
Pentacyclic Triterpenoids with Nitrogen-Containing Heterocyclic Moiety, Privileged Hybrids in Anticancer Drug Discovery




Abstract
:1. Introduction
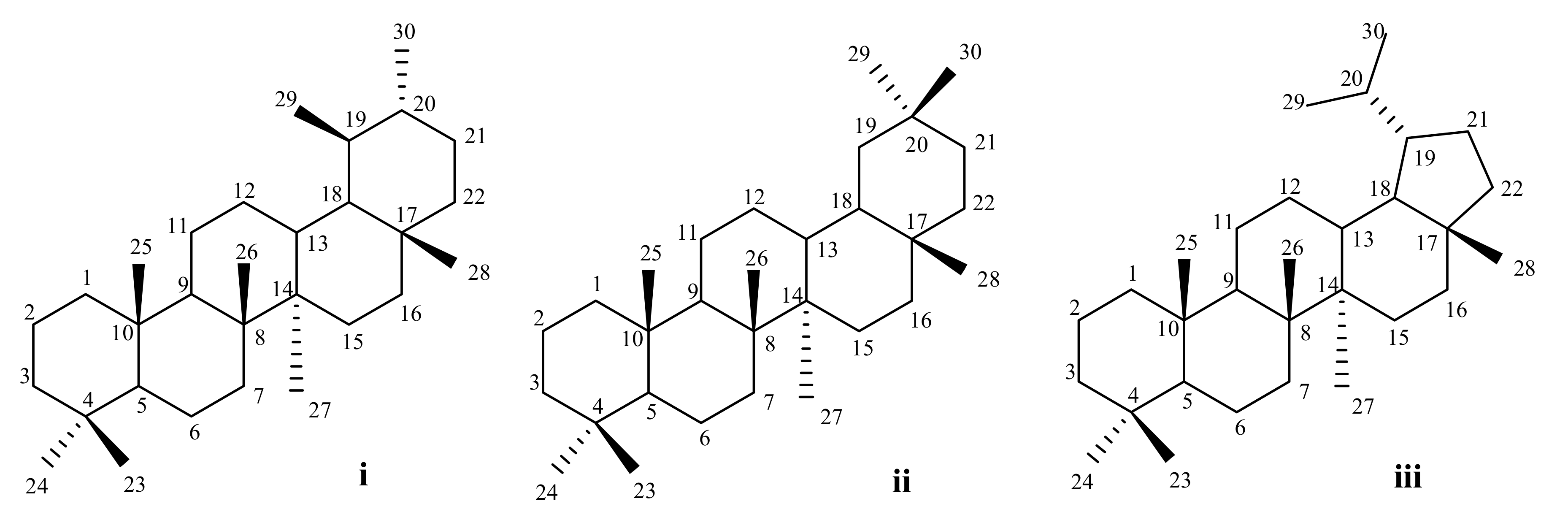
2. Anticancer Activity of Pentacyclic Triterpenoids
3. Triazole
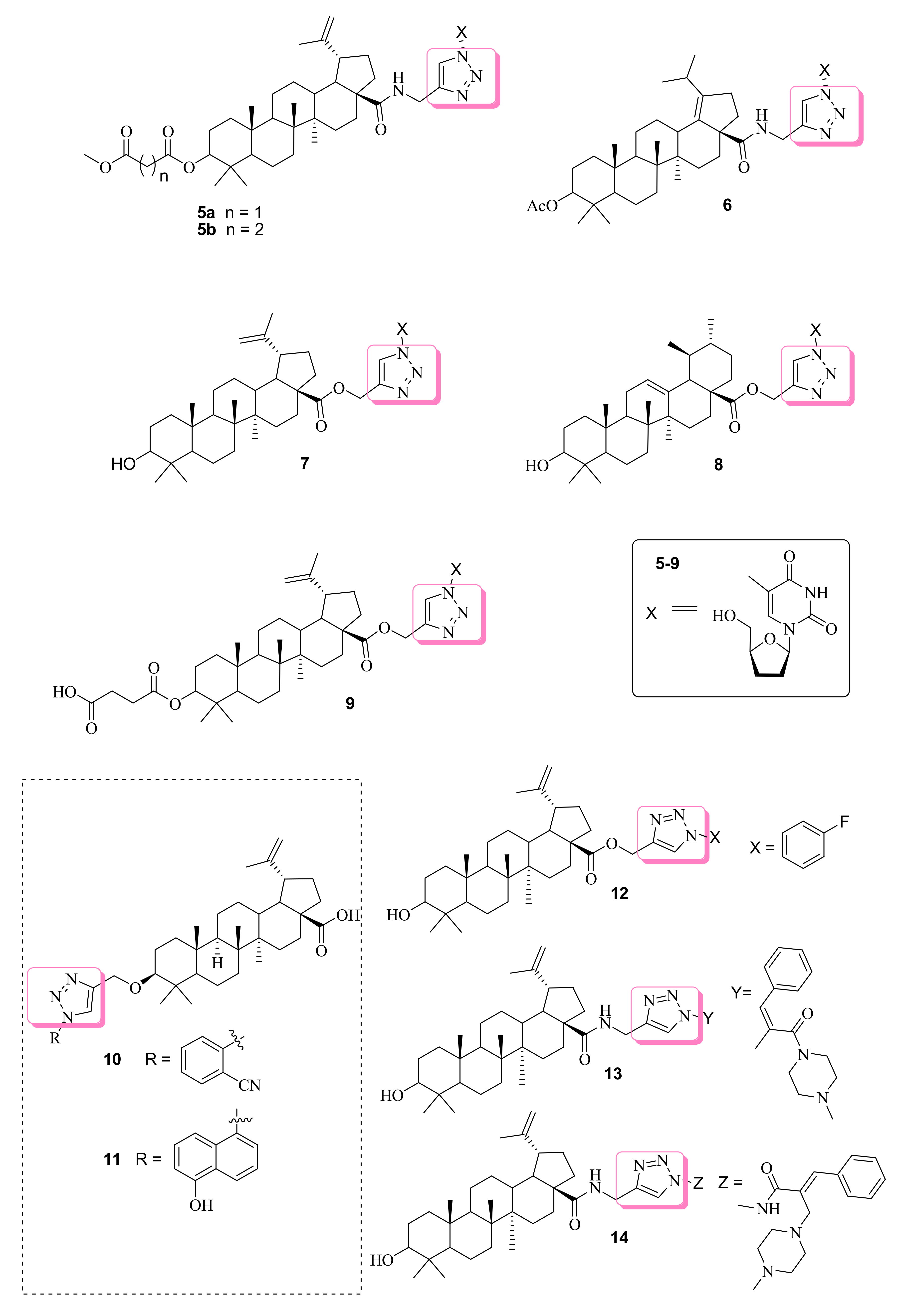
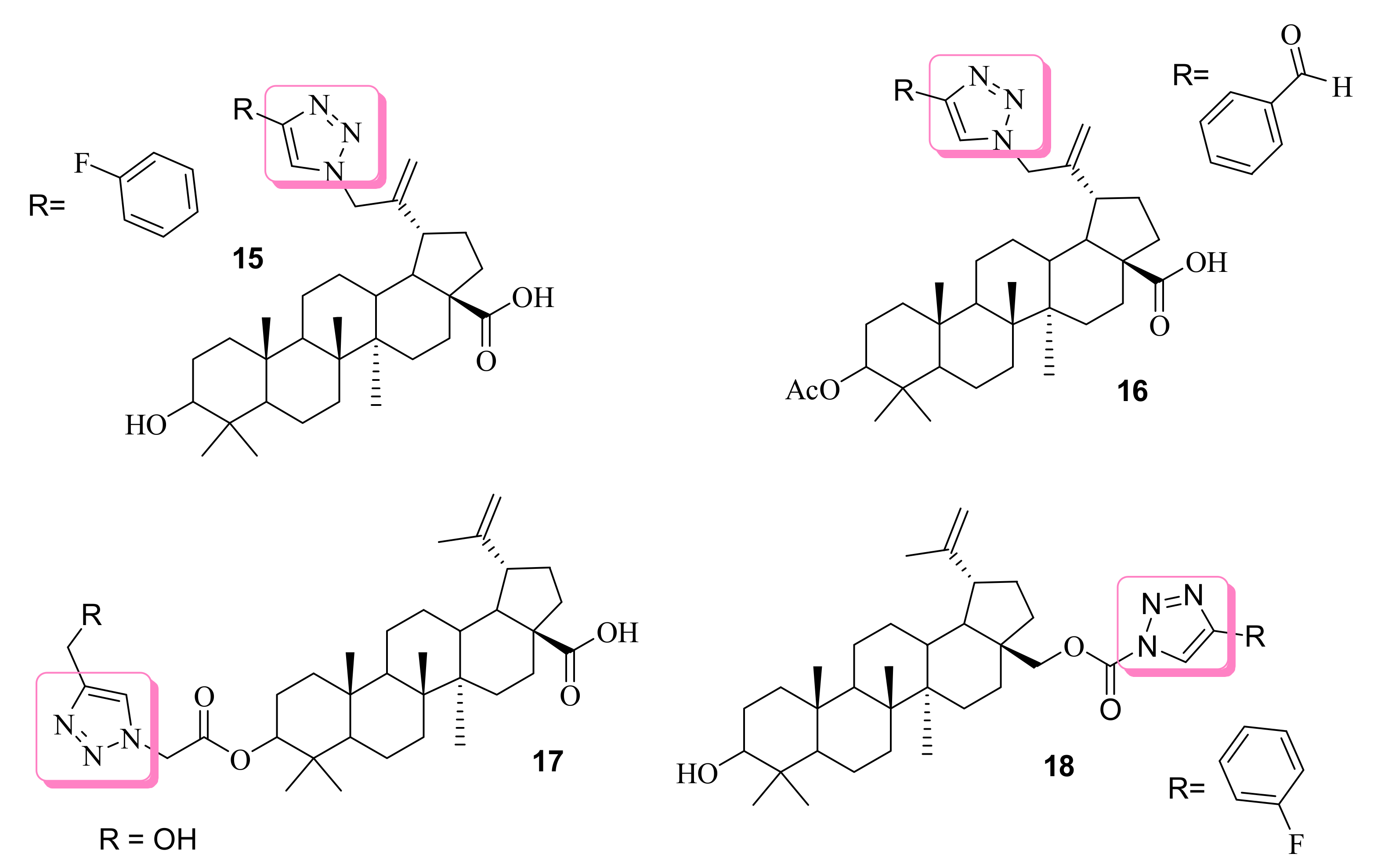
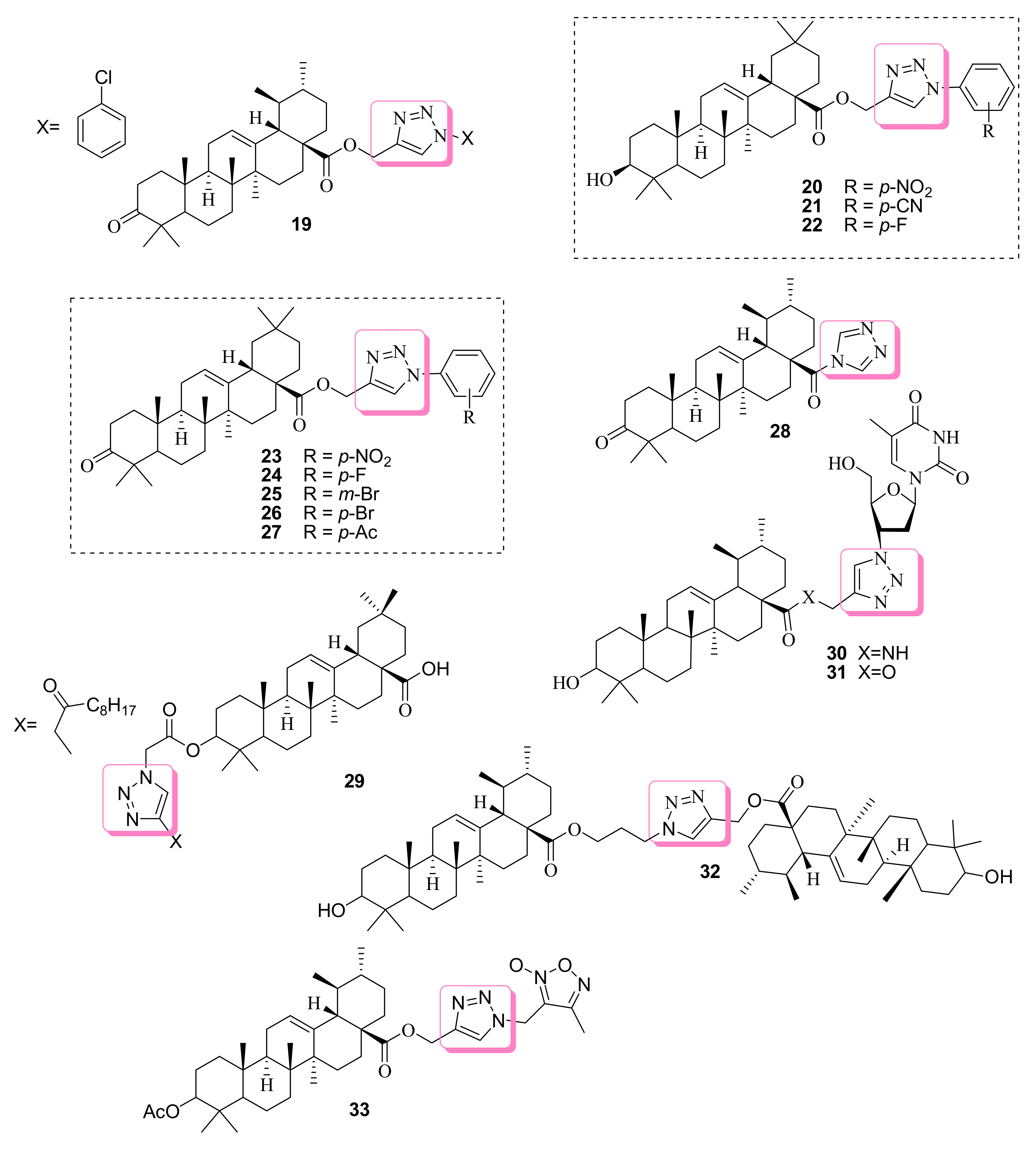
3.1. Anticancer Effects of Betulinic Acid-based 1,2,3-Triazole Molecules
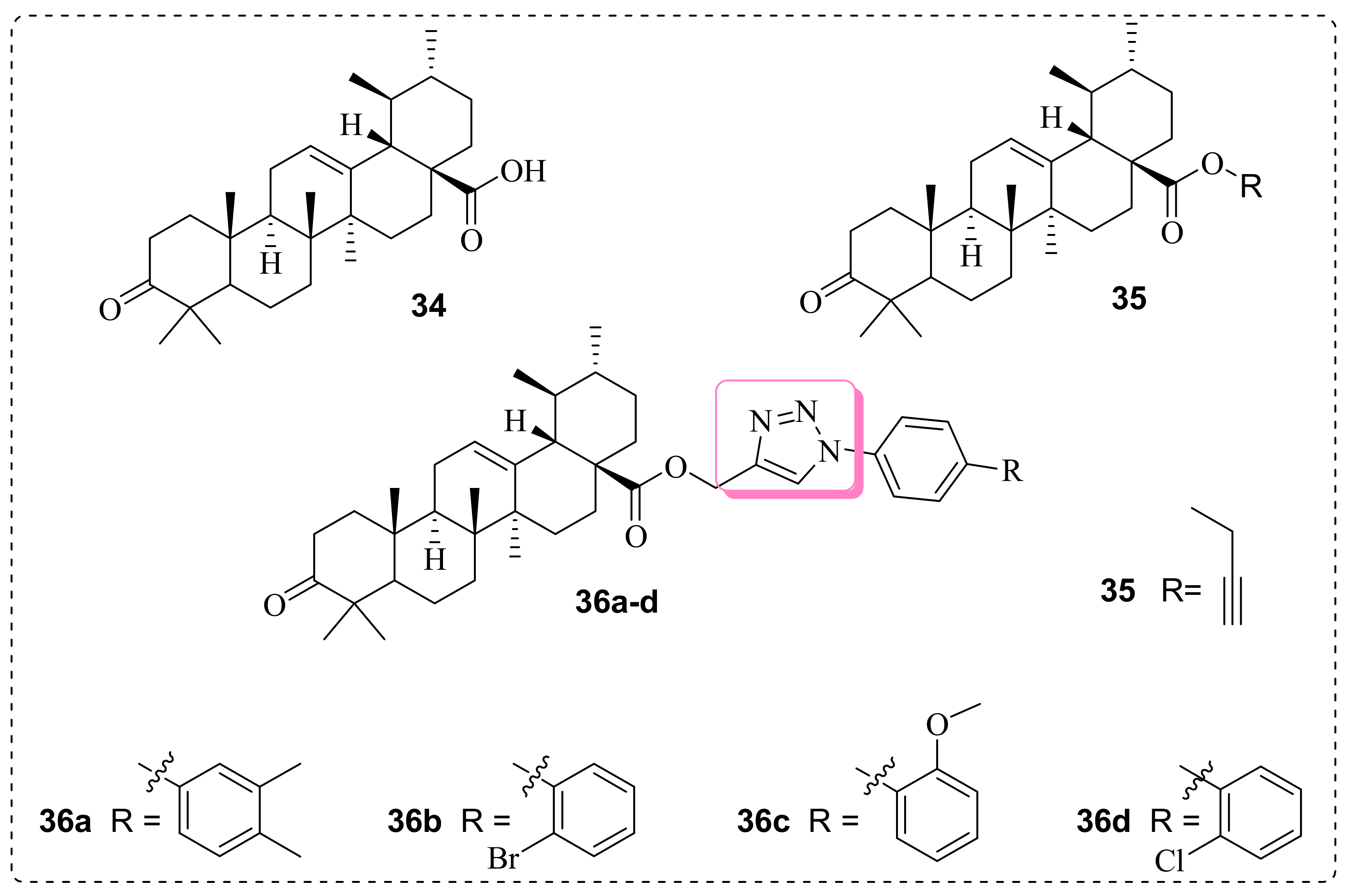
3.2. Anticancer Effects of Ursolic/oleanolic Acid-based 1,2,3-Triazole Molecules
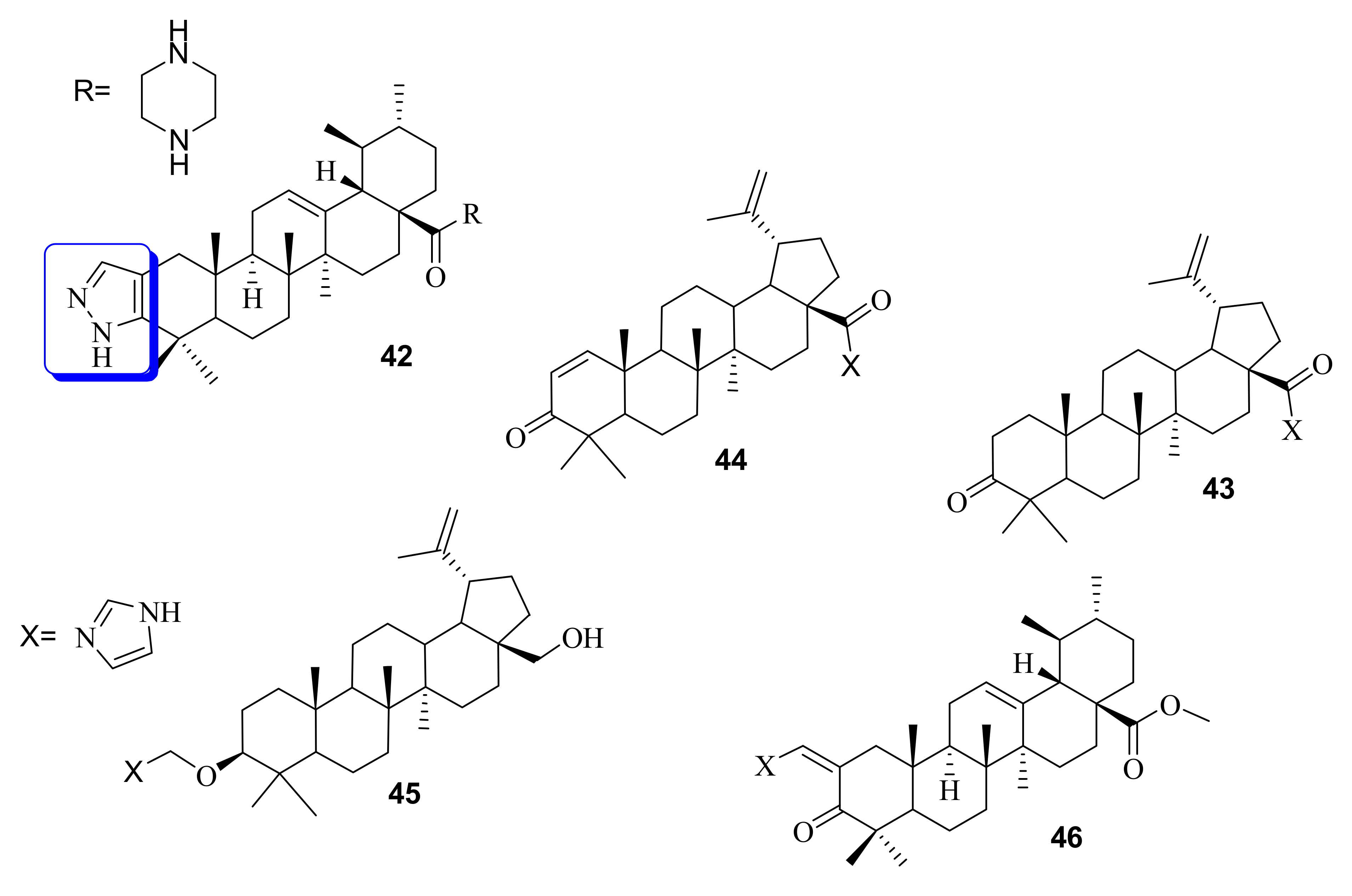
4. Pyrazole
Anticancer Effects of Triterpenoid-based Pyrole Molecules
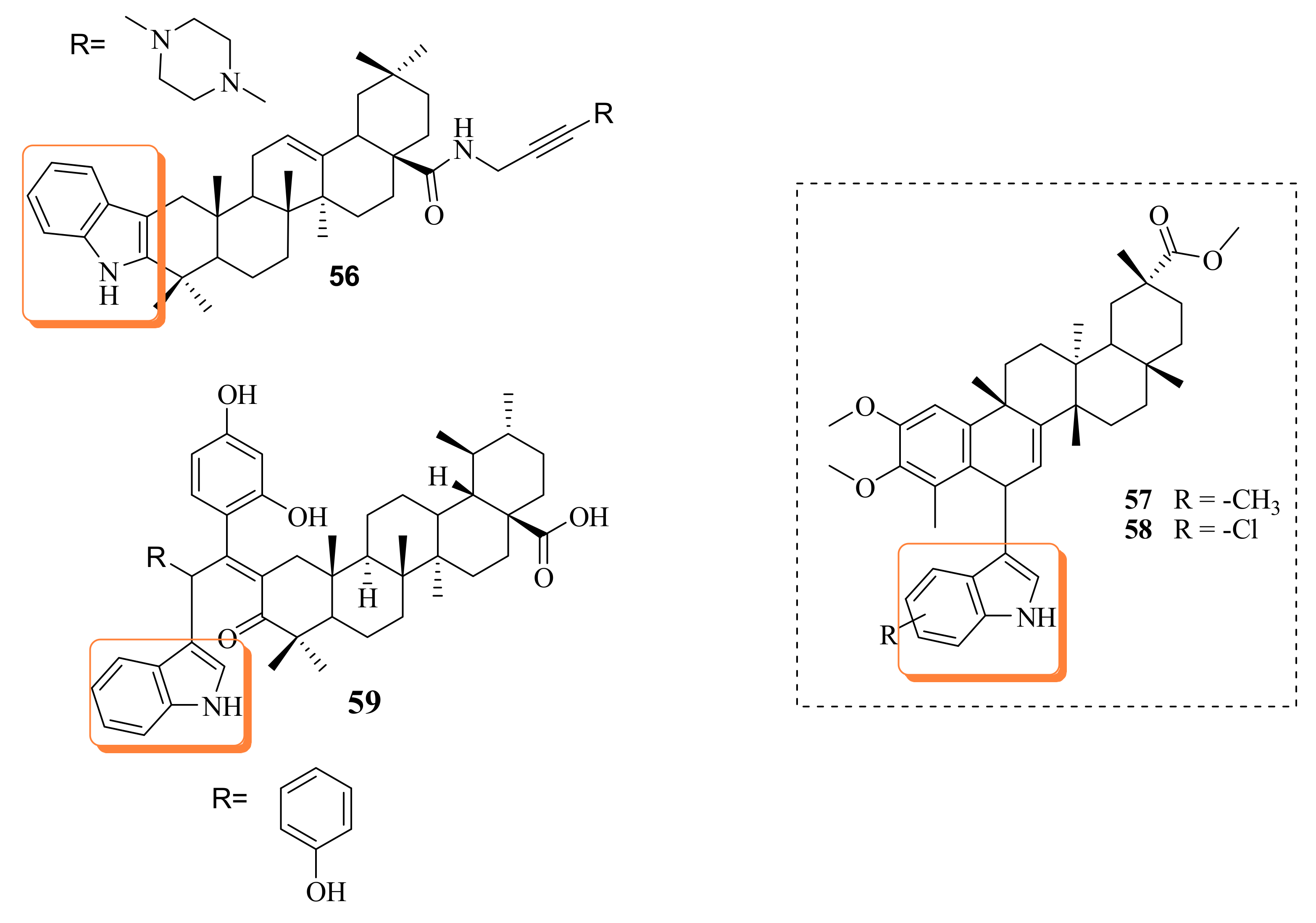
5. Indole
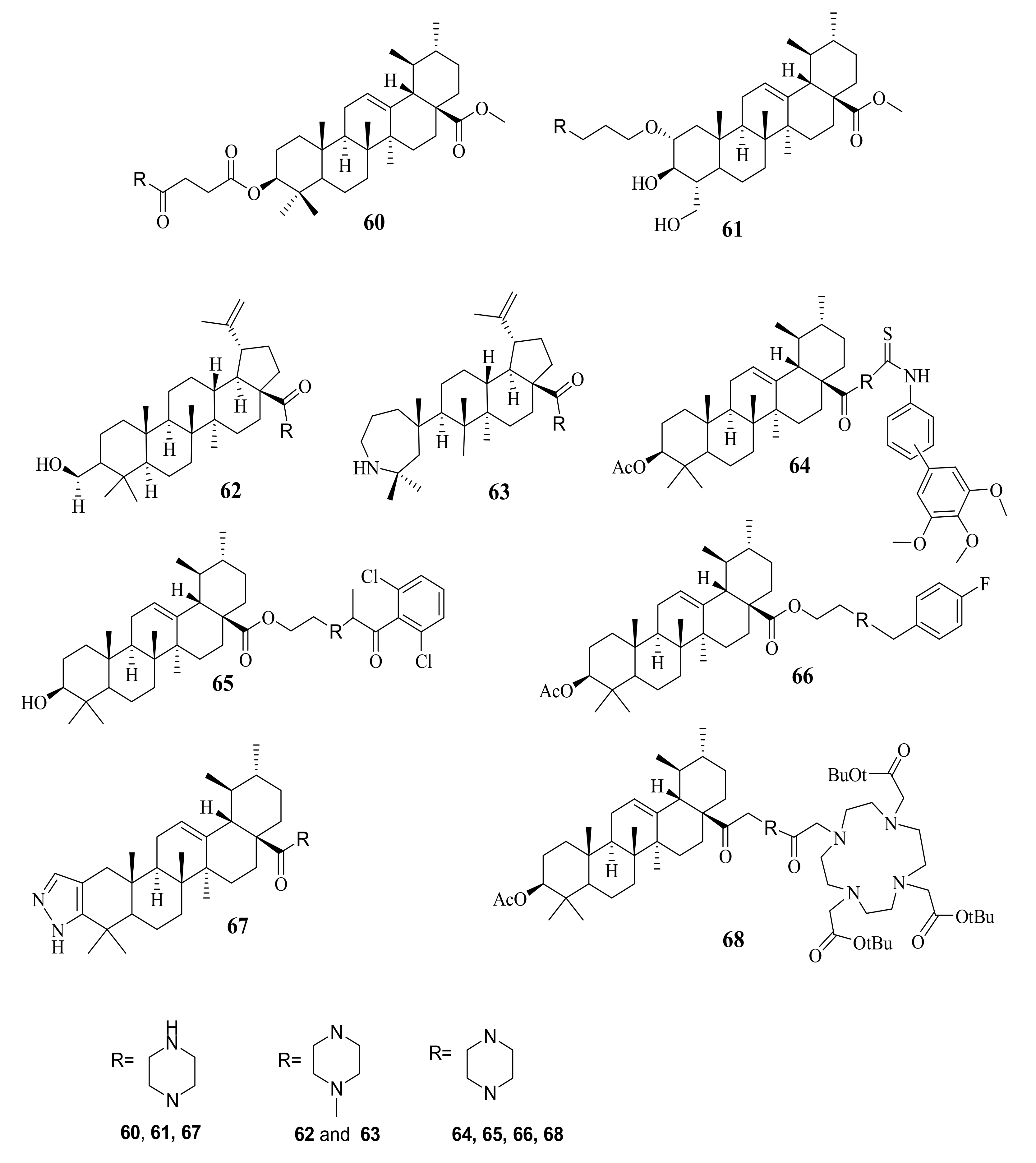
6. Piperazine
7. Aminoquinolines
8. Perspectives and Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Alves-Silva, J.M.; Romane, A.; Efferth, T.; Salgueiro, L. North African medicinal plants traditionally used in cancer therapy. Front. Pharmacol. 2017, 8, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tariq, A.; Sadia, S.; Pan, K.; Ullah, I.; Mussarat, S.; Sun, F.; Abiodun, O.O.; Batbaatar, A.; Li, Z.; Song, D.; et al. A systematic review on ethnomedicines of anti-cancer plants. Phyther. Res. 2017, 31, 202–264. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sharma, B.; Kanwar, S.S.; Kumar, A. Lead phytochemicals for anticancer drug development. Front. Plant Sci. 2016, 7, 1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, H.; Kumar, P.; Deshmukh, R.R.; Bishayee, A.; Kumar, S. Pentacyclic triterpenes: New tools to fight metabolic syndrome. Phytomedicine 2018, 50, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Borková, L.; Frydrych, I.; Jakubcová, N.; Adámek, R.; Lišková, B.; Gurská, S.; Medvedíková, M.; Hajdúch, M.; Urban, M. Synthesis and biological evaluation of triterpenoid thiazoles derived from betulonic acid, dihydrobetulonic acid, and ursonic acid. Eur. J. Med. Chem. 2020, 185, 111806. [Google Scholar] [CrossRef] [PubMed]
