
Theoretical Studies on the Reaction Mechanism for the Cycloaddition of Zwitterionic π-Allenyl Palladium Species: Substrate-Controlled Isomerization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
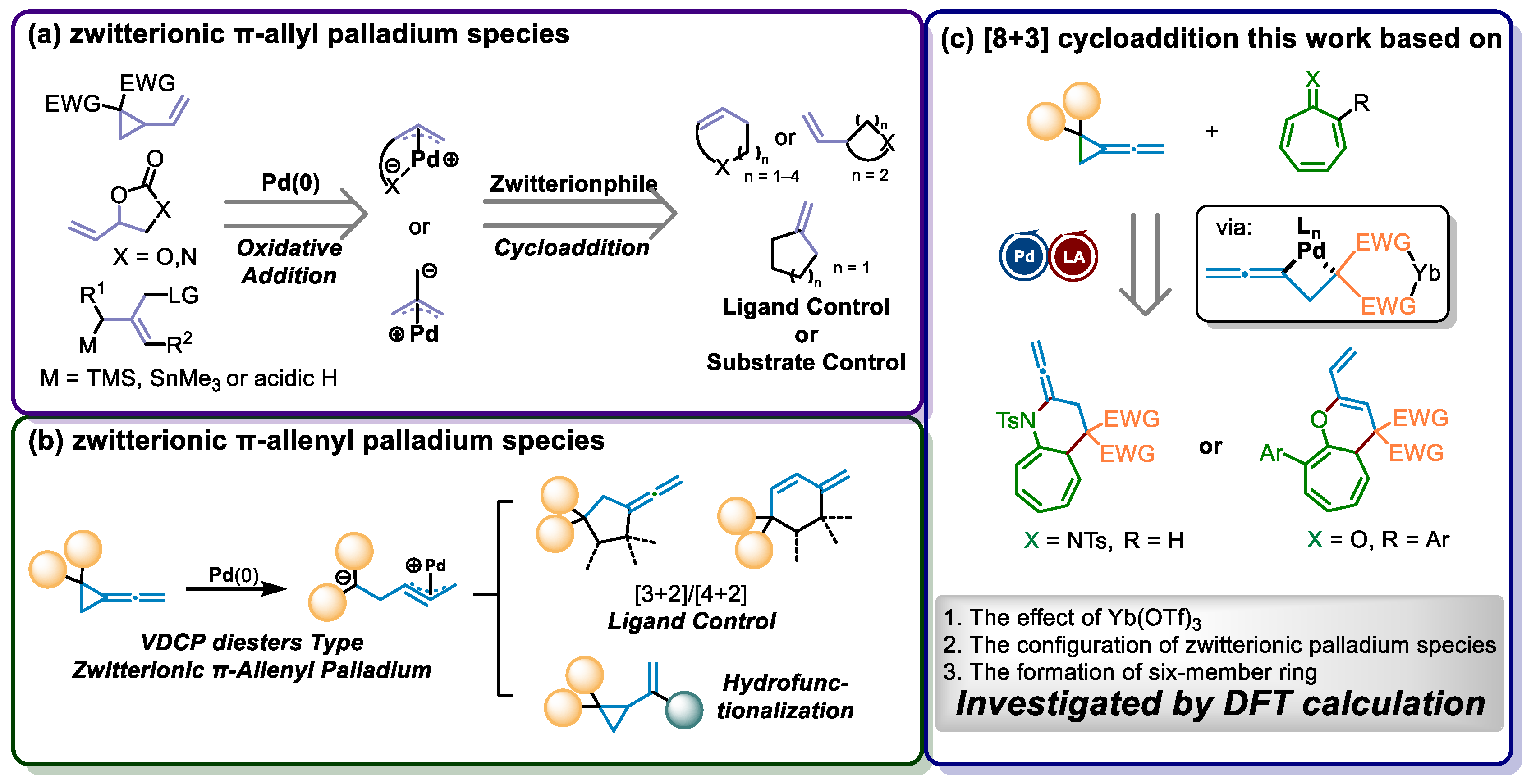
:1. Introduction
2. Results and Discussion
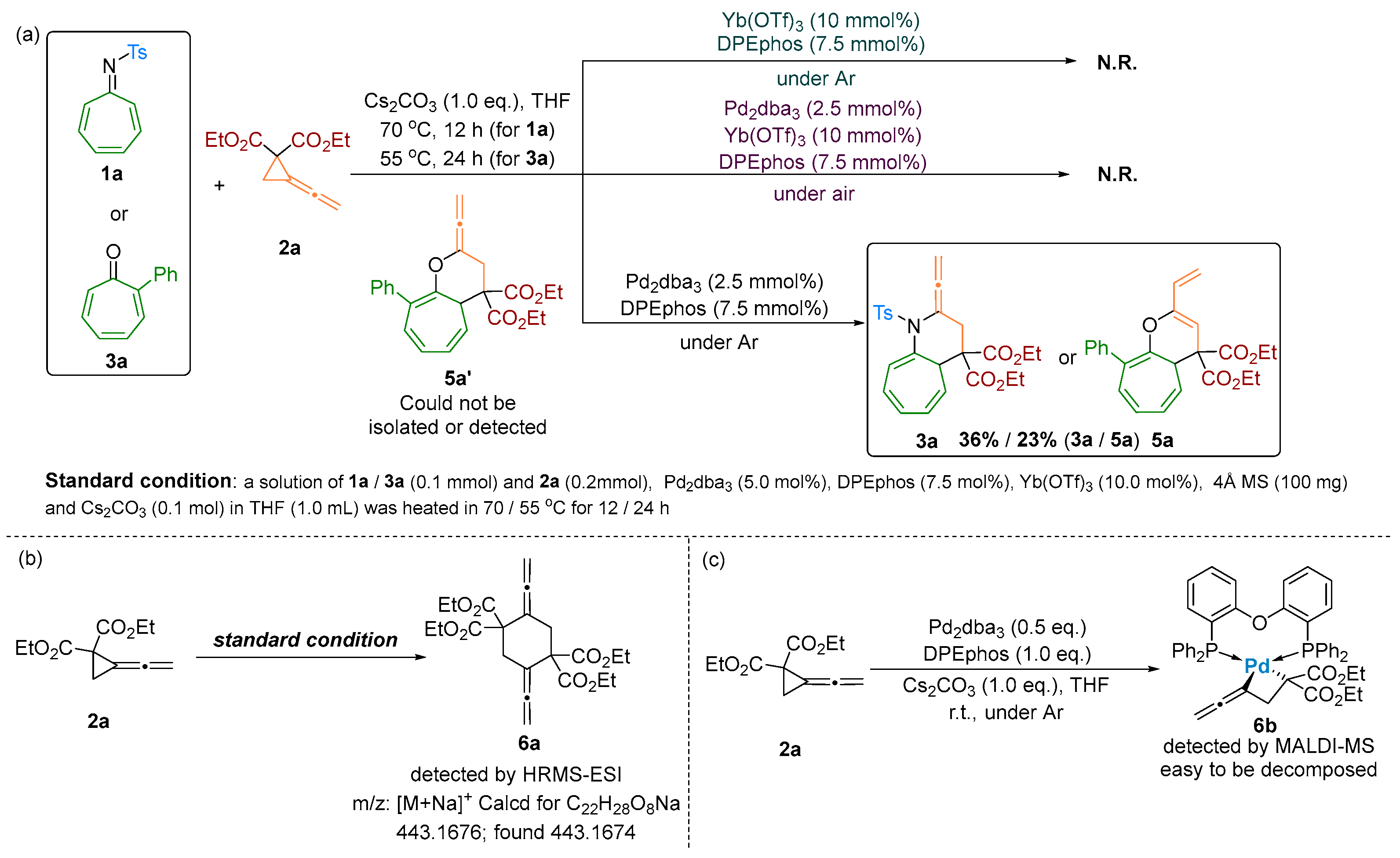
2.1. Mechanistic Studies by Experiments
2.2. Proposed Cycloaddition and Rearrangement Reaction Pathways
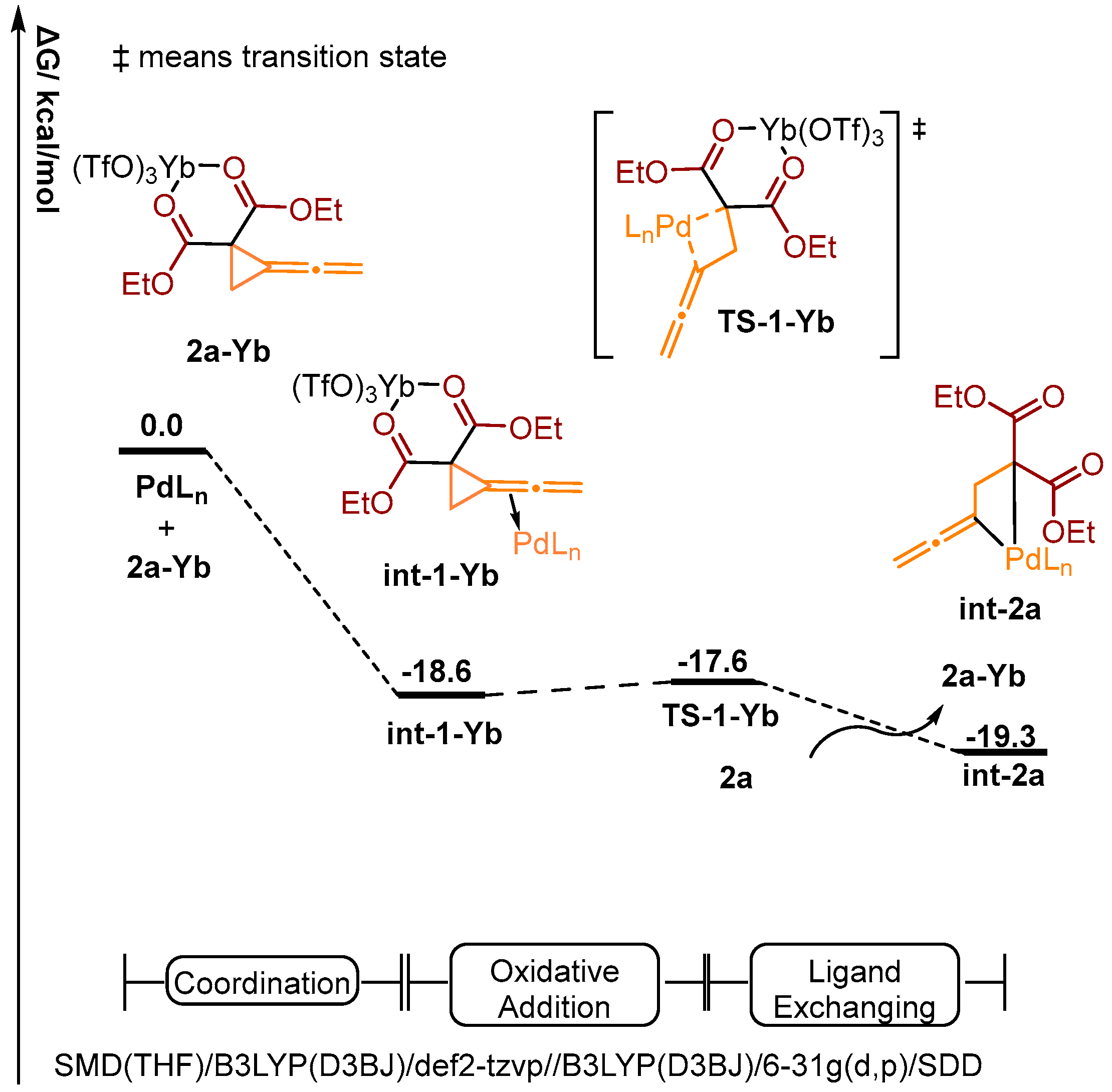
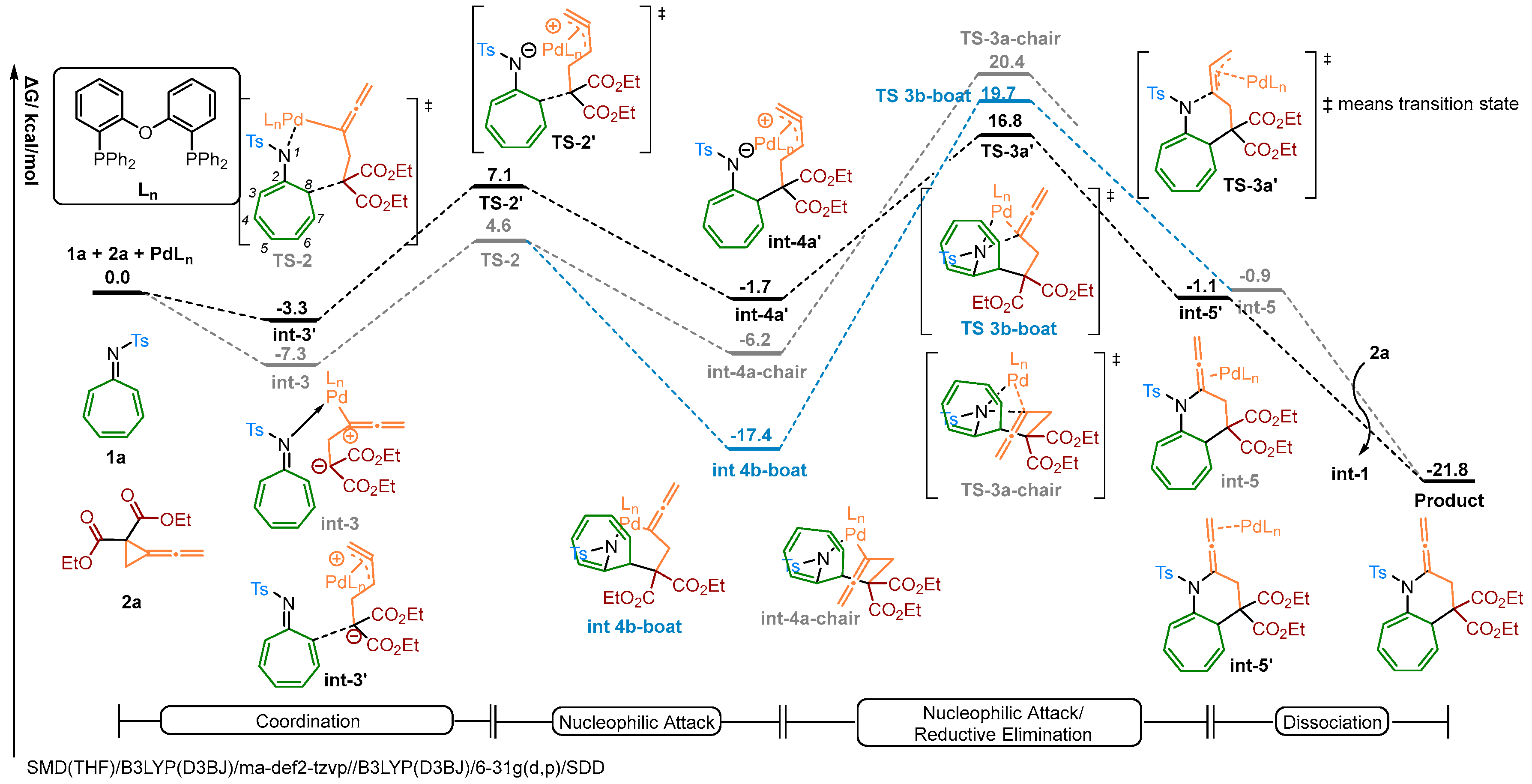
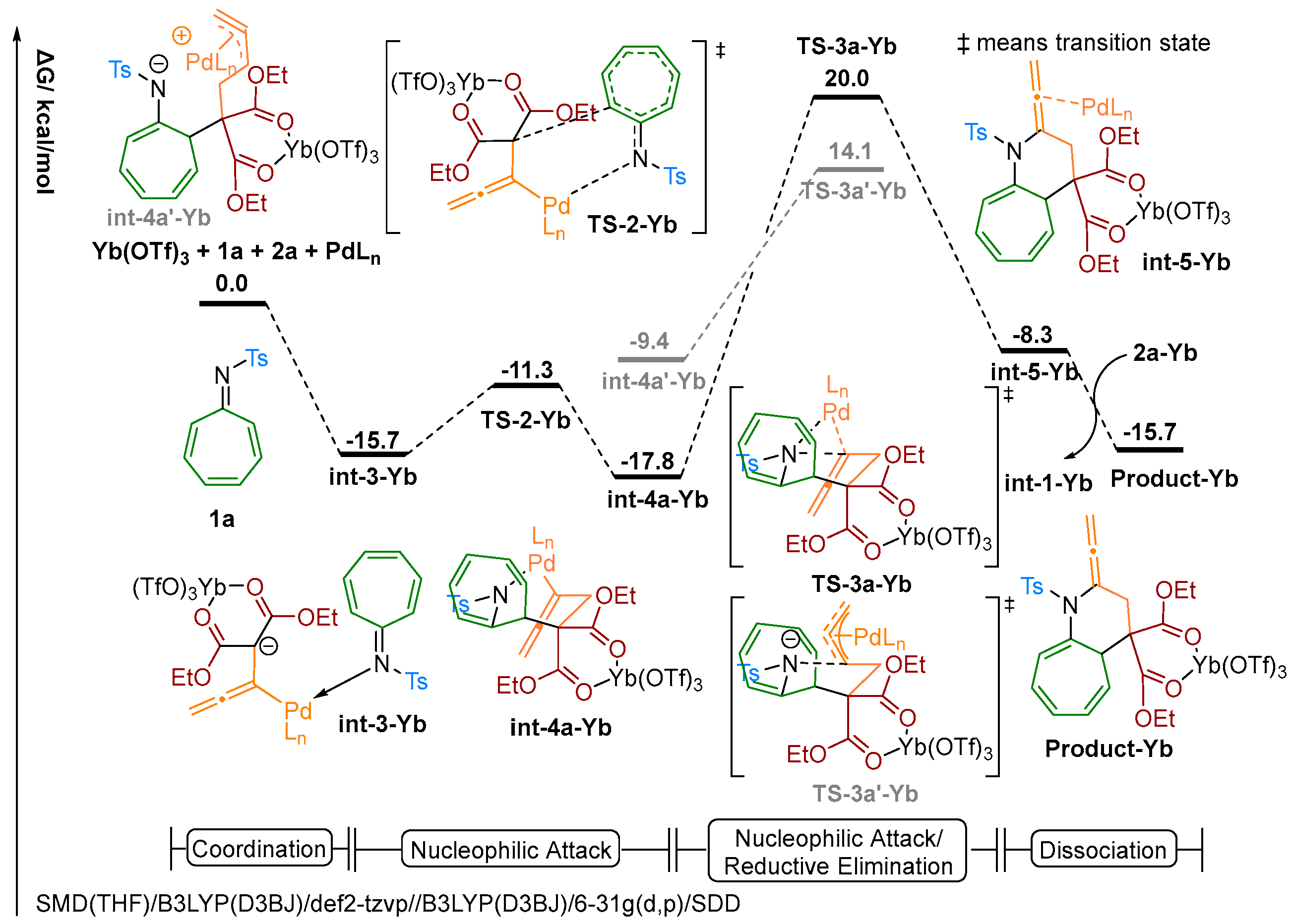
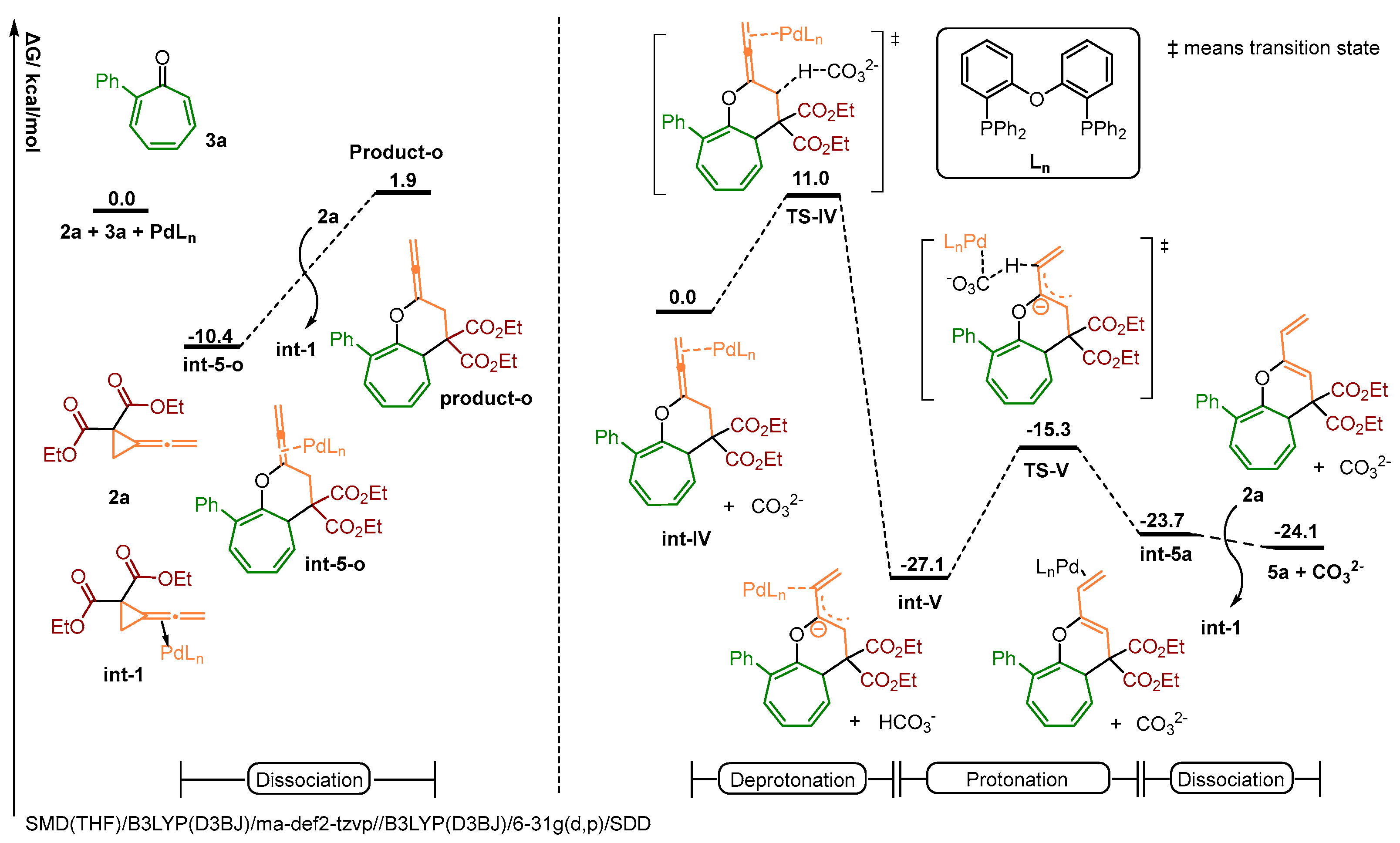
2.3. Theoretical Investigation on a Possible Reaction Pathway
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, J.-H.; Blaszczyk, S.A.; Li, X.-X.; Tang, W.-P. Transition Metal-Catalyzed Selective Carbon-Carbon Bond Cleavage of Vinylcyclopropanes in Cycloaddition Reactions. Chem. Rev. 2021, 121, 110. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Mata, G. Forging Odd-Membered Rings: Palladium-Catalyzed Asymmetric Cycloadditions of Trimethylenemethane. Acc. Chem. Res. 2020, 53, 1293. [Google Scholar] [CrossRef]
