
Iodine(III)-Containing Reagents in Photo-Assisted and Photo-Catalyzed Organic Synthesis

 ,
,  , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. General Considerations
3. Synthetic Methods Using Direct Photoexcitation and Iodine(III) Reagents


4. Photocatalytic Synthetic Methods Involving Transition Metals and Iodine(III) Reagents
5. Summary and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flores, A.; Cots, E.; Bergès, J.; Muñiz, K. Enantioselective Iodine(I/III) Catalysis in Organic Synthesis. Adv. Synth. Catal. 2018, 361, 2–25. [Google Scholar] [CrossRef]
- Singh, F.V.; Shetgaonkar, S.E.; Krishnan, M.; Wirth, T. Progress in Organocatalysis with Hypervalent Iodine Catalysts. Chem. Soc. Rev. 2022, 51, 8102–8139. [Google Scholar] [CrossRef] [PubMed]
- Shetgaonkar, S.E.; Singh, F.V. Hypervalent Iodine Reagents in Palladium-Catalyzed Oxidative Cross-Coupling Reactions. Front. Chem. 2020, 8, 705. [Google Scholar] [CrossRef]
- Segura-Quezada, L.A.; Torres-Carbajal, K.R.; Juárez-Ornelas, K.A.; Navarro-Santos, P.; Granados-López, A.J.; González-García, G.; Ortiz-Alvarado, R.; de León-Solis, C.; Solorio-Alvarado, C.R. Iodine(III)-Mediate Oxidative Cyanation, Azidation, Nitration, Sulfenylation and Slenization in Olefins and Aromatic Systems. Curr. Org. Chem. 2022, 26, 1954–1968. [Google Scholar] [CrossRef]
- Chávez-Rivera, R.; Navarro-Santos, P.; Chacón-García, L.; Ortiz-Alvarado, R.; Solorio Alvarado, C.R. Iodine(III)-Aluminum or -Ammonium Salts for the Oxidative Aromatic Inorganic Functionalization. ChemisrtySelect 2023, 8, e202303425. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhdankin, V.V. Recent Progress in Synthetic Applications of Hypervalent Iodine(III) Reagents. Chem. Rev. 2024, 124, 11108–11186. [Google Scholar] [CrossRef] [PubMed]
- Segura-Quezada, A.; Satkar, Y.; Patil, D.; Mali, N.; Wrobel, K.; González, G.; Zárraga, R.; Ortiz-Alvarado, R.; Solorio-Alvarado, C.R. Iodine(III)/AlX3-mediated electrophilic chlorination and bromination of arenes. Dual Role of AlX3 (X = Cl, Br) for (PhIO)n depolymerization and as the halogen source. Tetrahedron Lett. 2019, 60, 1551–1555. [Google Scholar] [CrossRef]
- Patil, D.B.; Gámez-Montaño, R.; Ordoñez, M.; Solis-Santos, M.; Jiménez-Halla, J.O.C.; Solorio-Alvarado, C.R. Iodine(III)-Mediated Electrophilic Chlorination and Catalytic Nitration of N-Tosyl Anilines. Eur. J. Org. Chem. 2022, e202201295. [Google Scholar] [CrossRef]
