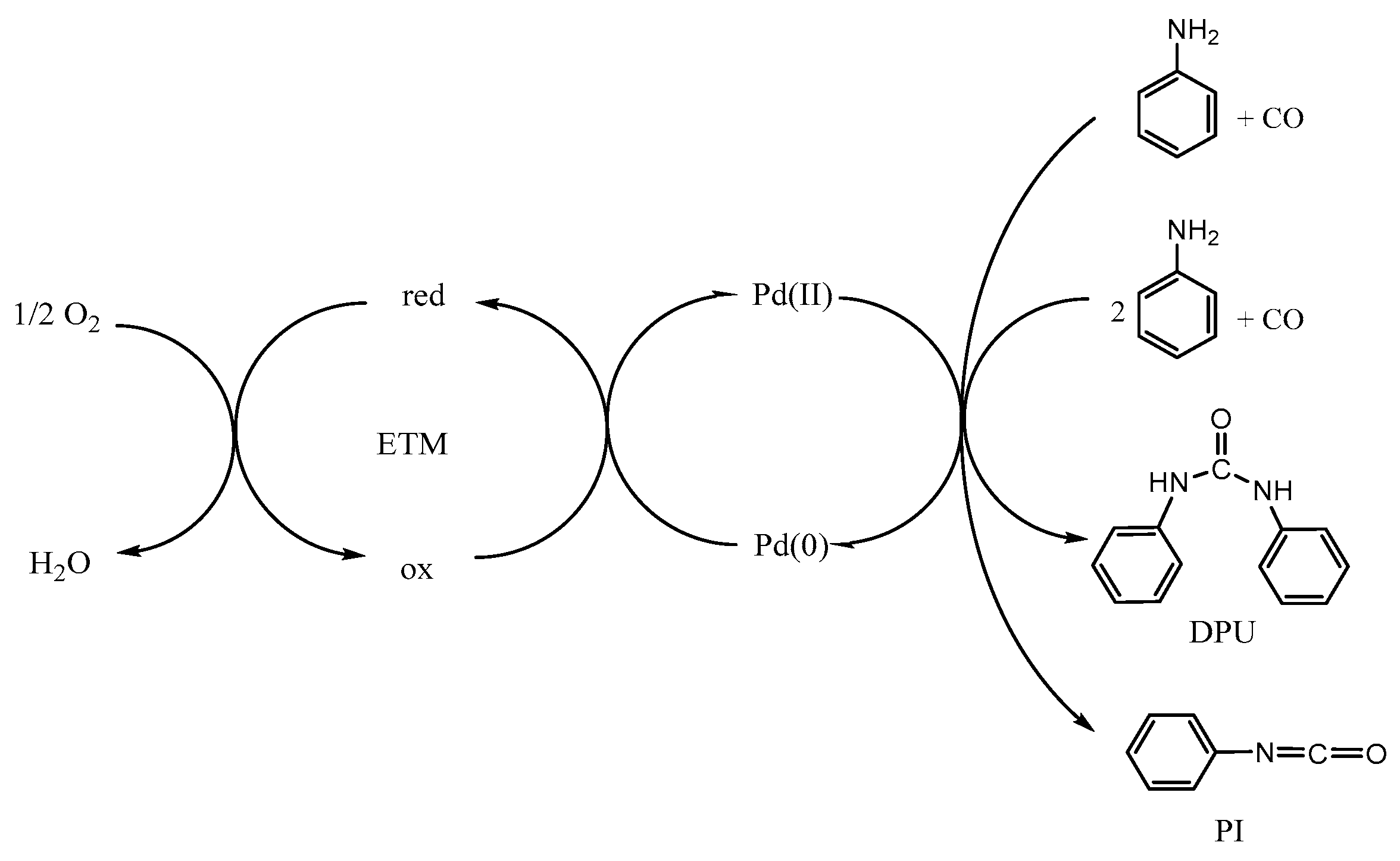
1. Introduction
2. Results and Discussion
2.1. Preliminary Experiments
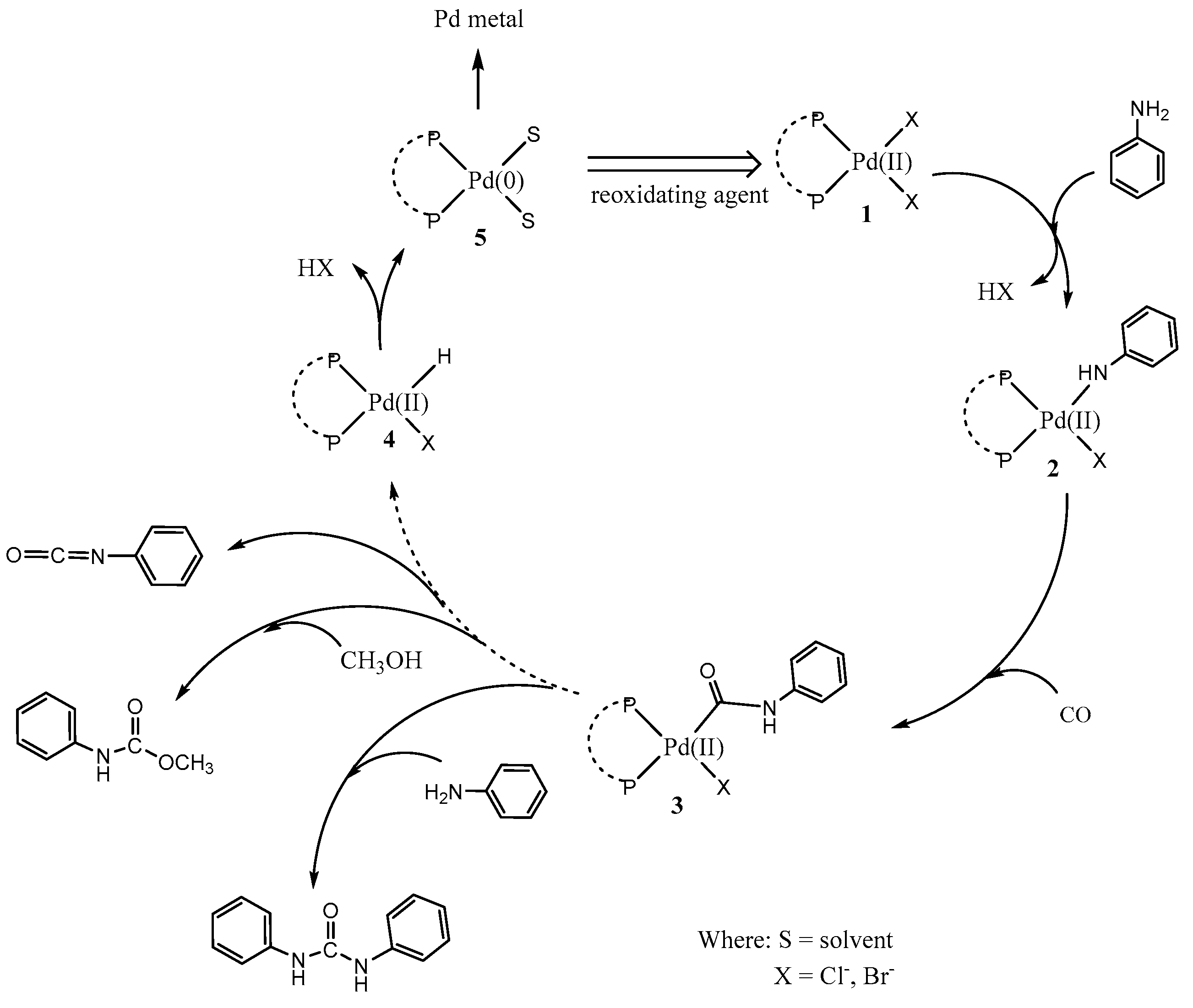
2.2. Influence of Diphosphine Ligands on the Catalytic Activity of Pd(II) Complexes
2.3. The Catalytic Activity of the FeCl3/[PdCl2(dppf)] Couple
2.4. Influence of Promoters on the Catalytic Activity of the FeCl3/[PdCl2(dppf)] Couple
2.5. Catalytic Activity of the [PdCl2(dppf)]/FeCl3/LiBr/O2 Electron Transfer System
3. Materials and Methods
3.1. Reagents
3.2. Equipment and Characterization
3.3. Experimental Setup
3.4. Catalytic Reactions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tonelli, F.; Evans, S.; Taticchi, P. Industrial sustainability: Challenges, perspectives, actions. Int. J. Business Innov. Res. 2013, 7, 143–163. [Google Scholar] [CrossRef]
- Mengistu, A.T.; Panizzolo, R. Indicators and Framework for Measuring Industrial Sustainability in Italian Footwear Small and Medium Enterprises. Sustainability 2021, 13, 5472. [Google Scholar] [CrossRef]
- Arena, M.; Ciceri, N.D.; Terzi, S.; Bengo, I.; Azzone, G.; Garetti, M.A. A state of the art of industrial sustainability: Definitions, tools and metrics. Int. J. Prod. Lifecycle Manag. 2009, 4, 207–251. [Google Scholar] [CrossRef]
- Babad, H.; Zeiler, A.G. The chemistry of phosgene. Chem Rev. 1973, 73, 75–91. [Google Scholar] [CrossRef]
- Cotarca, L.; Eckert, H. Phosgenations-a Handbook; Wiley-VCH: Weinheim, Germany, 2003; ISBN 3-527-29823-1. [Google Scholar]
- Sharma, A.K.; Kumar, N. Phosgene: Risk Assessment, Environmental, and Health Hazard, in Hazardous Gases; Singh, J., Kaushik, R.D., Chawla, M., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 313–325. ISBN 9780323898577. [Google Scholar] [CrossRef]
- Pauluhn, J. Phosgene inhalation toxicity: Update on mechanisms and mechanism-based treatment strategies. Toxicology 2021, 450, 152682. [Google Scholar] [CrossRef] [PubMed]
- Slocombe, R.J.; Hardy, E.E.; Saunders, J.H.; Jenkins, R.L. Phosgene derivatives. The preparation of isocyanates, carbamyl chlorides and cyanuric acid. J. Am. Chem. Soc. 1950, 72, 1888–1891. [Google Scholar] [CrossRef]
- Saunders, J.H.; Slocombe, R.J. The chemistry of the organic isocyanates. Chem. Rev. 1948, 43, 203–218. [Google Scholar] [CrossRef]
