
The Role of the Intrauterine Environment in Shaping Childhood and Adolescence Metabolic Outcomes

Abstract
:1. Introduction
2. Materials and Methods
3. Impact of Intrauterine Growth on the Development of MetS
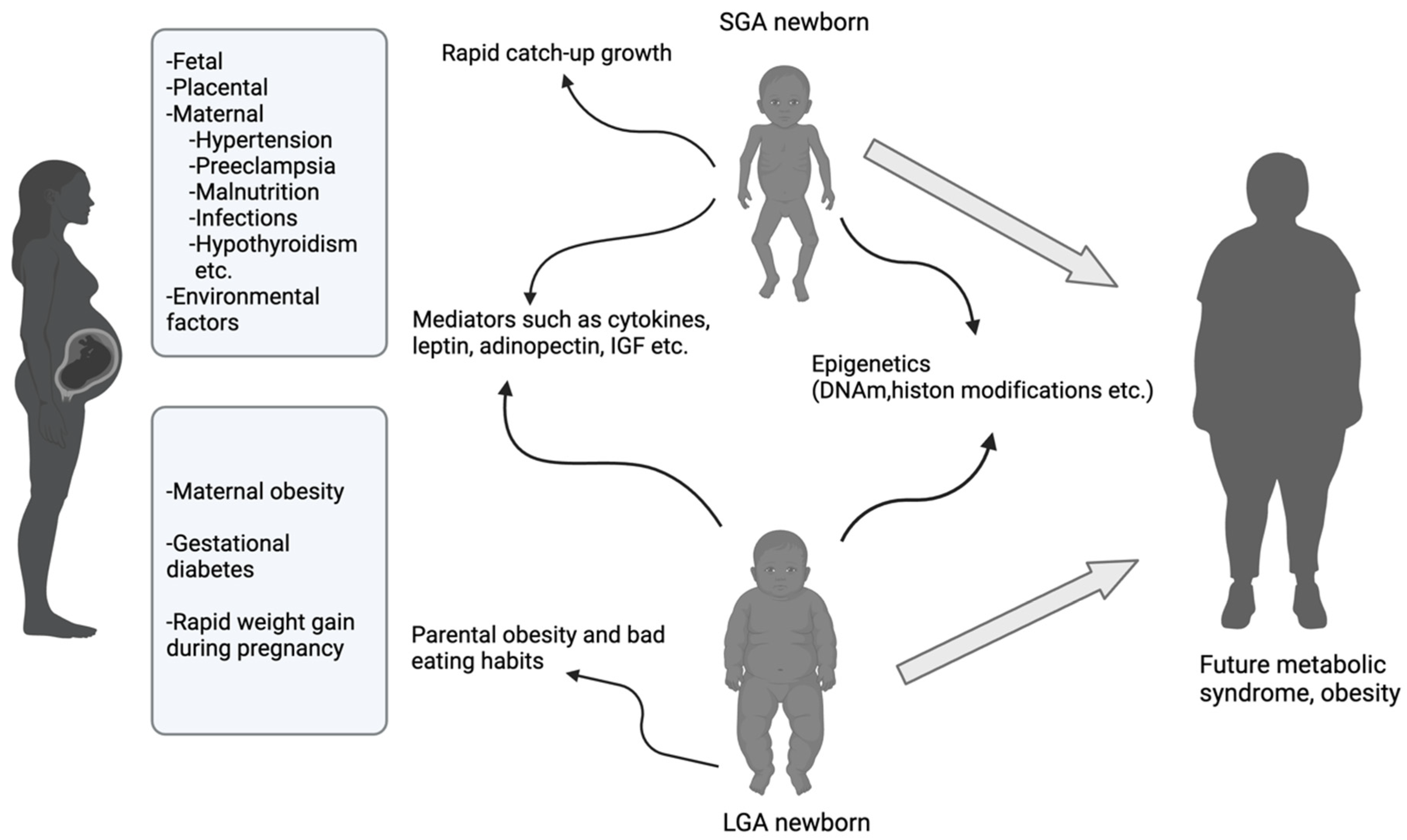
3.1. Intrauterine Growth Restriction (IUGR) and SGA
3.2. LGA
4. Impact of Maternal Obesity and Gestational Diabetes Mellitus on the Development of MetS
5. Maternal Nutrition
6. Maternal Hypertension and Preeclampsia
7. Impact of Epigenetic Modifications on the Development of MetS
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DeBoer, M.D. Assessing and Managing the Metabolic Syndrome in Children and Adolescents. Nutrients 2019, 11, 1788. [Google Scholar] [CrossRef] [PubMed]
- Weihe, P.; Weihrauch-Blüher, S. Metabolic Syndrome in Children and Adolescents: Diagnostic Criteria, Therapeutic Options and Perspectives. Curr. Obes. Rep. 2019, 8, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.D.; Ambrosio, F.; Scardigno, A.; Ricciardi, R.; Calabrò, G.E. Epidemiological Impact of Metabolic Syndrome in Overweight and Obese European Children and Adolescents: A Systematic Literature Review. Nutrients 2023, 15, 3895. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.C.; Blaha, M.J.; Blumenthal, R.S. Relation of C-reactive protein to abdominal adiposity. Am. J. Cardiol. 2010, 106, 56–61. [Google Scholar] [CrossRef]
- Paley, C.A.; Johnson, M.I. Abdominal obesity and metabolic syndrome: Exercise as medicine? BMC Sports Sci. Med. Rehabil. 2018, 10, 7. [Google Scholar] [CrossRef]
- GBD 2015 Obesity Collaborators; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar]
- GBD 2015 Risk Factors Collaborators. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1659–1724. [Google Scholar] [CrossRef]
- Herman, K.M.; Craig, C.L.; Gauvin, L.; Katzmarzyk, P.T. Tracking of obesity and physical activity from childhood to adulthood: The Physical Activity Longitudinal Study. Int. J. Pediatr. Obes. 2009, 4, 281–288. [Google Scholar] [CrossRef]
- Iafusco, D.; Franceschi, R.; Maguolo, A.; Guercio Nuzio, S.; Crinò, A.; Delvecchio, M.; Iughetti, L.; Maffeis, C.; Calcaterra, V.; Manco, M. From Metabolic Syndrome to Type 2 Diabetes in Youth. Children 2023, 10, 516. [Google Scholar] [CrossRef]
- Poyrazoglu, S.; Bas, F.; Darendeliler, F. Metabolic syndrome in young people. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 56–63. [Google Scholar] [CrossRef]
- Tropeano, A.; Corica, D.; Li Pomi, A.; Pepe, G.; Morabito, L.A.; Curatola, S.L.; Casto, C.; Aversa, T.; Wasniewska, M. The metabolic syndrome in pediatrics: Do we have a reliable definition? A systematic review. Eur. J. Endocrinol. 2021, 185, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.; Weitzman, M.; Auinger, P.; Nguyen, M.; Dietz, W.H. Prevalence of a metabolic syndrome phenotype in adolescents: Findings from the third National Health and Nutrition Examination Survey, 1988–1994. Arch. Pediatr. Adolesc. Med. 2003, 157, 821–827. [Google Scholar] [CrossRef]