- Kamble, S.M.; Goyal, S.N.; Patil, C.R. Multifunctional pentacyclic triterpenoids as adjuvants in cancer chemotherapy: A review. RSC Adv. 2014, 4, 33370–33382. [Google Scholar] [CrossRef]
- Herrera-España, A.D.; Us-Martín, J.; Hernández-Ortega, S.; Mirón-López, G.; Quijano, L.; Villanueva-Toledo, J.R.; Mena-Rejón, G.J. Synthesis, structure analysis and activity against breast and cervix cancer cells of a triterpenoid thiazole derived from ochraceolide A. J. Mol. Struct. 2020, 1204, 127555. [Google Scholar] [CrossRef]
- Han, B.; Peng, Z. Anti-HIV triterpenoid components. J. Chem. Pharm. Res. 2014, 6, 438–443. [Google Scholar]
- Medina-O’Donnell, M.; Rivas, F.; Reyes-Zurita, F.J.; Cano-Muñoz, M.; Martinez, A.; Lupiañez, J.A.; Parra, A. Oleanolic Acid Derivatives as Potential Inhibitors of HIV-1 Protease. J. Nat. Prod. 2019, 82, 2886–2896. [Google Scholar] [CrossRef]
- Kaur, R.; Sharma, P.; Gupta, G.K.; Ntie-Kang, F.; Kumar, D. Structure-activity-relationship and mechanistic insights for anti-HIV natural products. Molecules 2020, 25, 2070. [Google Scholar] [CrossRef]
- Bednarczyk-Cwynar, B.; Wachowiak, N.; Szulc, M.; Kamińska, E.; Bogacz, A.; Bartkowiak-Wieczorek, J.; Zaprutko, L.; Mikolajczak, P.L. Strong and long-lasting antinociceptive and anti-inflammatory conjugate of naturally occurring oleanolic acid and aspirin. Front. Pharmacol. 2016, 7, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumitsu, S.; Villareal, M.O.; Fujitsuka, T.; Aida, K.; Isoda, H. Anti-inflammatory and anti-arthritic effects of pentacyclic triterpenoids maslinic acid through NF-κB inactivation. Mol. Nutr. Food Res. 2016, 60, 399–409. [Google Scholar] [CrossRef]