- Arnold, R.G.; Nelson, J.A.; Verbanc, J.J. Recent advances in isocyanate chemistry. Chem. Rev. 1957, 57, 47–76. [Google Scholar] [CrossRef]
- Ozaki, S. Recent advances in isocyanate chemistry. Chem Rev. 1972, 72, 457–496. [Google Scholar] [CrossRef]
- Li, T.; Lv, S.Y.; Chen, J.; Gao, C.M.; Zhang, S.F.; Liu, M.Z. Progress in Polymer-based Environment-responsive Fertilizers. Acta Polym. Sin. 2018, 2018, 336–348. [Google Scholar] [CrossRef]
- Dhawan, G.; Sumana, G.; Malhotra, B.D. Recent developments in urea biosensors. Biochem. Eng. J. 2009, 44, 42–52. [Google Scholar] [CrossRef]
- Li, H.Q.; Lv, P.C.; Yan, T.; Zhu, H.L. Urea Derivatives as Anticancer Agents. Anti-Cancer Agents Med. Chem. 2009, 9, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.; Kirino, O.; Takayama, C.; Kamoshita, K. Studies on fungicidal activity of N-phenylcarbamates: IV. Determination of the hydrophobicity of N-phenylcarbamates by high-performance liquid chromatography. J. Chromat. A 1988, 436, 316–322. [Google Scholar] [CrossRef]
- Gama, N.; Ferreira, A.; Barros-Timmons, A. Polyurethane Foams: Past, Present, and Future. Materials 2018, 11, 1841. [Google Scholar] [CrossRef] [PubMed]
- Seymour, R.B.; Kauffman, G.B. Polyurethanes: A class of modern versatile materials. J. Chem. Ed. 1992, 69, 909–910. [Google Scholar] [CrossRef]
- Wang, X.-K.; Yan, S.-R.; Li, Z.-H.; Fan, K.-N.; Kang, M.-G.; Peng, S.-Y. A novel non-phosgene approach to the synthesis of methyl N-phenyl carbamate by a reaction of methanol with phenylurea. Korean J. Chem. Eng. 2004, 21, 378–380. [Google Scholar] [CrossRef]
- Giannoccaro, P. Palladium Catalysed Conversion of N, N′-Diphenylurea into Carbamate Esters. Inorg. Chim. Acta 1988, 142, 81–84. [Google Scholar] [CrossRef]
- Yang, B.; Wang, D.; Lin, H.; Sun, J.; Wang, X. Synthesis of dimethyl carbonate from urea and methanol catalyzed by the metallic compounds at atmospheric pressure. Catal. Comm. 2006, 7, 472–477. [Google Scholar] [CrossRef]
- Tsuda, A. In situ photo-on-demand phosgenation reactions with chloroform for syntheses of polycarbonates and polyurethanes. Polym. J. 2023, 55, 903–912. [Google Scholar] [CrossRef]
- Delledonne, D.; Rivetti, F.; Romano, U. Developments in the production and application of dimethylcarbonate. App. Catal. A Gen. 2001, 221, 241–251. [Google Scholar] [CrossRef]