- de Ferranti, S.D.; Gauvreau, K.; Ludwig, D.S.; Neufeld, E.J.; Newburger, J.W.; Rifai, N. Prevalence of the metabolic syndrome in American adolescents: Findings from the Third National Health and Nutrition Examination Survey. Circulation 2004, 110, 2494–2497. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Dziura, J.; Burgert, T.S.; Tamborlane, W.V.; Taksali, S.E.; Yeckel, C.W.; Allen, K.; Lopes, M.; Savoye, M.; Morrison, J.; et al. Obesity and the metabolic syndrome in children and adolescents. N. Engl. J. Med. 2004, 350, 2362–2374. [Google Scholar] [CrossRef]
- Cruz, M.L.; Weigensberg, M.J.; Huang, T.T.; Ball, G.; Shaibi, G.Q.; Goran, M.I. The metabolic syndrome in overweight Hispanic youth and the role of insulin sensitivity. J. Clin. Endocrinol. Metab. 2004, 89, 108–113. [Google Scholar] [CrossRef]
- Zimmet, P.; Alberti, K.G.; Kaufman, F.; Tajima, N.; Silink, M.; Arslanian, S.; Wong, G.; Bennett, P.; Shaw, J.; Caprio, S.; et al. The metabolic syndrome in children and adolescents—An IDF consensus report. Pediatr. diabetes. 2007, 8, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Winter, P.D.; Osmond, C.; Margetts, B.; Simmonds, S.J. Weight in infancy and death from ischaemic heart disease. Lancet 1989, 2, 577–580. [Google Scholar] [CrossRef]
- Barker, D.J.; Osmond, C.; Golding, J.; Kuh, D.; Wadsworth, M.E. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. Br. Med. J. 1989, 298, 564–567. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A.; Low, F.M. Evolutionary and developmental mismatches are consequences of adaptive developmental plasticity in humans and have implications for later disease risk. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2019, 374, 20180109. [Google Scholar] [CrossRef]
- Barker, D.J. The developmental origins of adult disease. J. Am. Coll. Nutr. 2004, 23 (Suppl. S6), 588S–595S. [Google Scholar] [CrossRef]
- Gillman, M.W. Developmental origins of health and disease. N. Engl. J. Med. 2005, 353, 1848–1850. [Google Scholar] [PubMed]
- Hokken-Koelega, A.C.S.; van der Steen, M.; Boguszewski, M.C.S.; Cianfarani, S.; Dahlgren, J.; Horikawa, R.; Mericq, V.; Rapaport, R.; Alherbish, A.; Braslavsky, D.; et al. International Consensus Guideline on Small for Gestational Age: Etiology and Management From Infancy to Early Adulthood. Endocr. Rev. 2023, 44, 539–565. [Google Scholar] [PubMed]
- Darendeliler, F. IUGR: Genetic influences, metabolic problems, environmental associations/triggers, current and future management. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101260. [Google Scholar] [PubMed]
- Giabicani, E.; Pham, A.; Brioude, F.; Mitanchez, D.; Netchine, I. Diagnosis and management of postnatal fetal growth restriction. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 523–534. [Google Scholar] [CrossRef]
- Finken, M.J.J.; van der Steen, M.; Smeets, C.C.J.; Walenkamp, M.J.E.; de Bruin, C.; Hokken-Koelega, A.C.S.; Wit, J.M. Children Born Small for Gestational Age: Differential Diagnosis, Molecular Genetic Evaluation, and Implications. Endocr. Rev. 2018, 39, 851–894. [Google Scholar] [CrossRef]
- Harder, T.; Rodekamp, E.; Schellong, K.; Dudenhausen, J.W.; Plagemann, A. Birth weight and subsequent risk of type 2 diabetes: A meta-analysis. Am. J. Epidemiol. 2007, 165, 849–857. [Google Scholar]