- Prachayasittikul, S.; Saraban, P.; Cherdtrakulkiat, R.; Ruchirawat, S.; Prachayasittikul, V. New bioactive triterpenoids and antimalarial activity of Diospyros rubra Lec. EXCLI J. 2010, 9, 1. [Google Scholar] [PubMed]
- Wang, G.W.; Deng, L.Q.; Luo, Y.P.; Liao, Z.H.; Chen, M. Hepatoprotective triterpenoids and lignans from the stems of Schisandra pubescens. Nat. Prod. Res. 2017, 31, 1855–1860. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.B.; Xiao, Y.H.; Zhang, Q.Y.; Zhou, M.; Liao, S.G. Hepatoprotective natural triterpenoids. Eur. J. Med. Chem. 2018, 145, 691–716. [Google Scholar] [CrossRef]
- Chung, P.Y. Novel targets of pentacyclic triterpenoids in Staphylococcus aureus: A systematic review. Phytomedicine 2020, 73, 152933. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Khalitova, R.R.; Nedopekina, D.A.; Gubaidullin, R.R. Antimicrobial properties of amine-and guanidine-functionalized derivatives of betulinic, ursolic and oleanolic acids: Synthesis and structure/activity evaluation. Steroids 2020, 154, 108530. [Google Scholar] [CrossRef]
- Beinke, C.; Scherthan, H.; Port, M.; Popp, T.; Hermann, C.; Eder, S. Triterpenoid CDDO-Me induces ROS generation and up-regulates cellular levels of antioxidative enzymes without induction of DSBs in human peripheral blood mononuclear cells. Radiat. Environ. Biophys. 2020, 59, 461–472. [Google Scholar] [CrossRef]
- Choi, C.W.; Jung, H.A.; Kang, S.S.; Choi, J.S. Antioxidant constituents and a new triterpenoid glycoside from Flos Lonicerae. Arch. Pharm. Res. 2007, 30, 1. [Google Scholar] [CrossRef]
- Banerjee, S.; Bose, S.; Mandal, S.C.; Dawn, S.; Sahoo, U.; Ramadan, M.A.; Mandal, S.K. Pharmacological Property of Pentacyclic Triterpenoids. Egypt. J. Chem. 2019, 62, 13–35. [Google Scholar] [CrossRef]