- Mali, N.; Ibarra-Gutiérrez, J.G.; Lugo Fuentes, L.I.; Ortíz-Alvarado, R.; Chacón-García, L.; Navarro-Santos, P.; Jiménez-Halla, J.O.C.; Solorio-Alvarado, C.R. Iodine(III)-Mediated Free-Aniline Iodination through Acetyl Hypoiodite Formation: Study of the Reaction Pathway. Eur. J. Org. Chem. 2022, 2022, e202201067. [Google Scholar] [CrossRef]
- Juárez-Ornelas, K.A.; Solís-Hernández, M.; Navarro-Santos, P.; Jiménez-Halla, J.O.C.; Solorio-Alvarado, C.R. Divergent role of PIDA and PIFA in the AlX3 (X = Cl, Br) halogenation of 2-naphthol: A mechanistic study. Beilstein J. Org. Chem. 2024, 20, 1580–1589. [Google Scholar] [CrossRef]
- Juárez-Ornelas, K.A.; Jiménez-Halla, J.O.C.; Kato, T.; Solorio-Alvarado, C.R.; Maruoka, K. Iodine(III)-Catalyzed Electrophilic Nitration of Phenols via Non-Brønsted Acidic NO2+ Generation. Org. Lett. 2019, 21, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Satkar, Y.; Wrobel, K.; Trujillo-González, D.E.; Ortiz-Alvarado, R.; Jiménez-Halla, J.O.C.; Solorio-Alvarado, C.R. The Diaryliodonium(III) Salts Reaction With Free-Radicals Enables One-Pot Double Arylation of Naphthols. Front. Chem. 2020, 8, 563470. [Google Scholar] [CrossRef] [PubMed]
- Naakajima, M.; Nagaswaa, S.; Matsumoto, K.; Matsuda, Y.; Nemoto, T. Synthesis of Visible-Light–Activated Hypervalent Iodine and Photooxidation under Visible Light Irradiation via a Direct S0→Tn Transition. Chem. Pharm. Bull. 2022, 70, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Mutlaq, D.Z.; Al-Shawi, A.A.A.; Al-Asadi, R.H. Synthesis, Characterization, Anticancer Activity, and Molecular Docking of Novel Maleimide–Succinimide Derivatives. Egypt Pharm. J. 2021, 20, 303–312. [Google Scholar] [CrossRef]
- Fernandes, R.A.; Kumar, P.; Choudhary, P. Advances in Catalytic and Protecting-Group-Free Total Synthesis of Natural Products: A Recent Update. Chem. Commun. 2020, 56, 8569–8590. [Google Scholar] [CrossRef]
- Casnati, A.; Motti, E.; Mancuso, R.; Gabriele, B.; Della Ca’, N. Recent Advances in the Catalytic Synthesis of Imidazolidin-2-Ones and Benzimidazolidin-2-Ones. Catalysts 2019, 9, 28. [Google Scholar] [CrossRef]
- Wu, Q.; Hou, J.; Gu, Q.; Gao, H.; Shi, M.; Zheng, L. PhI(OAc)2/Pd(OAc)2 Promoted the Formation of 8-Hydroxyquinoline Derivatives from Benzoxazoles and Alcohols. Org. Biomol. Chem. 2023, 21, 1148–1152. [Google Scholar] [CrossRef]