- Barker, D.J.; Gluckman, P.D.; Godfrey, K.M.; Harding, J.E.; Owens, J.A.; Robinson, J.S. Fetal nutrition and cardiovascular disease in adult life. Lancet 1993, 341, 938–941. [Google Scholar]
- Huang, L.; Yang, S.; Yang, F.; Xiong, F. A prospective study about physical growth of children from birth to 2 years old born full- term small-for-gestational-age. J. Paediatr. Child. Health. 2019, 55, 199–204. [Google Scholar]
- Leunissen, R.W.; Stijnen, T.; Hokken-Koelega, A.C. Influence of birth size on body composition in early adulthood: The programming factors for growth and metabolism (PROGRAM)-study. Clin. Endocrinol. 2009, 70, 245–251. [Google Scholar]
- Leunissen, R.W.J.; Kerkhof, G.F.; Stijnen, T.; Hokken-Koelega, A.C.S. Effect of birth size and catch-up growth on adult blood pressure and carotid intima-media thickness. Horm. Res. Paediatr. 2012, 77, 394–401. [Google Scholar]
- Leunissen, R.W.; Kerkhof, G.F.; Stijnen, T.; Hokken-Koelega, A. Timing and tempo of first-year rapid growth in relation to cardiovascular and metabolic risk profile in early adulthood. JAMA 2009, 301, 2234–2242. [Google Scholar] [CrossRef]
- Ong, K.K. Catch-up growth in small for gestational age babies: Good or bad? Curr. Opin. Endocrinol. Diabetes. Obes. 2007, 14, 30–34. [Google Scholar] [PubMed]
- Ibáñez, L.; López-Bermejo, A.; Díaz, M.; Marcos, M.V.; Casano, P.; de Zegher, F. Abdominal fat partitioning and high-molecular-weight adiponectin in short children born small for gestational age. J. Clin. Endocrinol. Metab. 2009, 94, 1049–1052. [Google Scholar] [CrossRef]
- Dulloo, A.G.; Jacquet, J.; Montani, J.P. Pathways from weight fluctuations to metabolic diseases: Focus on maladaptive thermogenesis during catch-up fat. Int. J. Obes. Relat. Metab. Disord. 2002, 26, S46–S57. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.K.; Potau, N.; Petry, C.J.; Jones, R.; Ness, A.R.; Honour, J.W.; de Zegher, F.; Ibáñez, L.; Dunger, D.B. Avon Longitudinal Study of Parents and Children Study Team. Opposing influences of prenatal and postnatal weight gain on adrenarche in normal boys and girls. J. Clin. Endocrinol. Metab. 2004, 89, 2647–2651. [Google Scholar]
- Ong, K.K.; Ahmed, M.L.; Emmett, P.M.; Preece, M.A.; Dunger, D.B. Association between postnatal catch-up growth and obesity in childhood: Prospective cohort study. BMJ. 2000, 320, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.K.; Petry, C.J.; Emmett, P.M.; Sandhu, M.S.; Kiess, W.; Hales, C.N.; Ness, A.R.; Dunger, D.B.; ALSPAC study team. Insulin sensitivity and secretion in normal children related to size at birth, postnatal growth, and plasma insulin-like growth factor-I levels. Diabetologia 2004, 47, 1064–1070. [Google Scholar] [CrossRef]
- Darendeliler, F.; Bas, F.; Bundak, R.; Coban, A.; Sancakli, O.; Eryilmaz, S.K.; Kucukemre, B.; Disci, R.; Gokcay, G.; Aki, S.; et al. Insulin resistance and body composition in preterm born children during prepubertal ages. Clin. Endocrinol. 2008, 68, 773–779. [Google Scholar] [CrossRef]
- Lihn, A.S.; Pederson, S.B.; Richeksen, B. Adiponectin: Action, regulation and association to insulin sensitivity. Obes. Rev. 2005, 6, 13–21. [Google Scholar] [CrossRef]