- Csuk, R. Betulinic acid and its derivatives: A patent review (2008–2013). Expert Opin. Ther. Pat. 2014, 24, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhang, X.; Qu, F.; Guo, Z.; Zhao, Y. Dammarane triterpenoids for pharmaceutical use: A patent review (2005–2014). Expert Opin. Ther. Pat. 2015, 25, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, D.; Sharma, A.; Tuli, H.S.; Punia, S.; Sharma, A.K. Ursolic acid and oleanolic acid: Pentacyclic terpenoids with promising anti-inflammatory activities. Recent Pat. Inflamm. Allergy Drug Discov. 2016, 10, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T.; Gribble, G.W.; Honda, T.; Kral, R.M.; Meyer, C.J. Dartmouth College and Reata Pharmaceuticals Inc. Synthetic Triterpenoids and Methods of Use in the Treatment of Disease. U.S. Patent 8,129,429, 06 March 2012. [Google Scholar]
- Yan, X.J.; Gong, L.H.; Zheng, F.Y.; Cheng, K.J.; Chen, Z.S.; Shi, Z. Triterpenoids as reversal agents for anticancer drug resistance treatment. Drug Discov. Today 2014, 19, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Kvasnica, M.; Urban, M.; Dickinson, N.J.; Sarek, J. Pentacyclic triterpenoids with nitrogen-and sulfur-containing heterocycles: Synthesis and medicinal significance. Nat. Prod. Rep. 2015, 32, 1303–1330. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, R.H.; Wang, M.; Xu, G.B.; Liao, S.G. Prodrugs of triterpenoids and their derivatives. Eur. J. Med. Chem. 2017, 131, 222–236. [Google Scholar] [CrossRef]
- Kerru, N.; Bhaskaruni, S.V.; Gummidi, L.; Maddila, S.N.; Maddila, S.; Jonnalagadda, S.B. Recent advances in heterogeneous catalysts for the synthesis of imidazole derivatives. Synth. Commun. 2019, 49, 2437–2459. [Google Scholar] [CrossRef]
- Kalaria, P.N.; Karad, S.C.; Raval, D.K. A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery. Eur. J. Med. Chem. 2018, 158, 917–936. [Google Scholar] [CrossRef]
- World Health Organisation. Internation Agency for Research on Cancer. 2018. Available online: https://www.who.int/cancer/PRGlobocanFinal.pdf (accessed on 8 October 2020).