- Romano, U.; Tesei, R.; Massi Mauri, M.; Rebora, P. Synthesis of Dimethyl Carbonate from Methanol, Carbon Monoxide, and Oxygen Catalyzed by Copper Compounds. Ind. Eng. Chem. Prod. Res. Dev. 1980, 19, 396–403. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, N.; Wei, W.; Sun, Y. Synthesis of Dimethyl Carbonate from Urea and Methanol over ZnO. Ind. Eng. Chem. Res. 2005, 44, 7596–7599. [Google Scholar] [CrossRef]
- Park, S.-E.; Chang, J.-S.; Lee, K.-W. Carbon Dioxide Utilization for Global Sustainability, 1st ed.; Park, S.-E., Chang, J.-S., Lee, K.-W., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2004; Volume 153, p. 197. ISBN 978-0-444-51600-8. [Google Scholar]
- de Groot, F.F.T.; Lammerink, R.R.G.J.; Heidemann, C.; van der Werff, M.P.M.; Garcia, T.C.; van der Ham, L.A.G.J.; van den Berg, H. The industrial production of dimethyl carbonate from methanol and carbon dioxide. Chem. Eng. Trans. 2014, 39, 1561–1566. [Google Scholar] [CrossRef]
- Sk, N.A.; Ali, S.I.; Bijani, S. Synthesis of aromatic carbamate via palladium catalyzed reductive carbonylation reaction of Nitro benzene: An alternative approach with different nucleophiles other than MeOH. J. Organomet. Chem. 2023, 990, 122651. [Google Scholar] [CrossRef]
- Vavasori, A.; Capponi, M.; Ronchin, L. A New Pd-Based Catalytic System for the Reductive Carbonylation of Nitrobenzene to Form N-(4-hydroxyphenyl)acetamide Selectively in One Pot. Reactions 2023, 4, 725–736. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Tran, A.V.; Lee, H.J.; Baek, J.; Kim, Y.J. Palladium-catalyzed reductive carbonylation of nitrobenzene for producing phenyl isocyanate. Tetrahedron Lett. 2019, 60, 151310. [Google Scholar] [CrossRef]
- Beller, M.; Steinhoff, B.A.; Zoeller, J.R.; Cole-Hamilton, D.J.; Drent, E.; Wu, X.F.; Neumann, H.; Ito, S.; Nozaki, K. Carbonylation, in Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Four Volumes, 3rd ed.; Cornils, B., Herrmann, W.A., Beller, M., Paciello, R., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar] [CrossRef]
- Vavasori, A.; Ronchin, L. Phosgene-free synthesis of 1,3-diphenylurea via catalyzed reductive carbonylation of nitrobenzene. Pure Appl. Chem. 2012, 84, 473–484. [Google Scholar] [CrossRef]