- Iniguez, G.; Soto, N.; Avila, A.; Salazar, T.; Ong, K.; Dunger, D.; Mericq, V. Adiponectin levels in the first two years of life in a prospective cohort: Relations with weight gain, leptin levels and insulin sensitivity. J. Clin. Endocrinol. Metab. 2004, 89, 5500–5503. [Google Scholar] [CrossRef]
- Cianfarani, S.; Martinez, C.; Maiorana, A.; Scire, G.; Spadoni, G.L.; Boemi, S. Adiponectin levels are reduced in children born small for gestational age and are inversely related to postnatal catch-up growth. J. Clin. Endocrinol. Metab. 2004, 89, 1346–1351. [Google Scholar] [PubMed]
- Lopez-Bermejo, A.; Casano-Sancho, P.; Fernandez-Real, J.M.; Kihara, S.; Funahashi, T.; Rodriquez-Hierro, F.; Ricart, W.; Ibanez, L. Both intrauterine growth restriction and postnatal catchup growth influence childhood serum concentrations of adiponectin. Clin. Endocrinol. 2004, 61, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Evagelidou, E.N.; Giapros, V.I.; Challa, A.S.; Kiortsis, D.N.; Tsatsoulis, A.A.; Andronikou, S.K. Serum adiponectin levels, insulin resistance, and lipid profile in children born small for gestational age are affected by the severity of growth retardation at birth. Eur. J. Endocrinol. 2007, 156, 271–277. [Google Scholar] [CrossRef]
- Kamoda, T.; Nozue, H.; Matsui, A. Serum levels of adiponectin and IGFBP-1 in short children born small for gestational age. Clin. Endocrinol. 2007, 66, 290–294. [Google Scholar]
- Yanni, D.; Darendeliler, F.; Bas, F.; Kucukemre Aydin, B.; Coban, A.; Ince, Z. The role of leptin, soluble leptin receptor, adiponectin and visfatin in insulin sensitivity in preterm born children in prepubertal ages. Cytokine 2013, 64, 448–453. [Google Scholar] [CrossRef]
- Sancakli, O.; Darendeliler, F.; Bas, F.; Gokcay, G.; Disci, R.; Aki, S.; Eskiyurt, N. Insulin, adiponectin, IGFBP-1 levels and body composition in small for gestational age born non-obese children during prepubertal ages. Clin. Endocrinol. 2008, 69, 88–92. [Google Scholar] [CrossRef]
- Saitoh, H.; Kamoda, T.; Nakahara, S.; Hirano, T.; Nakamura, N. Serum concentrations of insulin, insulin-like growth factor (IGF)-I, IGF binding protein (IGFBP)-1 and -3 and growth hormone binding protein in obese children: Fasting IGFBP-1 is suppressed in normoinsulinaemic obese children. Clin. Endocrinol. 1998, 48, 487–492. [Google Scholar]
- Woods, K.A.; van Helvoirt, M.; Ong, K.K.; Mohn, A.; Levy, J.; de Zegher, F.; Dunger, D.B. The somatotropic axis in short children born small for gestational age: Relation to insulin resistance. Pediatr. Res. 2002, 51, 76–80. [Google Scholar]
- Tenhola, S.; Halonen, P.; Jaaskelainen, J.; Voutilainen, R. Serum markers of GH and insulin action in 12-year-old children born small for gestational age. Eur. J. Endocrinol. 2005, 152, 335–340. [Google Scholar]
- Zhang, L.; Liu, J.; Gao, D.; Li, D. Role of ghrelin in promoting catch-up growth and maintaining metabolic homeostasis in small-for-gestational-age infants. Front. Pediatr. 2024, 12, 1395571. [Google Scholar] [CrossRef]
- Van der Lely, A.J.; Tschop, M.; Heiman, M.L.; Ghigo, E. Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocr Rev. 2004, 25, 426–457. [Google Scholar] [CrossRef]