- Camlik, G.; Bilakaya, B.; Küpeli Akkol, E.; Velaro, A.J.; Wasnik, S.; Muhar, A.M.; Degim, I.T.; Sobarzo-Sánchez, E. Oral Active Carbon Quantum Dots for Diabetes. Pharmaceuticals 2024, 17, 1395. [Google Scholar] [CrossRef]
- Hernández-Velázquez, E.D.; Herrera, M.D.; Alba-Betancourt, C.; Navarro-Santos, P.; Ortíz-Alvarado, R.; Solorio-Alvarado, C.R. Synthesis and in vivo Evaluation of Fluorobenzyl Metformin Derivatives as Potential Drugs in The Diabetes Treatment. Asian J. Org. Chem. 2023, 12, e202300200. [Google Scholar] [CrossRef]
- Segura-Quezada, L.A.; Hernández-Velázquez, E.D.; Corrales-Escobosa, A.R.; de León-Solis, C.; Solorio-Alvarado, C.R. Ningalins, Pyrrole-Bearing Metabolites Isolated from Didemnum spp. Synthesis and MDR-Reversion Activity in Cancer Therapy. Chem. Biodivers. 2024, 21, e202300883. [Google Scholar] [CrossRef]
- Segura-Quezada, L.A.; Alba-Betancourt, C.; Chacón-García, L.; Chávez-Rivera, R.; Navarro-Santos, P.; Ortiz-Alvarado, R.; Tapia-Juárez, M.; Negrete-Díaz, J.V.; Martínez-Morales, J.F.; Deveze-Álvarez, M.A.; et al. Synthesis and Anti-inflammatory Effect of Simple 2,3-Diarylindoles. On Route to New NSAID Scaffolds. ChemistrySelect 2024, 9, e202303803. [Google Scholar] [CrossRef]
- Amtul, Z.; Atta-ur-Rahman, B.S.P.; Siddiqui, R.; Choudhary, M. Chemistry and Mechanism of Urease Inhibition. Curr. Med. Chem. 2002, 9, 1323–1348. [Google Scholar] [CrossRef] [PubMed]
- Nahide, P.D.; Jiménez-Halla, J.O.C.; Wrobel, K.; Solorio-Alvarado, C.R.; Ortiz Alvarado, R.; Yahuaca-Juárez, B. Gold(I)-catalysed high-yielding synthesis of indenes by direct Csp3–H bond activation. Org. Biomol. Chem. 2018, 16, 7330–7335. [Google Scholar] [CrossRef]
- Segura-Quezada, L.A.; Torres-Carbajal, K.R.; Mali, N.; Patil, D.B.; Luna-Chagolla, M.; Ortiz-Alvarado, R.; Tapia-Juárez, M.; Fraire-Soto, I.; Araujo-Huitrado, J.G.; Granados-López, A.J.; et al. Gold(I)-Catalyzed Synthesis of 4H-Benzo[d][1,3]oxazines and Biological Evaluation of Activity in Breast Cancer Cells. ACS Omega 2022, 7, 6944–6955. [Google Scholar] [CrossRef]
- Moteki, S.A.; Usui, A.; Zhang, T.; Solorio Alvarado, C.R.; Maruoka, K. Site-Selective Oxidation of Unactivated Csp3-H Bonds with Hypervalent Iodine(III) Reagents. Angew. Chem. Int. Ed. 2013, 52, 13093–13096. [Google Scholar] [CrossRef] [PubMed]