- Ragaini, F.; Cenini, S. Mechanistic studies of palladium-catalysed carbonylation reactions of nitro compounds to isocyanates, carbamates and ureas. J. Mol. Catal. A Chem. 1996, 109, 1–25. [Google Scholar] [CrossRef]
- Weigert, F.J. Synthesis of aryl isocyanates from nitro compounds and carbon monoxide. J. Org. Chem. 1973, 38, 1316–1319. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Y.-H.; Yi, H.; Lei, A. An Update on Oxidative C-H Carbonylation with CO. ACS Catal. 2022, 12, 7470–7485. [Google Scholar] [CrossRef]
- Ferretti, F.; Barraco, E.; Gatti, C.; Ramadan, D.R.; Ragaini, F. Palladium/iodide catalyzed oxidative carbonylation of aniline to diphenylurea: Effect of ppm amounts of iron salts. J. Catal. 2019, 369, 257–266. [Google Scholar] [CrossRef]
- Giannoccaro, P.; Ferragina, C.; Gargano, M.; Quaranta, E. Pd-catalysed oxidative carbonylation of amino alcohols to N,N’-bis(hydroxyalkyl)ureas under mild conditions using molecular oxygen as the oxidant. App. Catal. A Gen. 2010, 375, 78–84. [Google Scholar] [CrossRef]
- Díaz, D.J.; Darko, A.K.; McElwee-White, L. Transition Metal-Catalyzed Oxidative Carbonylation of Amines to Ureas. Eur. J. Org. Chem. 2007, 2007, 4453–4465. [Google Scholar] [CrossRef]
- Orito, K.; Miyazawa, M.; Nakamura, T.; Horibata, A.; Ushito, H.; Nagasaki, H.; Yuguchi, M.; Yamashita, S.; Yamazaki, T.; Tokuda, M. Pd(OAc)2-Catalyzed Carbonylation of Amines. J. Org. Chem. 2006, 71, 5951–5958. [Google Scholar] [CrossRef]
- Gabriele, B.; Salerno, G.; Costa, M. Oxidative Carbonylations. In Catalytic Carbonylation Reactions. Topics in Organometallic Chemistry; Beller, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 18, pp. 239–272. [Google Scholar] [CrossRef]
- Gabriele, B.; Salerno, G.; Mancuso, R.; Costa, M. Efficient Synthesis of Ureas by Direct Palladium-Catalyzed Oxidative Carbonylation of Amines. J. Org. Chem. 2004, 69, 4741–4750. [Google Scholar] [CrossRef]
- Fukuoka, S.; Chono, M.; Kohno, M. A novel catalytic synthesis of carbamates by the oxidative alkoxycarbonylation of amines in the presence of platinum group metal and alkali metal halide or onium halide. J. Org. Chem. 1984, 49, 1458–1460. [Google Scholar] [CrossRef]
- Stern, E.W.; Spector, M.L. Carbonylation of Amines in the Presence of Palladium(II) Chloride. A New Route to Isocyanates. J. Org. Chem. 1966, 31, 596–597. [Google Scholar] [CrossRef]