- Korbonits, M.; Goldstone, A.P.; Gueorguev, M.; Grossman, A.B. Ghrelin—A hormone with multiple functions. Front. Neuroendocrinol. 2004, 25, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, B.C.; Huber, A.; Hecher, K.; Fimmers, R.; Bartmann, P.; Roth, C.L. Fetal insulin-like growth factor (IGF)-I, IGF-II, and ghrelin in association with birth weight and postnatal growth in monozygotic twins with discordant growth. J. Clin. Endocrinol. Metab. 2005, 90, 2270–2274. [Google Scholar] [CrossRef] [PubMed]
- Darendeliler, F.; Poyrazoglu, S.; Bas, F.; Sancakli, O.; Gokcay, G. Ghrelin levels are decreased in non-obese prepubertal children born large for gestational age. Eur. J. Endocrinol. 2009, 160, 951–956. [Google Scholar] [CrossRef]
- Rabinowicz, S.; Levkovitz, O.; Leibovitch, L.; Schushan-Eisen, I.; Morag, I.; Rosen, C.; Maayan-Metzger, A.; Strauss, T. Increased risk for early hypertriglyceridemia in small for gestational age preterm infants. Eur. J. Pediatr. 2020, 179, 1873–1879. [Google Scholar] [CrossRef]
- Chen, B.; Chen, Y.; Wang, Y.; Xin, Q.; Ma, D. The association between rapid growth and lipid profile: A systematic review and meta-analysis. Front. Endocrinol. 2024, 15, 1353334. [Google Scholar] [CrossRef] [PubMed]
- Faienza, M.F.; Brunetti, G.; Delvecchio, M.; Zito, A.; De Palma, F.; Cortese, F.; Nitti, A.; Massari, E.; Gesualdo, M.; Ricci, G.; et al. Vascular Function and Myocardial Performance Indices in Children Born Small for Gestational Age. Circ. J. 2016, 80, 958–963. [Google Scholar] [CrossRef]
- Leeson, C.P.; Kattenhorn, M.; Morley, R.; Lucas, A.; Deanfield, J.E. Impact of low birth weight and cardiovascular risk factors on endothelial function in early adult life. Circulation 2001, 103, 1264–1268. [Google Scholar] [CrossRef]
- Sebastiani, G.; Díaz, M.; Bassols, J.; Aragonés, G.; López-Bermejo, A.; de Zegher, F.; Ibáñez, L. The sequence of prenatal growth restraint and post-natal catch-up growth leads to a thicker intima-media and more pre-peritoneal and hepatic fat by age 3–6 years. Pediatr. Obes. 2016, 11, 251–257. [Google Scholar] [CrossRef]
- Hoy, W.E.; Bertram, J.F.; Denton, R.D.; Zimanyi, M.; Samuel, T.; Hughson, M.D. Nephron number, glomerular olüme, renal disease and hypertension. Curr. Opin. Nephrol. Hypertens. 2008, 17, 258–265. [Google Scholar] [CrossRef]
- Ruggajo, P.; Skrunes, R.; Svarstad, E.; Skjærven, R.; Reisæther, A.V.; Vikse, B.E. Familial Factors, Low Birth Weight, and Development of ESRD: A Nationwide Registry Study. Am. J. Kidney. Dis. 2016, 67, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Bilge, I.; Poyrazoglu, S.; Bas, F.; Emre, S.; Sirin, A.; Gokalp, S.; Eryilmaz, S.; Hekim, N.; Darendeliler, F. Ambulatory blood pressure monitoring and renal functions in term small-for-gestational age children. Pediatr. Nephrol. 2011, 26, 119–126. [Google Scholar] [CrossRef]
- Brathwaite, K.E.; Levy, R.V.; Sarathy, H.; Agalliu, I.; Johns, T.S.; Reidy, K.J.; Fadrowski, J.J.; Schwartz, G.J.; Kaskel, F.J.; Melamed, M.L. Reduced kidney function and hypertension in adolescents with low birth weight, NHANES 1999–2016. Pediatr. Nephrol. 2023, 38, 3071–3082. [Google Scholar] [CrossRef] [PubMed]