- Khotimchenko, M. Pectin polymers for colon-targeted antitumor drug delivery. Int. J. Biol. Macromol. 2020, 158, 1110–1124. [Google Scholar] [CrossRef]
- Rodrigues, F.C.; Kumar, N.A.; Thakur, G. Developments in the anticancer activity of structurally modified curcumin: An up-to-date review. Eur. J. Med. Chem. 2019, 177, 76–104. [Google Scholar] [CrossRef]
- Khwaza, V.; Oyedeji, O.O.; Aderibigbe, B.A. Ursolic acid-based derivatives as potential anti-cancer agents: An update. Int. J. Mol. Sci. 2020, 21, 5920. [Google Scholar] [CrossRef] [PubMed]
- Selvam, C.; Prabu, S.L.; Jordan, B.C.; Purushothaman, Y.; Umamaheswari, A.; Zare, M.S.H.; Thilagavathi, R. Molecular mechanisms of curcumin and its analogs in colon cancer prevention and treatment. Life Sci. 2019, 239, 117032. [Google Scholar] [CrossRef] [PubMed]
- Babalola, I.T.; Shode, F.O. Ubiquitous ursolic acid: A potential pentacyclic triterpene natural product. J. Pharmacogn. Phytochem. 2013, 2, 214–222. [Google Scholar]
- Khwaza, V.; Oyedeji, O.O.; Aderibigbe, B.A. Antiviral activities of oleanolic acid and its analogues. Molecules 2018, 23, 2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzubak, P.; Hajduch, M.; Vydra, D.; Hustova, A.; Kvasnica, M.; Biedermann, D.; Markova, L.; Urban, M.; Sarek, J. Pharmacological activities of natural triterpenoids and their therapeutic implications. Nat. Prod. Rep. 2006, 23, 394–411. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Nguyen, A.H.; Kumar, A.P.; Tan, B.K.; Sethi, G. Targeted inhibition of tumor proliferation, survival, and metastasis by pentacyclic triterpenoids: Potential role in prevention and therapy of cancer. Cancer Lett. 2012, 320, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.F.; Ogundele, A.; Forrest, A.; Wilton, J.; Salzwedel, K.; Doto, J.; Allaway, G.P.; Martin, D.E. Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single-dose 3-o-(3′,3′-dimethylsuccinyl) betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob. Agents Chemother. 2007, 51, 3574–3581. [Google Scholar] [CrossRef] [Green Version]
- Dang, Z.; Ho, P.; Zhu, L.; Qian, K.; Lee, K.H.; Huang, L.; Chen, C.H. New betulinic acid derivatives for bevirimat-resistant human immunodeficiency virus type-1. J. Med. Chem. 2013, 56, 2029–2037. [Google Scholar] [CrossRef]
- Xu, H.; Ji, L.; Yu, C.; Chen, Q.; Ge, Q.; Lu, Y. MiR-423-5p Regulates Cells Apoptosis and Extracellular Matrix Degradation via Nucleotide-Binding, Leucine-Rich Repeat Containing X1 (NLRX1) in Interleukin 1 beta (IL-1β)-Induced Human Nucleus Pulposus Cells. Med. Sci. Monit. 2020, 26, e922497. [Google Scholar] [CrossRef]
- Manu, K.A.; Kuttan, G. Ursolic acid induces apoptosis by activating p53 and caspase-3 gene expressions and suppressing NF-κB mediated activation of bcl-2 in B16F-10 melanoma cells. Int. Immunopharmacol. 2008, 8, 974–981. [Google Scholar] [CrossRef]
- Pengyue, Z.; Tao, G.; Hongyun, H.; Liqiang, Y.; Yihao, D. Breviscapine confers a neuroprotective efficacy against transient focal cerebral ischemia by attenuating neuronal and astrocytic autophagy in the penumbra. Biomed. Pharmacother. 2017, 90, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.S.; Zhou, W.; Park, I.; Kang, K.; Lee, S.R.; Piao, X.; Park, J.B.; Kwon, T.K.; Na, M.; Hur, G.M. C-27-carboxylated oleanane triterpenoids up-regulate TRAIL DISC assembly via p38 MAPK and CHOP-mediated DR5 expression in human glioblastoma cells. Biochem. Pharmacol. 2018, 158, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, C.; Jou, D.; Lü, J.; Zhang, C.; Lin, L.; Lin, J. Ursolic acid inhibits the growth of colon cancer-initiating cells by targeting STAT3. Anticancer Res. 2013, 33, 4279–4284. [Google Scholar] [PubMed]
- El Bourakadi, K.; Mekhzoum, M.E.M.; Saby, C.; Morjani, H.; Chakchak, H.; Merghoub, N.; Bouhfid, R. Synthesis, characterization and in vitro anticancer activity of thiabendazole-derived 1, 2, 3-triazole derivatives. New J. Chem. 2020, 44, 12099–12106. [Google Scholar] [CrossRef]
- Pokhodylo, N.; Shyyka, O.; Matiychuk, V. Synthesis and anticancer activity evaluation of new 1, 2, 3-triazole-4-carboxamide derivatives. Med. Chem. Res. 2014, 23, 2426–2438. [Google Scholar] [CrossRef]
- Louie, T.; Goodman, C.D.; Holloway, G.A.; McFadden, G.I.; Mollard, V.; Watson, K.G. Dimeric cyclohexane-1, 3-dione oximes inhibit wheat acetyl-CoA carboxylase and show anti-malarial activity. Bioorg. Med. Chem. Lett. 2010, 20, 4611–4613. [Google Scholar] [CrossRef]
- Batra, N.; Rajendran, V.; Wadi, I.; Lathwal, A.; Dutta, R.K.; Ghosh, P.C.; Gupta, R.D.; Nath, M. Synthesis, characterization, and antiplasmodial efficacy of sulfonamide-appended [1,2,3]-triazoles. J. Heterocycl. Chem. 2020, 57, 1625–1636. [Google Scholar] [CrossRef]
- Tan, W.; Li, Q.; Wang, H.; Liu, Y.; Zhang, J.; Dong, F.; Guo, Z. Synthesis, characterization, and antibacterial property of novel starch derivatives with 1, 2, 3-triazole. Carbohydr. Polym. 2016, 142, 1–7. [Google Scholar] [CrossRef]
- Nejadshafiee, V.; Naeimi, H.; Zahraei, Z. Efficient synthesis and antibacterial evaluation of some substituted β-hydroxy-1, 2, 3-triazoles. Chem. Data Collect. 2020, 28, 100443. [Google Scholar] [CrossRef]
- Dai, Z.; Chen, Y.; Zhang, M.; Li, S.; Yang, T.; Shen, L.; Wang, J.; Qian, S.; Zhu, H.; Ye, Y. Synthesis and Antifungal Activity of 1,2,3-Triazole Phenylhydrazone Derivatives. Org. Biomol. Chem. 2015, 13, 10715–10722. [Google Scholar] [CrossRef]
- Aher, N.G.; Pore, V.S.; Mishra, N.N.; Kumar, A.; Shukla, P.K.; Sharma, A.; Bhat, M.K. Synthesis and antifungal activity of 1,2,3-triazole containing fluconazole analogues. Bioorg. Med. Chem. Lett. 2009, 19, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Chandrika, K.M.; Sharma, S. Promising antifungal agents: A minireview. Bioorganic Med. Chem. 2020, 28, 115398. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Haque, A.; Hsieh, M.F.; Imran Hassan, S.; Faizi, M.; Haque, S.; Dege, N.; Khan, M.S. 1, 4-Disubstituted 1H-1,2,3-Triazoles for renal diseases: Studies of viability, anti-inflammatory, and antioxidant activities. Int. J. Mol. Sci. 2020, 21, 3823. [Google Scholar] [CrossRef] [PubMed]
- He, Y.W.; Dong, C.Z.; Zhao, J.Y.; Ma, L.L.; Li, Y.H.; Aisa, H.A. 1,2,3-Triazole-containing derivatives of rupestonic acid: Click-chemical synthesis and antiviral activities against influenza viruses. Eur. J. Med. Chem. 2014, 76, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Wang, W.; Wang, S.; Bao, L. Asymmetric synthesis of novel triazole derivatives and their in vitro antiviral activity and mechanism of action. Eur. J. Med. Chem. 2017, 139, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhao, S.J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef]