- Li, M.-B.; Bäckvall, J.-E. Efficient Heterogeneous Palladium Catalysts in Oxidative Cascade Reactions. Acc. Chem. Res. 2021, 54, 2275–2286. [Google Scholar] [CrossRef]
- Vavasori, A.; Fantinel, B.; Ronchin, L.; Zanrosso, F.; Bulybayev, M.; Kudaibergenov, N.; Shalmagambetov, K.; Zhaksylykova, G. New Sustainable Pd(II)/Fe(III) Catalytic System Very Efficient in the Hydromethoxycarbonylation of 1-octene. Period. Polytech. Chem. Eng. 2024, 68, 16–22. [Google Scholar] [CrossRef]
- Liu, J.; Guomundsson, A.; Bäckvall, J.-E. Efficient Aerobic Oxidation of Organic Molecules by Multistep Electron Transfer. Angew. Chem. 2021, 133, 15818–15836. [Google Scholar] [CrossRef]
- Ronchin, L.; Vavasori, A.; Amadio, E.; Cavinato, G.; Toniolo, L. Oxidative carbonylation of phenols catalyzed by homogeneous and heterogeneous Pd precursors. J. Mol. Catal. A Chem. 2009, 298, 23–30. [Google Scholar] [CrossRef]
- Vavasori, A.; Toniolo, L. Multistep electron transfer catalytic system for the oxidative carbonylation of phenol to diphenyl carbonate. J. Mol. Catal. A Chem. 1999, 139, 109–119. [Google Scholar] [CrossRef]
- Bäckvall, J.E.; Hopkins, R.B.; Greenberg, H.; Mader, M.M.; Awasthi, A.K. Multistep electron transfer in palladium-catalyzed aerobic oxidations via a metal macrocycle-quinone system. J. Am. Chem. Soc. 1990, 112, 5160–5166. [Google Scholar] [CrossRef]
- Chen, B.; Chuang, S.S.C. CuCl2 and PdCl2 catalysts for oxidative carbonylation of aniline with methanol. J. Mol. Catal. A Chem. 2003, 195, 37–45. [Google Scholar] [CrossRef]
- Ragaini, F.; Ferretti, F.; Gatti, C.; Ramadan, D.R. Rebuttal to: Polemic against conclusions drawn in “Palladium/iodide catalyzed oxidative carbonylation of aniline to diphenylurea: Effect of ppm amounts of iron salts” (J. Catal. 2019, 369, 257–266). J. Catal. 2019, 380, 391–395. [Google Scholar] [CrossRef]
- Nataro, C.; Fosbenner, S.M. Synthesis and Characterization of Transition-Metal Complexes Containing 1,1′-Bis(diphenylphosphino)ferrocene. J. Chem. Educ. 2009, 86, 1412–1415. [Google Scholar] [CrossRef]
- Brown, J.M.; Guiry, P. Bite angle dependence of the rate of reductive elimination from diphosphine palladium complexes. Inorg. Chim. Acta 1994, 220, 249–259. [Google Scholar] [CrossRef]