- Yahuaca-Juárez, B.; González, G.; Ramírez-Morales, M.A.; Alba-Betancourt, C.; Deveze-Álvarez, M.A.; Mendoza-Macías, C.L.; Ortiz-Alvarado, R.; Juárez-Ornelas, K.A.; Solorio-Alvarado, C.R.; Maruoka, K. Iodine(III)-catalyzed benzylic oxidation by using the (PhIO)n/Al(NO3)3 system. Synth. Commun. 2020, 50, 539–548. [Google Scholar] [CrossRef]
- Xue, X.-S.; Ji, P.; Zhou, B.; Cheng, J.P. The Essential Role of Bond Energetics in C–H Activation/Functionalization. Chem. Rev. 2017, 117, 8622–8648. [Google Scholar] [CrossRef]
- Francis, A.C.; Richard, J.S. Advanced Organic Chemistry, 5th ed.; Springer US: New York, NY, USA, 2007; p. 1074. [Google Scholar] [CrossRef]
- Concepión, J.I.; Francisco, C.G.; Hernández, R.; Salazar, J.A.; Suárez, E. Intramolecular hydrogen abstraction. Iodosobenzene diacetate, an efficient and convenient reagent for alkoxy radical generation. Tetrahedron Lett. 1984, 25, 1953–1956. [Google Scholar] [CrossRef]
- Hernández, R.; León, E.I.; Moreno, P.; Riesco-Fagundo, C.; Suárez, E. Synthesis of Highly Functionalized Chiral Nitriles by Radical Fragmentation of β-Hydroxy Azides. Convenient Transformation of Aldononitriles into 1,4- and 1,5-Iminoalditols. J. Org. Chem. 2004, 69, 8437–8444. [Google Scholar] [CrossRef]
- Boto, A.; Hernández, D.; Hernández, R.; Suárez, E. β-Fragmentation of Primary Alkoxyl Radicals versus Hydrogen Abstraction: Synthesis of Polyols and α,ω-Differently Substituted Cyclic Ethers from Carbohydrates. J. Org. Chem. 2003, 68, 5310–5319. [Google Scholar] [CrossRef]
- Francisco, C.G.; González, C.C.; Paz, N.R.; Suárez, E. Fragmentation of Carbohydrate Anomeric Alkoxyl Radicals: A New Synthesis of 1,1-Difluoro-1-iodo Alditols. Org. Lett. 2003, 5, 4171–4173. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, M.; Spyroudis, S.; Varvoglis, A. 1,3-Oxathiole-2-Thiones from the Reaction of Carbon Disulfide with Zwitterionic Iodonium Compounds. J. Org. Chem. 1985, 50, 1509–1511. [Google Scholar] [CrossRef]
- Georgiev, G.; Spyroudis, S.; Varvoglis, A. Diacetoxyiodobenzene and Bis(Trifluoroacetoxy)Iodobenzene as Photoinitiators for Cationic Polymerisations. Polym. Bull. 1985, 14, 523–526. [Google Scholar] [CrossRef]