- Niu, B.; Wei, Y.; Shi, M. Recent Advances in Annulation Reactions based on Zwitterionic π-Allyl Palladium and Propargyl Palladium Complexes. Org. Chem. Front. 2021, 8, 3475. [Google Scholar] [CrossRef]
- Allen, B.D.W.; Lakeland, C.P.; Harrity, J.P.A. Utilizing Palladium-Stabilized Zwitterions for the Construction of N-Heterocycles. Chem. Eur. J. 2017, 23, 13830. [Google Scholar] [CrossRef]
- Khan, S.; Ahmad, T.; Rasheed, T.; Ullah, N. Recent Applications of Vinylethylene Carbonates in Pd-Catalyzed Allylic Substitution and Annulation Reactions: Synthesis of Multifunctional Allylic and Cyclic Structural Motifs. Coord. Chem. Rev. 2022, 462, 214526. [Google Scholar] [CrossRef]
- You, Y.; Li, Q.; Zhang, Y.-P.; Zhao, J.-Q.; Wang, Z.-H.; Yuan, W.-C. Advances in Palladium-Catalyzed Decarboxylative Cycloadditions of Cyclic Carbonates, Carbamates and Lactones. ChemCatChem 2022, 14, e202101887. [Google Scholar] [CrossRef]
- Wang, Y.-N.; Chai, J.-L.; You, C.; Zhang, J.; Mi, X.-L.; Zhang, L.; Luo, S.-Z. π-Coordinating Chiral Primary Amine/Palladium Synergistic Catalysis for Asymmetric Allylic Alkylation. J. Am. Chem. Soc. 2020, 142, 3184. [Google Scholar] [CrossRef]
- Huang, Q.-W.; Qi, T.; Liu, X.; Zhang, X.; Li, Q.-Z.; Gou, C.; Tao, Y.-M.; Leng, H.-J.; Li, J.-L. Lewis Acid/Brønsted Base-Assisted Palladium Catalysis: Stereoselective Construction of Skeletally Diverse Spiro-Ketolactams from Vinylethylene Carbonates. ACS Catal. 2021, 11, 10148. [Google Scholar] [CrossRef]
- Wu, H.-L.; Hu, L.-F.; Shen, Z.; Huang, G.-P. Computational Insights into Palladium/Boron-Catalyzed Allylic Substitution of Vinylethylene Carbonates with Water: Outer-Sphere versus Inner-Sphere Pathway and Origins of Regio- and Enantioselectivities. ACS Catal. 2022, 12, 2722. [Google Scholar] [CrossRef]
- Li, Q.-Z.; Guan, Y.-L.; Huang, Q.-W.; Qi, T.; Xiang, P.; Zhang, X.; Leng, H.-J.; Li, J.-L. Temperature-Controlled Divergent Asymmetric Synthesis of Indole-Based Medium-Sized Heterocycles through Palladium Catalysis. ACS Catal. 2023, 13, 1164. [Google Scholar] [CrossRef]