- van Leeuwen, P.W.N.M.; Kamer, P.C.J.; Reek, J.N.H. The bite angle makes the catalyst. Pure Appl. Chem. 1999, 71, 1443–1452. [Google Scholar] [CrossRef]
- Kamer, P.C.J.; van Leeuwen, P.W.N.M. Phosphorus (III) Ligands in Homogeneous Catalysis: Design and Synthesis; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2012; ISBN 978-0-470-66627-2. [Google Scholar]
- Mansell, S.M. Catalytic applications of small bite-angle diphosphorus ligands with single-atom linkers. Dalton Trans. 2017, 46, 15157–15174. [Google Scholar] [CrossRef]
- Messinis, A.M.; Luckham, S.L.J.; Wells, P.P.; Gianolio, D.; Gibson, E.K.; O’Brien, H.M.; Sparkes, H.A.; Davis, S.A.; Callison, J.; Elorriaga, D.; et al. The highly surprising behaviour of diphosphine ligands in iron-catalysed Negishi cross-coupling. Nat. Catal. 2019, 2, 123–133. [Google Scholar] [CrossRef]
- Barclay, J.E.; Hills, A.; Hughes, D.L.; Leigh, G.J. Crystal and molecular structures of four bis(diphosphine) complexes of iron(ii): Bis[1.2-bis(diethylphosphino)ethane]di-iodoiron(ii), dichlorobis[o-phenylenebis(diphenylphosphine)]iron(ii), bis(acetonitrile)bis[o-phenylenebis(diphenylphosphine)]iron(ii)di-iodide, and lodobis-[o-phenylenebis(diphenylphosphine)]iron(ii) iodide. J. Chem. Soc. Dalton Trans. 1988, 2871–2877. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Okada, Y.; Yoshimoto, Y.; Nakamura, M. Tuning chemoselectivity in iron-catalyzed Sonogashira-type reactions using a bisphosphine ligand with peripheral steric bulk: Selective alkynylation of nonactivated alkyl halides. Angew. Chem. Int. Ed. 2011, 50, 10973–10976. [Google Scholar] [CrossRef]
- Özkar, S.; Finke, R.G. Palladium (0) Nanoparticle Formation, Stabilization, and Mechanistic Studies: Pd(acac)2 as a Preferred Precursor, [Bu4N]2HPO4 Stabilizer, plus the Stoichiometry, Kinetics, and Minimal, Four-Step Mechanism of the Palladium Nanoparticle Formation and Subsequent Agglomeration Reactions. Langmuir 2016, 32, 3699–3716. [Google Scholar] [CrossRef] [PubMed]
- Widegren, J.A.; Finke, R.G. A Review of the Problem of Distinguishing True Homogeneous Catalysis from Soluble or Other Metal-Particle Heterogeneous Catalysis under Reducing Conditions. J. Mol. Catal. A Chem. 2003, 198, 3177341. [Google Scholar] [CrossRef]