- Minisci, F.; Vismara, E.; Fontana, F.; Claudia Nogueira Barbosa, M. A New General Method of Homolytic Alkylation of Protonated Heteroaromatic Bases by Carboxylic Acids and Iodosobenzene Diacetate. Tetrahedron Lett. 1989, 30, 4569–4572. [Google Scholar] [CrossRef]
- Togo, H.; Muraki, T.; Yokoyama, M. Preparation of Adamantyl Sulfides with [Bis(1-Adamantanecarboxy)Iodo]Arenes and Disulfides. Synthesis 1995, 1995, 155–157. [Google Scholar] [CrossRef]
- Togo, H.; Muraki, T.; Hoshina, Y.; Yamaguchi, K.; Yokoyama, M. Formation and Synthetic Use of Oxygen-Centred Radicals with (Diacetoxyiodo)Arenes. J. Chem. Soc. Perkin Trans. 1997, 1, 787–794. [Google Scholar] [CrossRef]
- Gogonas, E.P.; Hadjiarapoglou, L.P. [3+2]-Cycloaddition Reactions of 2-Phenyliodonio-5,5-Dimethyl-1,3-Dioxacyclohexanemethylide. Tetrahedron Lett. 2000, 41, 9299–9303. [Google Scholar] [CrossRef]
- Matveeva, E.D.; Podrugina, T.A.; Pavlova, A.S.; Mironov, A.V.; Borisenko, A.A.; Gleiter, R.; Zefirov, N.S. Heterocycles from Phosphonium−Iodonium Ylides. Photochemical Synthesis of λ5-Phosphinolines. J. Org. Chem. 2009, 74, 9428–9432. [Google Scholar] [CrossRef]
- Matveeva, E.D.; Podrugina, T.A.; Taranova, M.A.; Borisenko, A.A.; Mironov, A.V.; Gleiter, R.; Zefirov, N.S. Photochemical Synthesis of Phosphinolines from Phosphonium–Iodonium Ylides. J. Org. Chem. 2010, 76, 566–572. [Google Scholar] [CrossRef]
- Nekipelova, T.D.; Kuzmin, V.A.; Matveeva, E.D.; Gleiter, R.; Zefirov, N.S. On the Mechanism of Photoinduced Addition of Acetonitrile to Phosphonium–Iodonium Ylides. J. Phys. Org. Chem. 2012, 26, 137–143. [Google Scholar] [CrossRef]
- Nekipelova, T.D.; Taranova, M.A.; Matveeva, E.D.; Podrugina, T.A.; Kuzmin, V.A.; Zefirov, N.S. Mechanism and Remarkable Features of Photoinduced Cycloaddition of Phenylacetylene to Mixed Phosphonium-Iodonium Ylide. Dokl. Akad. Nauk 2012, 447, 262–265. [Google Scholar] [CrossRef]
- Moteki, S.A.; Usui, A.; Selvakumar, S.; Zhang, T.; Maruoka, K. Metal-Free C–H Bond Activation of Branched Aldehydes with a Hypervalent Iodine(III) Catalyst under Visible-Light Photolysis: Successful Trapping with Electron-Deficient Olefins. Angew. Chem. Int. Ed. 2014, 53, 11060–11064. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Li, H.; Ji, W.; Wang, L. Sunlight-Driven Decarboxylative Alkynylation of α-Keto Acids with Bromoacetylenes by Hypervalent Iodine Reagent Catalysis: A Facile Approach to Ynones. Angew. Chem. Int. Ed. 2015, 54, 8374–8377. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Tan, H.; Wang, M.; Li, P.; Wang, L. Photocatalyst-Free Hypervalent Iodine Reagent Catalyzed Decarboxylative Acylarylation of Acrylamides with α-Oxocarboxylic Acids Driven by Visible-Light Irradiation. Chem. Commun. 2016, 52, 1462–1465. [Google Scholar] [CrossRef] [PubMed]