- Xiao, Y.-Q.; Li, M.-M.; Zhou, Z.-L.; Li, Y.-J.; Cao, M.-Y.; Liu, X.-P.; Lu, H.-H.; Rao, L.; Lu, L.-Q.; Beauchemin, A.M.; et al. Taming Chiral Quaternary Stereocenters via Remote H-BondingStereoinduction in Palladium-Catalyzed (3 + 2) Cycloadditions. Angew. Chem. Int. Ed. 2023, 62, e202212444. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Xiao, M.; Zhao, J.-P.; Zhang, Z.-H.; Xiao, W.-J.; Lu, L.-Q. Synthesis of Chiral Endocyclic Allenes and Alkynes via Pd-Catalyzed Asymmetric Higher-Order Dipolar Cycloaddition. J. Am. Chem. Soc. 2024, 146, 26622. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Qu, B.-L.; Xiao, M.; Yu, J.-Q.; Yu, J.; Cheng, Y.; Zhang, Z.-H.; Xiao, W.-J.; Lu, L.-Q. Synthesis of Chiral Spiro-Indenes via Pd-Catalyzed Asymmetric (4 + 2) Dipolar Cyclization. Chem. Synth. 2024, 4, 53. [Google Scholar] [CrossRef]
- Qin, T.-Z.; He, M.-Y.; Zi, W.-W. Palladium-Catalysed [2σ + 2π] Cycloaddition Reactions of Bicyclo[1.1.0]butanes with Aldehydes. Nat. Synth. 2024. [Google Scholar] [CrossRef]