- Jin, R.; Zeng, C.; Zhou, M.; Chen, Y. Atomically precise colloidal metal nanoclusters and nanoparticles: Fundamentals and opportunities. Chem. Rev. 2016, 116, 10346–10413. [Google Scholar] [CrossRef]
- Seitkalieva, M.M.; Samoylenko, D.E.; Lotsman, K.A.; Rodygin, K.S.; Ananikov, V.P. Metal nanoparticles in ionic liquids: Synthesis and catalytic applications. Coord. Chem. Rev. 2021, 445, 213982. [Google Scholar] [CrossRef]
- Zamani, S.; van der Voort, S.H.E.; Lange, J.-P.; Kersten, S.R.A.; Ruiz, M.P. Polyurethane Recycling: Thermal Decomposition of 1,3-Diphenyl Urea to Isocyanates. Polymers 2023, 15, 2522. [Google Scholar] [CrossRef]
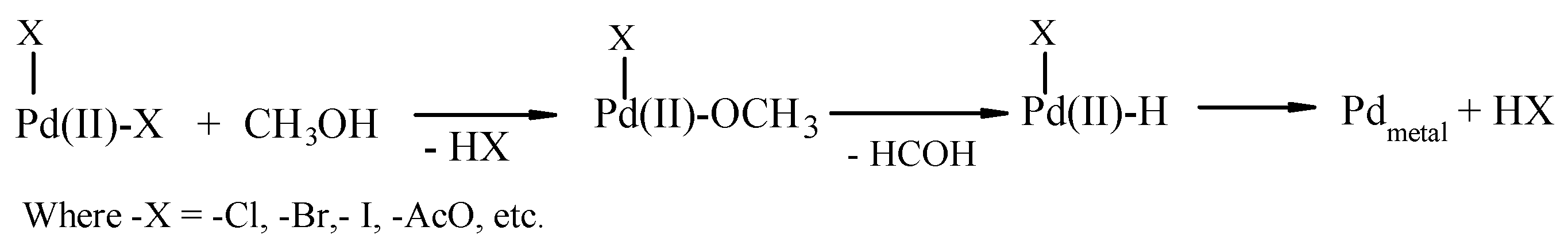

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | T | Catalyst/Cocatalyst | TOF | Selectivity | ||
|---|---|---|---|---|---|---|
| n° | K | h−1 | PI% a | DPU% b | PMC% c | |
| 1 | 373 | PdCl2/CuCl2 | 9 | 2 | 90 | 8 |
| 2 | 373 | PdCl2/FeCl3 | 26 | 8 | 70 | 22 |
| 3 | 373 | Pd(AcO)2/FeCl3 | 22 | 2 | 78 | 20 |
| 4 | 373 | PdCl2/NiCl2 | 12 | 4 | 75 | 21 |
| 5 | 373 | PdCl2/CeBr3 | 5 | 18 | 50 | 32 |
| 6 | 373 | PdCl2/Mn(AcO)2 | 10 | 2 | 80 | 18 |
| 7 | 373 | Pd(AcO)2/Co(AcO)2 | 7 | 8 | 85 | 7 |
| 8 | 373 | Pd(AcO)2/NiCl2 | 11 | n.d. | 88 | 12 |
| 9 | 393 | PdCl2/CuCl2 | 16 | n.d. | 95 | 5 |
| 10 | 393 | PdCl2/FeCl3 | 40 | 5 | 85 | 10 |
| 11 | 393 | Pd(AcO)2/FeCl3 | 38 | 10 | 80 | 10 |
| 12 | 393 | PdCl2/NiCl2 | 23 | 2 | 86 | 12 |
| 13 | 393 | PdCl2/CeBr3 | 8 | 11 | 63 | 26 |
| 14 | 393 | PdCl2/Mn(AcO)2 | 12 | n.d. | 82 | 18 |
| 15 | 393 | Pd(AcO)2/Co(AcO)2 | 15 | 3 | 90 | 7 |
| 16 | 393 | Pd(AcO)2/NiCl2 | 22 | n.d. | 88 | 12 |
| Entry | Pd-Catalyst | TOF | Selectivity | Notes: | ||
|---|---|---|---|---|---|---|
| n° | h−1 | PI molar% | DPU molar% | PMC molar% | ||
| 1 | PdCl2 | 40 | 5 | 85 | 10 | Pd metal |
| 2 | [PdCl2(PPh3)2] | 60 | 1 | 95 | 4 | traces of Pd metal |
| 3 | [PdCl2(dppm)] | 65 | 1 | 96 | 3 | traces of Pd metal |
| 4 | [PdCl2(dppe)] | 71 | traces | 100 | n.d. | / |
| 5 | [PdCl2(dppp)] | 90 | traces | 100 | n.d. | / |
| 6 | [PdCl2(dppb)] | 120 | traces | 100 | n.d. | / |
| 7 | [PdCl2(dppf)] | 180 | traces | 100 | n.d. | / |
| 8 | [PdCl2(Xantphos)] | 80 | traces | 100 | n.d. | / |
| 9 | [Pd(OAc)2(dppf)] | 181 | traces | 100 | n.d. | / |
| 10 | [Pd(TsO)2(dppf)] | 183 | traces | 100 | n.d. | / |
| Entry | T | CO | FeCl3/Pd | TOF |