- Thi, T.A.D.; Tuyet, N.T.K.; The, C.P.; Nguyen, H.T.; Thi, C.B.; Phuong, H.T.; Van Boi, L.; Van Nguyen, T.; D’hooghe, M. Synthesis and cytotoxic evaluation of novel amide–triazole-linked triterpenoid–AZT conjugates. Tetrahedron Lett. 2015, 56, 218–224. [Google Scholar]
- Thi, T.A.D.; Tuyet, N.T.K.; The, C.P.; Nguyen, H.T.; Thi, C.B.; Duy, T.D.; D’hooghe, M.; Van Nguyen, T. Synthesis and cytotoxic evaluation of novel ester-triazole-linked triterpenoid–AZT conjugates. Bioorg. Med. Chem. 2014, 24, 5190–5194. [Google Scholar]
- Majeed, R.; Sangwan, P.L.; Chinthakindi, P.K.; Khan, I.; Dangroo, N.A.; Thota, N.; Hamid, A.; Sharma, P.R.; Saxena, A.K.; Koul, S. Synthesis of 3-O-propargylated betulinic acid and its 1,2,3-triazoles as potential apoptotic agents. Eur. J. Med. Chem. 2013, 63, 782–792. [Google Scholar] [CrossRef]
- Khan, I.; Guru, S.K.; Rath, S.K.; Chinthakindi, P.K.; Singh, B.; Koul, S.; Bhushan, S.; Sangwan, P.L. A novel triazole derivative of betulinic acid induces extrinsic and intrinsic apoptosis in human leukemia HL-60 cells. Eur. J. Med. Chem. 2016, 108, 104–116. [Google Scholar] [CrossRef]
- Suman, P.; Patel, A.; Solano, L.; Jampana, G.; Gardner, Z.S.; Holt, C.M.; Jonnalagadda, S.C. Synthesis and cytotoxicity of Baylis-Hillman template derived betulinic acid-triazole conjugates. Tetrahedron 2017, 73, 4214–4226. [Google Scholar] [CrossRef]
- Shi, W.; Tang, N.; Yan, W.D. Synthesis and cytotoxicity of triterpenoids derived from betulin and betulinic acid via click chemistry. J. Asian Nat. Prod. Res. 2015, 17, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Sidova, V.; Zoufaly, P.; Pokorny, J.; Dzubak, P.; Hajduch, M.; Popa, I.; Urban, M. Cytotoxic conjugates of betulinic acid and substituted triazoles prepared by Huisgen Cycloaddition from 30-azidoderivatives. PLoS ONE 2017, 12, 0171621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, B.; Dutta, D.; Mukherjee, S.; Das, S.; Maiti, N.C.; Das, P.; Chowdhury, C. Synthesis and biological evaluation of a novel betulinic acid derivative as an inducer of apoptosis in human colon carcinoma cells (HT-29). Eur. J. Med. Chem. 2015, 102, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Bębenek, E.; Jastrzębska, M.; Kadela-Tomanek, M.; Chrobak, E.; Orzechowska, B.; Zwolińska, K.; Latocha, M.; Mertas, A.; Czuba, Z.; Boryczka, S. Novel triazole hybrids of betulin: Synthesis and biological activity profile. Molecules 2017, 22, 1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, R.X.; Zhao, L.; Zhang, Y.K.; Luo, P.; Liu, S.H.; Wang, J.G. Sequential treatment with ursolic acid chlorophenyl triazole followed by 5-fluorouracil shows synergistic activity in small cell lung cancer cells. Bangladesh J. Pharmacol. 2015, 10, 197–204. [Google Scholar] [CrossRef]
- Wei, G.; Luan, W.; Wang, S.; Cui, S.; Li, F.; Liu, Y.; Liu, Y.; Cheng, M. A library of 1,2,3-triazole-substituted oleanolic acid derivatives as anticancer agents: Design, synthesis, and biological evaluation. Org. Biomol. Chem. 2015, 13, 1507–1514. [Google Scholar] [CrossRef]
- Li, F.; Liu, Y.; Wang, S.; Wei, G.; Cheng, M. Synthesis and tumor cytotoxicity of novel 1, 2, 3-triazole-substituted 3-oxo-oleanolic acid derivatives. Chem. Res. Chin. Univ. 2016, 32, 938–942. [Google Scholar] [CrossRef]
- Leal, A.S.; Wang, R.; Salvador, J.A.; Jing, Y. Synthesis of novel ursolic acid heterocyclic derivatives with improved abilities of antiproliferation and induction of p53, p21waf1 and NOXA in pancreatic cancer cells. Bioorg. Med. Chem. 2012, 20, 5774–5786. [Google Scholar] [CrossRef]
- Pertino, M.W.; Lopez, C.; Theoduloz, C.; Schmeda-Hirschmann, G. 1,2,3-Triazole-substituted oleanolic acid derivatives: Synthesis and antiproliferative activity. Molecules 2013, 18, 7661. [Google Scholar] [CrossRef] [Green Version]
- Lakshmi, J.K.; Pattnaik, B.; Kavitha, R.; Mallavadhani, U.V.; Jagadeesh, B. Conformation of flexibly linked triterpene dimers by using RDC-enhanced NMR spectroscopy. J. Mol. Struct 2018, 1162, 26–30. [Google Scholar] [CrossRef]
- Pattnaik, B.; Lakshmi, J.K.; Kavitha, R.; Jagadeesh, B.; Bhattacharjee, D.; Jain, N.; Mallavadhani, U.V. Synthesis, structural studies, and cytotoxic evaluation of novel ursolic acid hybrids with capabilities to arrest breast cancer cells in mitosis. J. Asian Nat. Prod. Res. 2017, 19, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Popov, S.A.; Semenova, M.D.; Baev, D.S.; Frolova, T.S.; Shestopalov, M.A.; Wang, C.; Qi, Z.; Shults, E.E.; Turks, M. Synthesis and cytotoxicity of hybrids of 1, 3, 4-or 1, 2, 5-oxadiazoles tethered from ursane and lupane core with 1, 2, 3-triazole. Steroids 2020, 162, 108698. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Dar, B.A.; Majeed, R.; Hamid, A.; Bhat, B.A. Synthesis and biological evaluation of ursolic acid-triazolyl derivatives as potential anti-cancer agents. Eur. J. Med. Chem. 2013, 66, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Fustero, S.; Sanchez-Rosello, M.; Barrio, P.; Simon-Fuentes, A. From 2000 to mid-2010: A fruitful decade for the synthesis of pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A. Synthesis and pharmacological activities of pyrazole derivatives: A review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [Green Version]
- Ansari, A.; Ali, A.; Asif, M. Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Balbi, A.; Anzaldi, M.; Macciò, C.; Aiello, C.; Mazzei, M.; Gangemi, R.; Castagnola, P.; Miele, M.; Rosano, C.; Viale, M. Synthesis and biological evaluation of novel pyrazole derivatives with anticancer activity. Eur. J. Med. Chem. 2011, 46, 5293–5309. [Google Scholar] [CrossRef]
- Puneeth, H.R.; Ananda, H.; Kumar, K.S.S.; Rangappa, K.S.; Sharada, A.C. Synthesis and antiproliferative studies of curcumin pyrazole derivatives. Med. Chem. Res. 2016, 25, 1842–1851. [Google Scholar] [CrossRef]
- Ravula, P.; Vamaraju, H.B.; Paturi, M.; JN, N.S.C.; Kolli, S. Design, synthesis, in silico toxicity prediction, molecular docking, and evaluation of novel pyrazole derivatives as potential antiproliferative agents. EXCLI J. 2016, 15, 187. [Google Scholar]
- Nitulescu, G.M.; Draghici, C.; Missir, A.V. Synthesis of new pyrazole derivatives and their anticancer evaluation. Eur. J. Med. Chem 2010, 45, 4914–4919. [Google Scholar] [CrossRef] [PubMed]
- Insuasty, B.; Tigreros, A.; Orozco, F.; Quiroga, J.; Abonía, R.; Nogueras, M.; Sanchez, A.; Cobo, J. Synthesis of novel pyrazolic analogues of chalcones and their 3-aryl-4-(3-aryl-4, 5-dihydro-1H-pyrazol-5-yl)-1-phenyl-1H-pyrazole derivatives as potential antitumor agents. Bioorg. Med. Chem. 2010, 18, 4965–4974. [Google Scholar] [CrossRef] [PubMed]