- Niu, B.; Wei, Y.; Shi, M. Palladium Catalyzed Divergent Cycloadditions of Vinylidenecyclopropane-Diesters with MethyleneIndolinones enabled by Zwitterionic π-Propargyl Palladium Species. Chem. Commun. 2021, 57, 4783. [Google Scholar] [CrossRef]
- Yang, Z.-R.; Zhang, B.; Long, Y.-J.; Wei, Y.; Shi, M. Palladium-Catalyzed Hydroamination of Vinylidenecyclopropane-Diester with Pyrroles and Indoles: An Approach to Azaaromatic Vinylcyclopropanes. Chem. Commun. 2022, 58, 9926. [Google Scholar] [CrossRef]
- Shen, J.-H.; Long, Y.-J.; Shi, M.; Wei, Y. Palladium-Catalyzed and Ligand-Controlled Divergent Cycloadditions of Vinylidenecyclopropane-Diesters with Para-Quinone Methides enabled by Zwitterionic π-Propargyl Palladium Species. Org. Chem. Front. 2024, 11, 3214. [Google Scholar] [CrossRef]
- Zhong, X.-H.; Long, Y.-J.; Wei, Y.; Shi, M. Palladium-Catalyzed Sc(OTf)3/Cs2CO3-Assisted C−O Cross-Coupling of Vinylidenecyclopropane-Diesters with Phenolic Compounds. Adv. Synth. Catal. 2024, 366, 3387. [Google Scholar] [CrossRef]