|---|---|---|---|---|
| n° | K | MPa | mol/mol | h−1 |
| 1 | 393 | 5.0 | 100 | 95 |
| 2 | 393 | 5.0 | 200 | 180 |
| 3 | 393 | 5.0 | 500 | 480 |
| 4 | 393 | 5.0 | 800 | 596 |
| 5 | 393 | 5.0 | 1200 | 637 |
| 6 | 393 | 2.0 | 1200 | 225 |
| 7 | 393 | 3.0 | 1200 | 350 |
| 8 | 393 | 4.0 | 1200 | 498 |
| 9 | 393 | 6.0 | 1200 | 757 |
| 10 | 373 | 5.0 | 1200 | 320 |
| 11 | 383 | 5.0 | 1200 | 455 |
| 12 | 415 | 5.0 | 1200 | 233 |
| Entry | Promoter | TOF |
|---|---|---|
| h−1 | ||
| 1 | / | 637 |
| 2 | NaI | 695 |
| 3 | KI | 700 |
| 4 | LiI | 815 |
| 5 | TBAI | 733 |
| 6 | NaBr | 685 |
| 7 | KBr | 695 |
| 8 | LiBr | 1177 |
| 9 | TBAB | 882 |
| 10 | NaCl | 645 |
| 11 | KCl | 650 |
| 12 | LiCl | 760 |
| 13 | TBAC | 807 |
| Entry | LiBr/[PdCl2(dppf)] | TOF |
|---|---|---|
| h−1 | ||
| 1 | / | 637 |
| 2 | 50/1 | 715 |
| 3 | 100/1 | 950 |
| 4 | 150/1 | 1165 |
| 5 | 200/1 | 1177 |
| 6 | 300/1 | 1170 |
| 7 | 500/1 | 1175 |
| Entry | O2 | TOF | DPU | PI | Byproducts |
|---|---|---|---|---|---|
| n° | MPa | h−1 | mol% | mol% | mol% |
| 1 | 0 | 1177 | 100 | 0 | n.d. |
| 2 | 0.05 | 1900 | 90 | 10 | traces |
| 3 | 0.10 | 2150 | 76 | 24 | traces |
| 4 | 0.20 | 3120 | 65 | 34 | 1 |
| 5 | 0.25 | 3500 | 40 | 58 | 2 |
| 6 | 0.30 | 3930 | 31 | 65 | 4 |
| 7 a | 0.30 | / | n.d. | n.d. | n.d. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vavasori, A.; Ronchin, L.; Pietrobon, L.; Bravo, S. Influence of Electron Transfer Mediators in the Pd(II)-Catalyzed Oxidative Carbonylation of Aniline. Molecules 2025, 30, 2027. https://doi.org/10.3390/molecules30092027
Vavasori A, Ronchin L, Pietrobon L, Bravo S. Influence of Electron Transfer Mediators in the Pd(II)-Catalyzed Oxidative Carbonylation of Aniline. Molecules. 2025; 30(9):2027. https://doi.org/10.3390/molecules30092027
Chicago/Turabian StyleVavasori, Andrea, Lucio Ronchin, Luca Pietrobon, and Sara Bravo. 2025. "Influence of Electron Transfer Mediators in the Pd(II)-Catalyzed Oxidative Carbonylation of Aniline" Molecules 30, no. 9: 2027. https://doi.org/10.3390/molecules30092027
APA StyleVavasori, A., Ronchin, L., Pietrobon, L., & Bravo, S. (2025). Influence of Electron Transfer Mediators in the Pd(II)-Catalyzed Oxidative Carbonylation of Aniline. Molecules, 30(9), 2027. https://doi.org/10.3390/molecules30092027