- Lucchetti, N.; Tkacheva, A.; Fantasia, S.; Muñiz, K. Radical C−H-Amination of Heteroarenes Using Dual Initiation by Visible Light and Iodine. Adv. Synth. Catal. 2018, 360, 3889–3893. [Google Scholar] [CrossRef]
- Chidley, T.; Jameel, I.; Rizwan, S.; Peixoto, P.A.; Pouységu, L.; Quideau, S.; Hopkins, W.S.; Murphy, G.K. Blue LED Irradiation of Iodonium Ylides Gives Diradical Intermediates for Efficient Metal-free Cyclopropanation with Alkenes. Angew. Chem. Int. Ed. 2019, 58, 16959–16965. [Google Scholar] [CrossRef]
- Voutyritsa, E.; Garreau, M.; Kokotou, M.G.; Triandafillidi, I.; Waser, J.; Kokotos, C.G. Photochemical Functionalization of Heterocycles with EBX Reagents: C−H Alkynylation versus Deconstructive Ring Cleavage. Chem. Eur. J. 2020, 26, 14453–14460. [Google Scholar] [CrossRef]
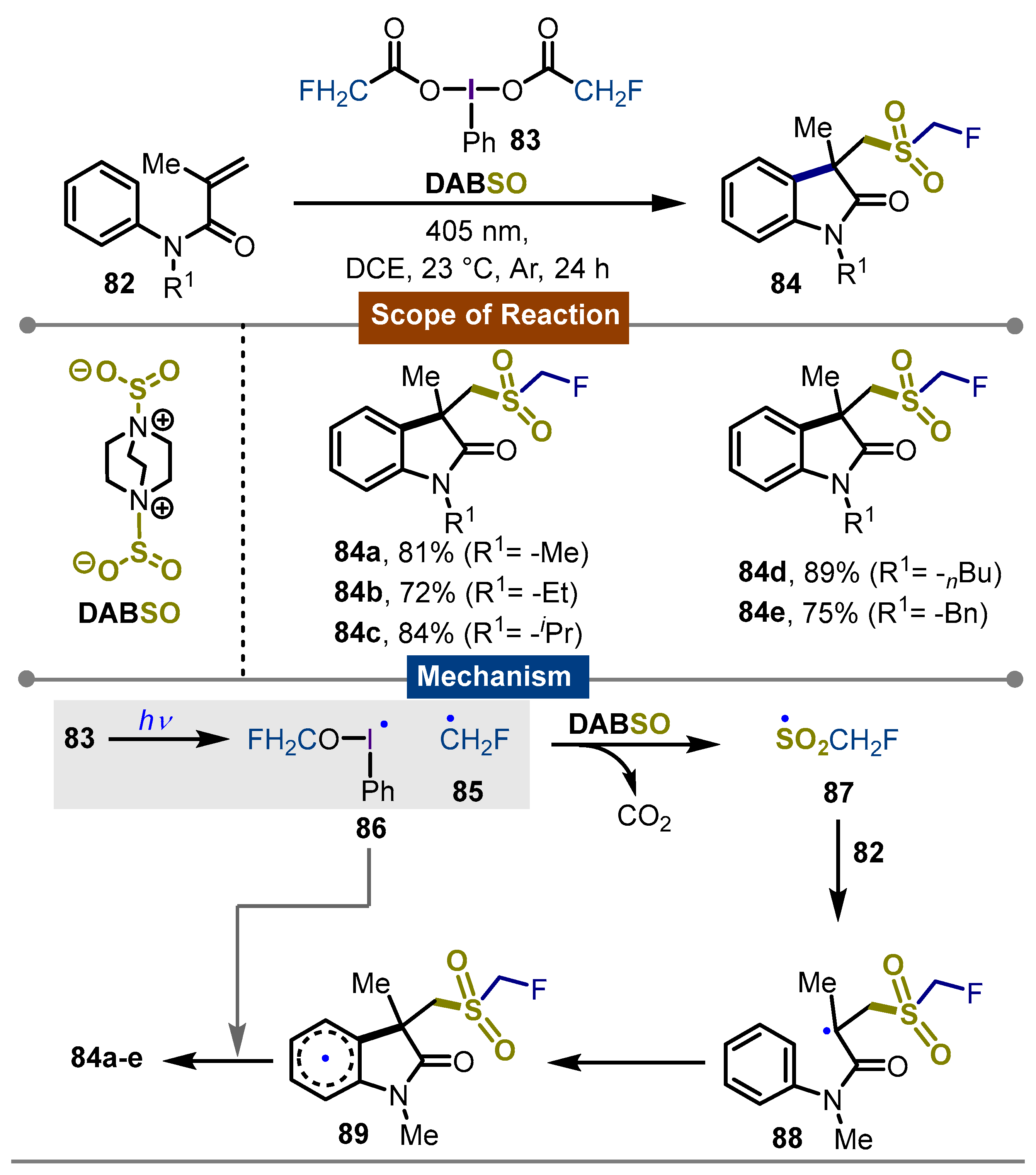
- Induced, Catalyst-Free Monofluoromethyl Sulfonylation of Alkenes with Iodine(III) Reagent and DABSO. Org. Lett. 2023, 25, 7062–7066. [CrossRef]
- Ou, C.; Cai, Y.; Ma, Y.; Zhang, H.; Ma, X.; Liu, C. Aliphatic Sulfonyl Fluoride Synthesis via Decarboxylative Fluorosulfonylation of Hypervalent Iodine(III) Carboxylates. Org. Lett. 2023, 25, 6751–6756. [Google Scholar] [CrossRef]
- Han, X.; Yue, W.; Wang, Z.; Xu, H.; Yang, M.; Zhu, J. Iodine(III)-Mediated Photochemical C–H Azolation. Org. Lett. 2024, 26, 9305–9310. [Google Scholar] [CrossRef]
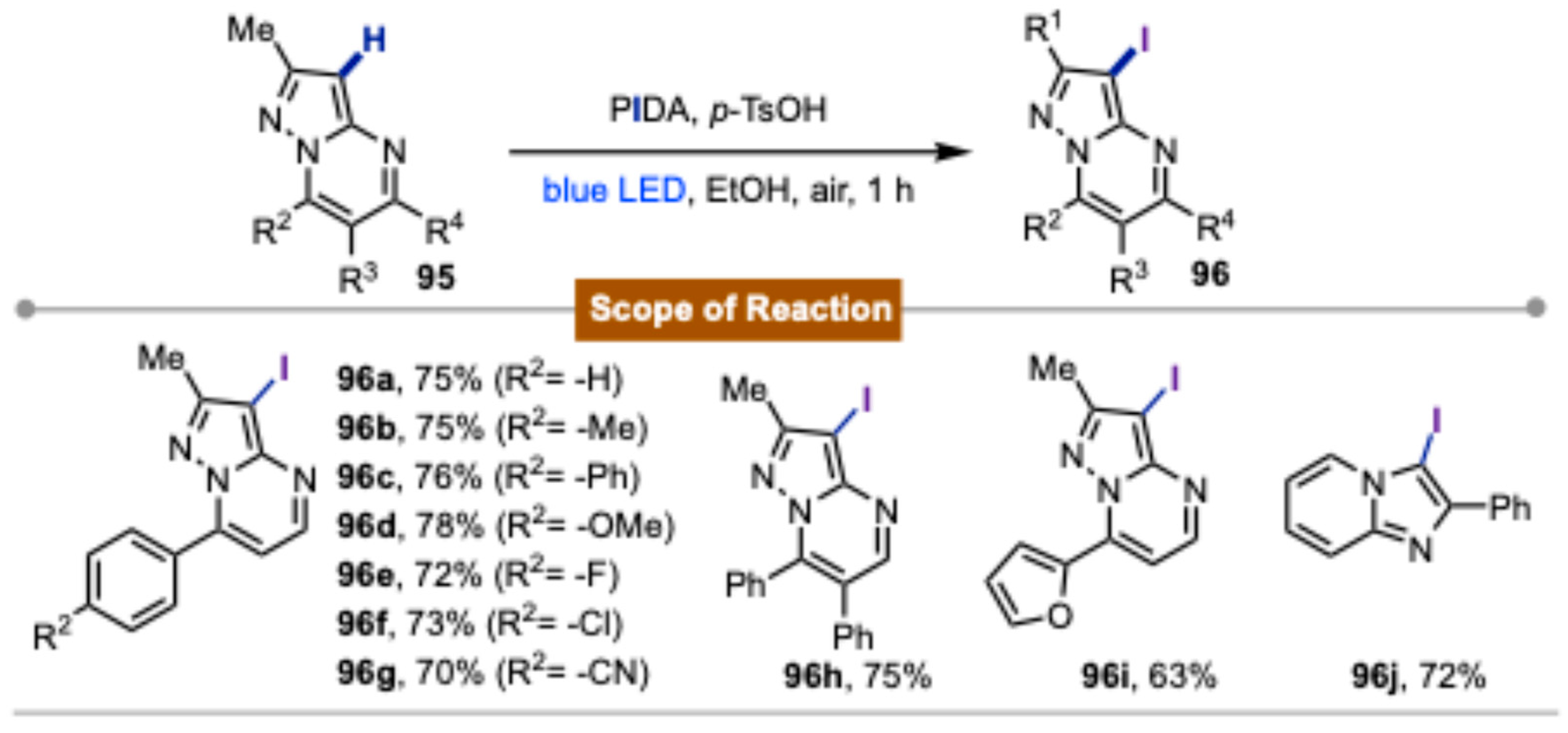
- Paul, S.; Das, S.; Choudhuri, T.; Sikdar, P.; Bagdi, A.K. PIDA as an Iodinating Reagent: Visible-Light-Induced Iodination of Pyrazolo[1,5-a]Pyrimidines and Other Heteroarenes. Chem. Asian J. 2024, 20, e202401101. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Xie, J.; Xue, Q.; Pan, C.; Cheng, Y.; Zhu, C. Visible-Light-Induced Trifluoromethylation of N-Aryl Acrylamides: A Convenient and Effective Method To Synthesize CF3-Containing Oxindoles Bearing a Quaternary Carbon Center. Chem. Eur. J. 2013, 19, 14039–14042. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Jia, K.; Chen, Y. Hypervalent Iodine Reagents Enable Chemoselective Deboronative/Decarboxylative Alkenylation by Photoredox Catalysis. Angew. Chem. 2014, 127, 1901–1904. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, G.; Gong, L.; Zhang, S.; Chen, Y. Visible-Light-Induced Chemoselective Deboronative Alkynylation under Biomolecule-Compatible Conditions. J. Am. Chem. Soc. 2014, 136, 2280–2283. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, G.; Chen, Y. Dual Hypervalent Iodine(III) Reagents and Photoredox Catalysis Enable Decarboxylative Ynonylation under Mild Conditions. Angew. Chem. Int. Ed. 2015, 54, 7872–7876. [Google Scholar] [CrossRef]
- Jia, K.; Zhang, F.; Huang, H.; Chen, Y. Visible-Light-Induced Alkoxyl Radical Generation Enables Selective C(sp3)–C(sp3) Bond Cleavage and Functionalizations. J. Am. Chem. Soc. 2016, 138, 1514–1517. [Google Scholar] [CrossRef]
- Li, G.-X.; Morales-Rivera, C.A.; Wang, Y.; Gao, F.; He, G.; Liu, P.; Chen, G. Photoredox-Mediated Minisci C–H Alkylation of N-Heteroarenes Using Boronic Acids and Hypervalent Iodine. Chem. Sci. 2016, 7, 6407–6412. [Google Scholar] [CrossRef]
- Jia, K.; Pan, Y.; Chen, Y. Selective Carbonyl−C(Sp3) Bond Cleavage To Construct Ynamides, Ynoates, and Ynones by Photoredox Catalysis. Angew. Chem. 2017, 129, 2518–2521. [Google Scholar] [CrossRef]
- Wang, Z.; Herraiz, A.G.; del Hoyo, A.M.; Suero, M.G. Generating carbyne equivalents with photoredox catalysis. Nature 2018, 554, 86–91. [Google Scholar] [CrossRef]
- Wang, C.; Tu, Y.; Ma, D.; Bolm, C. Photocatalytic Fluoro Sulfoximidations of Styrenes. Angew. Chem. Int. Ed. 2020, 59, 14134–14137. [Google Scholar] [CrossRef]
- Dong, J.-Y.; Wang, H.; Mao, S.; Wang, X.; Zhou, M.-D.; Li, L. Visible Light-Induced [3+2] Cyclization Reactions of Hydrazones with Hypervalent Iodine Diazo Reagents for the Synthesis of 1-Amino-1,2,3-Triazoles. Adv. Synth. Catal. 2021, 363, 2133–2139. [Google Scholar] [CrossRef]
- Wen, J.; Zhao, W.; Gao, X.; Ren, X.; Dong, C.; Wang, C.; Liu, L.; Li, J. Synthesis of [1,2,3]Triazolo-[1,5-a]quinoxalin-4(5H)-ones through Photoredox-Catalyzed [3 + 2] Cyclization Reactions with Hypervalent Iodine(III) Reagents. J. Org. Chem. 2022, 87, 4415–4423. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, Z.; Zou, P.; Chen, Y.; Chen, Y. Hypervalent Iodine Reagents Enable C(sp2)–H Amidation of (Hetero)Arenes with Iminophenylacetic Acids. Org. Lett. 2022, 24, 6681–6685. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhang, Q.; Rolka, A.B.; Suero, M.G. Alkoxy Diazomethylation of Alkenes by Photoredox-Catalyzed Oxidative Radical-Polar Crossover. J. Am. Chem. Soc. 2024, 146, 12294–12299. [Google Scholar] [CrossRef]