- Labbozzetta, M.; Baruchello, R.; Marchetti, P.; Gueli, M.C.; Poma, P.; Notarbartolo, M.; Simoni, D.; D’Alessandro, N. Lack of nucleophilic addition in the isoxazole and pyrazole diketone modified analogs of curcumin; implications for their antitumor and chemosensitizing activities hem. Biol. Interact. 2009, 181, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Bennani, F.E.; Doudach, L.; Cherrah, Y.; Ramli, Y.; Karrouchi, K.; Faouzi, M.E.A. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorg. Chem. 2020, 97, 103470. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, B.; Su, X.; Chen, G.; Li, Y.; Yu, L.; Li, L.; Wei, W. An ursolic acid derived small molecule triggers cancer cell death through hyperstimulation of macropinocytosis. J. Med. Chem. 2017, 60, 6638–6648. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, H.; Nie, M.; Wang, W.; Liu, Z.; Chen, C.; Chen, H.; Liu, R.; Baloch, Z.; Ma, K. A novel synthetic ursolic acid derivative inhibits growth and induces apoptosis in breast cancer cell lines. Oncol. Lett. 2018, 15, 2323–2329. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Li, C.; Zheng, Y.; Gao, Y.; Hu, J.; Chen, H. Discovery of FZU-03,010 as a self-assembling anticancer amphiphile for acute myeloid leukemia. Bioorg. Med. Chem. Lett. 2017, 27, 1007–1011. [Google Scholar] [CrossRef]
- Santos, R.C.; Salvador, J.A.; Cortés, R.; Pachón, G.; Marín, S.; Cascante, M. New betulinic acid derivatives induce potent and selective antiproliferative activity through cell cycle arrest at the S phase and caspase dependent apoptosis in human cancer cells. Biochimie 2011, 93, 1065–1075. [Google Scholar] [CrossRef]
- Leal, A.S.; Wang, R.; Salvador, J.A.; Jing, Y. Synthesis of novel heterocyclic oleanolic acid derivatives with improved antiproliferative activity in solid tumor cells. Org. Biomol. Chem. 2013, 11, 1726–1738. [Google Scholar] [CrossRef]
- Han, Y.; Dong, W.; Guo, Q.; Li, X.; Huang, L. The importance of indole and azaindole scaffold in the development of antitumor agents. Eur. J. Med. Chem. 2020, 112506. [Google Scholar] [CrossRef]
- Khusnutdinova, E.F.; Petrova, A.V.; Apryshko, G.N.; Kukovinets, O.S.; Kazakova, O.B. Synthesis and cytotoxicity of indole derivatives of betulin, erythrodiol, and uvaol. Russ. J. Bioorg. Chem. 2018, 44, 322–329. [Google Scholar] [CrossRef]
- Khusnutdinova, E.F.; Petrova, A.V.; Kukovinets, O.S.; Kazakova, O.B. Synthesis and cytotoxicity of 28-N-propargylaminoalkylated 2,3-indolotriterpenic acids. Nat. Prod. Commun. 2018, 13, 1934578X1801300603. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.; Huang, J.; Pan, J.; Zhang, X.; Lu, W. Design, synthesis and biological evaluation of C (6)-indole celastrol derivatives as potential antitumor agents. RSC Adv. 2015, 5, 19620–19623. [Google Scholar] [CrossRef]
- Fan, H.; Geng, L.; Yang, F.; Dong, X.; He, D.; Zhang, Y. Ursolic acid derivative induces apoptosis in glioma cells through down-regulation of cAMP. Eur. J. Med. Chem. 2019, 176, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Walayat, K.; Mohsin, N.U.A.; Aslam, S.; Ahmad, M. An insight into the therapeutic potential of piperazine-based anticancer agents. Turkish J. Chem. 2019, 43, 1–23. [Google Scholar] [CrossRef]
- Rathi, A.K.; Syed, R.; Shin, H.S.; Patel, R.V. Piperazine derivatives for therapeutic use: A patent review (2010-present). Expert Opin. Ther. Pat. 2016, 26, 777–797. [Google Scholar] [CrossRef]
- Zhao, C.H.; Zhang, C.L.; Shi, J.J.; Hou, X.Y.; Feng, B.; Zhao, L.X. Design, synthesis, and biofunctional evaluation of novel pentacyclic triterpenes bearing O-[4-(1-piperazinyl)-4-oxo-butyryl moiety as antiproliferative agents. Bioorg. Med. Chem. Lett. 2015, 25, 4500–4504. [Google Scholar] [CrossRef]
- Yamansarov, E.Y.; Osterman, I.A.; Komarova, E.S.; Skvortsov, D.A.; Saltikova, I.V.; Majouga, A.G.; Ivanenkov, Y.A.; Giniyatullina, G.V.; Kazakova, O.B.; Baikova, I.P. Synthesis and cytotoxicity of a-azepanobetulinic acid N-methyl-piperazinylamide. Nat. Prod. Commun. 2019, 14. [Google Scholar] [CrossRef] [Green Version]
- Hua, S.X.; Huang, R.Z.; Ye, M.Y.; Pan, Y.M.; Yao, G.Y.; Zhang, Y.; Wang, H.S. Design, synthesis and in vitro evaluation of novel ursolic acid derivatives as potential anticancer agents. Eur. J. Med. Chem. 2015, 95, 435–452. [Google Scholar] [CrossRef]
- Liu, M.C.; Yang, S.J.; Jin, L.H.; Hu, D.Y.; Xue, W.; Song, B.A.; Yang, S. Synthesis and cytotoxicity of novel ursolic acid derivatives containing an acyl piperazine moiety. Eur. J. Med. Chem. 2012, 58, 128–135. [Google Scholar] [CrossRef]
- Wang, W.; Lei, L.; Liu, Z.; Wang, H.; Meng, Q. Design, synthesis, and biological evaluation of novel nitrogen heterocycle-containing ursolic acid analogs as antitumor agents. Molecules 2019, 24, 877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahnt, M.; Hoenke, S.; Fischer, L.; Al-Harrasi, A.; Csuk, R. Synthesis and cytotoxicity evaluation of DOTA-conjugates of ursolic acid. Molecules 2019, 24, 2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, S.; Singh, P.K.; Verma, H.; Singh, H.; Silakari, O. Success stories of natural product-based hybrid molecules for multi-factorial diseases. Eur. J. Med. Chem. 2018, 151, 62–97. [Google Scholar] [CrossRef] [PubMed]
- Brandão, G.C.; Missias, F.C.R.; Arantes, L.M.; Soares, L.F.; Roy, K.K.; Doerksen, R.J.; de Oliveira, A.B.; Pereira, G.R. Antimalarial naphthoquinones. Synthesis via click chemistry, in vitro activity, docking to PfDHODH and SAR of lapachol-based compounds. Eur. J. Med. Chem. 2018, 145, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Sommerwerk, S.; Heller, L.; Kuhfs, J.; Csuk, R. Selective killing of cancer cells with triterpenoic acid amides-The substantial role of an aromatic moiety alignment. Eur. J. Med. Chem. 2016, 122, 452–464. [Google Scholar] [CrossRef]
- Hoenke, S.; Heise, N.V.; Kahnt, M.; Deigner, H.P.; Csuk, R. Betulinic acid derived amides are highly cytotoxic, apoptotic and selective. Eur. J. Med. Chem. 2020, 207, 112815. [Google Scholar] [CrossRef] [PubMed]
- Kadela-Tomanek, M.; Bębenek, E.; Chrobak, E.; Marciniec, K.; Latocha, M.; Kuśmierz, D.; Jastrzębska, M.; Boryczka, S. Betulin-1, 4-quinone hybrids: Synthesis, anticancer activity and molecular docking study with NQO1 enzyme. Eur. J. Med. Chem. 2019, 177, 302–315. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybrid Compounds | Mechanism/Pathways | Cancer Cell Lines | Hybrid Compounds (IC50 µM) | Reference Molecules (IC50 µM) | Bibliography | ||
|---|---|---|---|---|---|---|---|
| 5a | n.r | KB Hep-G2 | (8.1) (6.6) | Betulinic acid (27.5) (23.9) | Ellipticine (1.2) (1.2) | [59] | |
| 5b | (4.6) (3.5) | ||||||
| 6 | (8.1) (5.9) | ||||||
| 7 | (5.9) (7.0) | [60] | |||||
| 8 | (6.3) (7.4) | ||||||
| 9 | (7.7) (11.2) | ||||||
| 10 | Induced apoptosis and inhibit cell migration, disruption of mitochondrial membrane. | THP-1 HL-60 | (4.5) (2.5) | Betulinic acid (20) (17) | ------ | [61] | |
| 11 | (8) (3.5) | ||||||