- Long, Y.-J.; Shen, J.-H.; Wei, Y.; Shi, M. Substrate-Controlled [8 + 3] Cycloaddition of Tropsulfimides and Tropones with Zwitterionic Allenyl Palladium Species Derived from Vinylidenecyclopropane-diesters. J. Org. Chem. 2024, 89, 14831. [Google Scholar] [CrossRef]
- Yang, L.-C.; Wang, Y.-N.; Liu, R.-Y.; Luo, Y.-X.; Ng, X.-Q.; Yang, B.-M.; Rong, Z.-Q.; Lan, Y.; Shao, Z.-H.; Zhao, Y. Stereoselective Access to [5.5.0] and [4.4.1] Bicyclic Compounds Through Pd-Catalysed Divergent Higher-order Cycloadditions. Nat. Chem. 2020, 12, 860. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.-F.; Sun, Z.-M.; Chen, L.-A. Palladium-Catalyzed Regiodivergent Synthesis of 1,3-Dienyl and Allyl Esters from Propargyl Esters. Angew. Chem. Int. Ed. 2022, 61, e202203835. [Google Scholar] [CrossRef] [PubMed]
- Baize, M.W.; Blosser, P.W.; Plantevin, V.; Schimpff, D.G.; Gallucci, J.C.; Wojcicki, A. η3-Propargyl/Allenyl Complexes of Platinum and Palladium of the Type [(PPh3)2M(η3-CH2CCR)]. Organometallics 1996, 15, 164. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Yabukami, T.; Fujimoto, K.; Kawase, T.; Morimoto, T.; Kakiuchi, K. Effects of a Bidentate Phosphine Ligand on Palladium-Catalyzed Nucleophilic Substitution Reactions of Propargyl and Allyl Halides with Thiol. Organometallics 2003, 22, 2996–2999. [Google Scholar] [CrossRef]
- Manzano, R.; Romaniega, A.; Prieto, L.; Díaz, E.; Reyes, E.; Uria, U.; Carrillo, L.; Vicario, J.L. γ-Substituted Allenic Amides in the Phosphine-Catalyzed Enantioselective Higher Order Cycloaddition with Azaheptafulvenes. Org. Lett. 2020, 22, 4721. [Google Scholar] [CrossRef]
- Chen, J.-C.; Gao, S.; Chen, M. Cu-Catalyzed Silylation and Borylation of Vinylidene Cyclopropanes. Org. Lett. 2019, 21, 8800. [Google Scholar] [CrossRef]
- Ononye, S.N.; VanHeyst, M.D.; Oblak, E.Z.; Zhou, W.-D.; Ammar, M.; Anderson, A.C.; Wright, D.L. Tropolones As Lead-Like Natural Products: The Development of Potent and Selective Histone Deacetylase Inhibitors. ACS Med. Chem. Lett. 2013, 4, 757. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C. 01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, Y.; Shen, J.; Shi, M.; Wei, Y. Theoretical Studies on the Reaction Mechanism for the Cycloaddition of Zwitterionic π-Allenyl Palladium Species: Substrate-Controlled Isomerization. Molecules 2025, 30, 103. https://doi.org/10.3390/molecules30010103
Long Y, Shen J, Shi M, Wei Y. Theoretical Studies on the Reaction Mechanism for the Cycloaddition of Zwitterionic π-Allenyl Palladium Species: Substrate-Controlled Isomerization. Molecules. 2025; 30(1):103. https://doi.org/10.3390/molecules30010103
Chicago/Turabian StyleLong, Yongjie, Jiahao Shen, Min Shi, and Yin Wei. 2025. "Theoretical Studies on the Reaction Mechanism for the Cycloaddition of Zwitterionic π-Allenyl Palladium Species: Substrate-Controlled Isomerization" Molecules 30, no. 1: 103. https://doi.org/10.3390/molecules30010103
APA StyleLong, Y., Shen, J., Shi, M., & Wei, Y. (2025). Theoretical Studies on the Reaction Mechanism for the Cycloaddition of Zwitterionic π-Allenyl Palladium Species: Substrate-Controlled Isomerization. Molecules, 30(1), 103. https://doi.org/10.3390/molecules30010103