- Xie, J.; Xu, P.; Li, H.; Xue, Q.; Jin, H.; Cheng, Y.; Zhu, C. A Room Temperature Decarboxylation/C–H Functionalization Cascade by Visible-Light Photoredox Catalysis. Chem. Commun. 2013, 49, 5672. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibarra-Gutiérrez, J.G.; Segura-Quezada, L.A.; Hernández-Velázquez, E.D.; García-Dueñas, A.K.; Millán-Cortés, J.A.; Mondragón-Hernández, K.; Miranda-Navarrete, L.K.; Valtierra-Camarena, E.M.; Yebra-Rivera, S.Y.; Alférez-Carmona, O.E.; et al. Iodine(III)-Containing Reagents in Photo-Assisted and Photo-Catalyzed Organic Synthesis. Molecules 2025, 30, 784. https://doi.org/10.3390/molecules30040784
Ibarra-Gutiérrez JG, Segura-Quezada LA, Hernández-Velázquez ED, García-Dueñas AK, Millán-Cortés JA, Mondragón-Hernández K, Miranda-Navarrete LK, Valtierra-Camarena EM, Yebra-Rivera SY, Alférez-Carmona OE, et al. Iodine(III)-Containing Reagents in Photo-Assisted and Photo-Catalyzed Organic Synthesis. Molecules. 2025; 30(4):784. https://doi.org/10.3390/molecules30040784
Chicago/Turabian StyleIbarra-Gutiérrez, Jaime G., Luis A. Segura-Quezada, Edson D. Hernández-Velázquez, Ana K. García-Dueñas, José A. Millán-Cortés, Kevin Mondragón-Hernández, Luz K. Miranda-Navarrete, Evelyn M. Valtierra-Camarena, Steffi Y. Yebra-Rivera, Omar E. Alférez-Carmona, and et al. 2025. "Iodine(III)-Containing Reagents in Photo-Assisted and Photo-Catalyzed Organic Synthesis" Molecules 30, no. 4: 784. https://doi.org/10.3390/molecules30040784
APA StyleIbarra-Gutiérrez, J. G., Segura-Quezada, L. A., Hernández-Velázquez, E. D., García-Dueñas, A. K., Millán-Cortés, J. A., Mondragón-Hernández, K., Miranda-Navarrete, L. K., Valtierra-Camarena, E. M., Yebra-Rivera, S. Y., Alférez-Carmona, O. E., Ávalos-Otero, O. E., Chávez-Rivera, R., de León-Solís, C., Ortíz-Alvarado, R., & Solorio-Alvarado, C. R. (2025). Iodine(III)-Containing Reagents in Photo-Assisted and Photo-Catalyzed Organic Synthesis. Molecules, 30(4), 784. https://doi.org/10.3390/molecules30040784