| 12 | Induced apoptosis through both intrinsic and extrinsic pathways. | HL-60 MiaPaCa-2 PC-43 A549 | (7) (5) (7) (7) | Betulinic acid (8) (7) (7) (8) | ------ | [62] | |
| 13 | n.r | 4T1 MIAPaCa-2 | (2.38 ± 0.45) (1.36 ± 0.21) | Betulinic acid (6.29 ± 0.96) (25.63 ± 3.79) | ------ | [63] | |
| 14 | (2.62 ± 0.24) (1.64 ± 0.20) | ||||||
| 15 | nr | HL-60 | (1.3 ± 0.1) | Betulinic acid 11.5 ± 2.8 | ------ | [64] | |
| 16 | Inhibit DNA and RNA, cause block in G0/G1 cell cycle phase similar to actinomycin D. | CCRF-CEM CEM-DNR K562 K562-TAX A549 HCT116 HCT116p53-/- U2OS | (3.3) (4.0) (3.6) (3.9) (14.8) (6.4) (9.5) (12.8) | Betulinic acid (45.5) (45.4) (40.0) (43.1) (43.4) (38.0) (>50.0) (>50.0) | ------ | [65] | |
| 17 | Induce apoptosis, acts as a minor groove binder to the DNA | HT29 | (14.9 ± 1.3) | Betulinic acid (14.9 ± 1.3) | ------ | [66] | |
| 18 | n.r | T47D2 MCF-7 SNB-19 | (0.05 ± 0.01) (0.09 ± 0.01) (0.08 ± 0.01) | Betulinic acid (neg) (17.7 ± 1.2) (neg) | Cisplatin (4.9 ± 1.1) (5.5 ± 1.0) (2.3 ± 0.05) | [67] | |
| 19 | Activate NF-kB protein, repression of TNF-induced NF-kB-dependent reporter gene expression, and TNF-induced COX-2, MMP-9 and Cyclin D1 activation | n.r | n.r | n.r | n.r | [68] | |
| 20 | Apoptosis inducer in HT1080 cells | HeLa HepG2 HCT116 A375-S2 HT1080 | (10.85) (24.15) (12.28) (4.97) (3.51) | Oleanolic acid (>200) (>200) (>200) (>200) (>200) | 5-Fu (26.18) (67.64) (35.16) (90.74) (25.46) | [69] | |
| 23 | n.r | A375-S2 HT1080 | (2.82±0.11) (1.69±0.26) | Taxol (59.57±0.17) (35.18±0.44) | 5-Fu (59.57±0.17) (35.18±0.44) | [70] | |
| 24 | (36.54±0.39) (1.86±0.17) | ||||||
| 25 | (13.98±0.78) (1.73±0.45) | ||||||
| 26 | (34.87±0.62) (1.82±0.16) | ||||||
| 27 | (4.76±0.11) (1.84±0.16) | ||||||
| 28 | Induction of p21waf1, p53 and NOXA which leads to cell cycle arrest and AsPC-1 apoptosis | AsPC-1 | (1.9 ± 0.02) | Ursolic acid (15.2 ± 0.1) | ------ | [71] | |
| 29 | Inhibits proliferation of Gastric epithelial adenocarcinoma(AGS) | MRC-5 AGS SK-MES-1 J82 HL-60 | (>100) (8.9 ± 0.4) (50.4 ± 3.5) (35.4 ± 2.8) (35.8 ± 4.1) | ----- | Etoposide (0.33 ± 0.02) (0.58 ± 0.02) (1.83 ± 0.09) (3.49 ± 0.16) (2.23 ± 0.09) | [72] | |
| 33 | Affinity to Mdm2 binding sites | MCF-7 U-87 MG A549 HepG2 | (1.55 ± 0.08) (>100) (>100) (>100) | Ursolic acid (25.05 ± 3.17) (43.82 ± 3.88) (41.02 ± 3.77) (37.28 ± 5.02) | Doxorubicin (4.51 ± 1.12) (2.05 ± 0.22) (6.17 ± 1.17) (10.02 ± 1.67) | [75] | |
| 36a | A-549 MCF-7 HCT-116 THP-1 FR-2 | (0.5 ± 0.05) (5.5 ± 0.08) (˂0.1 ± 0.09) (0.9 ± 0.02) (10 ± 0.04) | Ursolic acid (33 ± 0.03) (37 ± 0.07) (42 ± 0.08) (9.1 ± 0.07) (31 ± 0.08) | ------ | [76] | ||
| 36b | (2.9 ± 0.05) (˂0.1 ± 0.05) (15 ± 0.06) (˂0.1 ± 0.03) (69 ± 0.05) | ||||||
| 36c | (˂0.1 ± 0.001) (˂0.1 ± 0.09) (0.3 ± 0.001) (˂0.1 ± 0.001) (˃50 ± 4.1) | ||||||
| 36d | (0.15 ± 0.01) (˂0.1 ± 0.001) (9.1 ± 1.0) (˂0.1 ± 0.001) (˃50 ± 3.9) | ||||||
| Hybrid Compounds | Mechanism /Pathways | Cancer Cell Lines | Hybrid Compounds (IC50 µM) | Reference Molecules (IC50 µM) | Bibliography | |
|---|---|---|---|---|---|---|
| 41 | Induced apoptosis by hyperstimulation of macropinocytosis | Hela HepG2 HT1080 MCF-7 SK-N-MC | (18.63 ± 2.34) (27.87 ± 2.98) (26.7 ± 0.07) (25.25 ± 0.07) (28.63 ± 1.03) | Ursolic acid (43.30 ± 2.22) (34.12 ± 0.68) (39.43 ± 0.52) (57.64 ± 5.75) (67.64 ± 1.78) | ---- | [87] |
| 42 | Hindered the breast cancer cell progression by inducing apoptosis and cell cycle arrest at S and G0/G1 phase | HL-60 | (0.91±0.05) | Ursolic acid (>40) | Doxorubicin (<0.63) | [88,89] |
| 43 | Induced apoptosis and cell cycle arrest in HepG2, HeLa, and Jurkat cell lines | Jurkat HeLa HepG2 | (1.4 ± 0.2) (2.0 ± 0.3) (0.8 ± 0.05) | Betulinic acid (26.9 ± 2.2) (26.0 ± 2.1) (36.4 ± 1.5) | ---- | [90] |
| 44 | n.r | (2.3 ± 0.3) (3.0 ± 0.2) (1.7 ± 0.2) | ||||
| 45 | n.r | (11.1 ± 1.3) (3.0 ± 0.2) (2.0 ± 0.4) | ||||
| 46 | Antiproliferative, Apoptosis induction abilities correlated with upregulation of NOXA and downregulation of Bcl-xL | AsPC-1 | (0.9 ± 0.01) | (˃100) | ---- | [91] |
| Hybrid Compounds | Mechanism /Pathways | Cancer Cell Lines | Hybrid Compounds (IC50 µM) | Reference Molecules (IC50 µM) | Bibliography | ||
|---|---|---|---|---|---|---|---|
| 60 | n.r | MCF-7 Hela A549 | (7.05 ± 1.45) (7.75 ± 1.09) (9.91 ± 2.11) | ----- | Gefitini (17.83 ± 7.85)(15.40 ± 4.63)(11.02 ± 3.27) | [99] | |
| 61 | (7.58 ± 1.25) (8.13 ± 1.69) (13.13 ± 4.37) | ||||||
| 62 | n.r | LNCAP PC3 22RW1 Huh7 VA13 | (1.12) (1.11) (0.37) (0.74) (0.19) | ----- | Paclitaxel (0.01) (0.015) (0.016) (0.002) (0.001) | [100] | |
| 63 | (n.r) (3.58) (n.r) (2.75) (n.r) | ||||||
| 64 | Inhibits proliferation, Induce cell apoptosis by G1 cell-cycle arrest and through intrinsic and extrinsic apoptosis pathway. | MGC-803 HCT-116 T24 HepG2 A549 HL-7702 | (9.82 ± 0.29) (18.97 ± 0.53) (13.64 ± 0.43) (5.40 ± 0.79) (11.06 ± 0.37) (>100) | Ursolic acid (27.08 ± 0.29) (38.78 ± 0.16) (29.29 ± 0.80) (30.21 ± 0.58) (35.79 ± 0.37) (>100) | 5-FU (40.94 ± 0.95) (29.58 ± 1.31) (37.56 ± 0.49) (30.79 ± 0.82) (36.34 ± 0.57) (58.74 ± 2.31) | [101] | |
| 65 | Induced cell apoptosis in MGC-803 cells. | MGC-803 Bcap-37 | (2.50 ± 0.25) (9.24 ± 0.53) | Ursolic acid (24.32 ± 0.57) (28.69 ± 0.35) | Hydroxycamptothecin (>20) (>20) | [102] | |
| 66 | Decreased the apoptosis regulator (BCL2/BAX) ratio, disrupted mitochondrial potential and induced apoptosis, and suppressed the growth of Hela xenografts in nude mice | Hela MKN45 | (2.6 ± 1.1) (2.1 ± 0.3) | Ursolic acid (15.1 ± 2.7) (16.7 ± 1.4) | Cisplatin (15.1± 0.9) (2.8±0.1) | [103] | |
| 68 | Cytotoxicity through fluorescence microscopy, annexin V assays and cell cycle analysis. | A375 A2780 HT29 MCF-7 FaDu NIH 3T3 | (1.5 ± 0.4) (1.9 ± 0.3) (5.7 ± 0.5) (4.4 ± 0.7) (3.7 ± 0.6) (4.6 ± 1.0) | Ursolic acid (n.r) (11.7 ± 0.6) (10.6 ±0.7) (12.7±0.1) (n.r) (13.1±1.1) | Doxorubici (n.r) (0.01 ± 0.01) (0.9 ± 0.2) (1.1 ± 0.3) (n.r) (0.06 ± 0.03) | [104] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khwaza, V.; Mlala, S.; Oyedeji, O.O.; Aderibigbe, B.A. Pentacyclic Triterpenoids with Nitrogen-Containing Heterocyclic Moiety, Privileged Hybrids in Anticancer Drug Discovery. Molecules 2021, 26, 2401. https://doi.org/10.3390/molecules26092401
Khwaza V, Mlala S, Oyedeji OO, Aderibigbe BA. Pentacyclic Triterpenoids with Nitrogen-Containing Heterocyclic Moiety, Privileged Hybrids in Anticancer Drug Discovery. Molecules. 2021; 26(9):2401. https://doi.org/10.3390/molecules26092401
Chicago/Turabian StyleKhwaza, Vuyolwethu, Sithenkosi Mlala, Opeoluwa O. Oyedeji, and Blessing A. Aderibigbe. 2021. "Pentacyclic Triterpenoids with Nitrogen-Containing Heterocyclic Moiety, Privileged Hybrids in Anticancer Drug Discovery" Molecules 26, no. 9: 2401. https://doi.org/10.3390/molecules26092401