- Derraik, J.G.B.; Maessen, S.E.; Gibbins, J.D.; Cutfield, W.S.; Lundgren, M.; Ahlsson, F. Large-for-gestational-age phenotypes and obesity risk in adulthood: A study of 195,936 women. Sci. Rep. 2020, 10, 2157. [Google Scholar] [CrossRef]
- Mahindra, M.P.; Sampurna, M.T.A.; Mapindra, M.P.; Sutowo Putri, A.M. Maternal lipid levels in pregnant women without complications indeveloping risk of large for gestational age newborns: A study of meta-analysis. F1000Research 2020, 9, 1213. [Google Scholar] [CrossRef]
- Lorenzo-Almorós, A.; Hang, T.; Peiró, C.; Soriano-Guillén, L.; Egido, J.; Tuñón, J.; Lorenzo, Ó. Predictive and diagnostic biomarkers for gestational diabetes and its associated metabolic and cardiovascular diseases. Cardiovasc. Diabetol. 2019, 18, 140. [Google Scholar] [CrossRef] [PubMed]
- Nordman, H.; Jaaskelainen, J.; Voutilainen, R. Birth size as a determinant of cardiometabolic risk factors in children. Horm. Res. Paediatr. 2020, 93, 144–153. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, S.F.; Mu, M.; Sheng, J. Birth weight and overweight/obesityin adults: A meta-analysis. Eur. J. Pediatr. 2012, 171, 1737–1746. [Google Scholar] [CrossRef]
- Schellong, K.; Schulz, S.; Harder, T.; Plagemann, A. Birth weight and long-term overweight risk: Systematic review and a meta-analysis includ-ing 643,902 persons from 66 studies and 26 countries globally. PLoS ONE 2012, 7, e477. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, H.; Liu, S.J.; Fu, G.J.; Zhao, Y.; Xie, Y.J.; Zhang, Y.; Wang, Y.X. The associations of high birth weight with blood pressure and hypertension in later life: A systematic review and meta-analysis. Hypertens. Res. 2013, 36, 725–735. [Google Scholar] [CrossRef]
- Johannsson, E.; Arngrimsson, S.A.; Thorsdottir, I.; Sveinsson, T. Tracking of overweight from early childhood to adolescence in cohorts born 1988 and 1994: Overweight in a high birth weight population. Int. J. Obes. 2006, 30, 1265–1271. [Google Scholar]
- Gunnarsdottir, I.; Birgisdottir, B.E.; Benediktsson, R.; Gudnason, V.; Thorsdottir, I. Association between size at birth, truncal fat and obesity in adult life and its contribution to blood pressure and coronary heart disease; study in a high birth weight population. Eur. J. Clin. Nutr. 2004, 58, 812–818. [Google Scholar]
- Kampmann, F.B.; Thuesen, A.C.B.; Hjort, L.; Olsen, S.F.; Pires, S.M.; Tetens, I.; Grunnet, L.G. Exposure to Gestational Diabetes Is a Stronger Predictor of Dysmetabolic Traits in Children Than Size at Birth. J. Clin. Endocrinol. Metab. 2019, 104, 1766–1776. [Google Scholar] [CrossRef]
- Hong, Y.H.; Lee, J.E. Large for Gestational Age and Obesity-Related Comorbidities. J. Obes. Metab. Syndr. 2021, 30, 124–131. [Google Scholar]
- Bueno, A.C.; Espiñeira, A.R.; Fernandes-Rosa, F.L.; de Souza, R.M.; de Castro, M.; Moreira, A.C.; Bettiol, H.; Barbieri, M.A.; Antonini, S.R. Adiponectin: Serum levels, promoter polymorphism, and associations with birth size and cardiometabolic outcome in young adults born large for ges- tational age. Eur. J. Endocrinol. 2010, 162, 53–60. [Google Scholar] [PubMed]
- Griffiths, L.J.; Dezateux, C.; Cole, T.J. Differential parental weight andheight contributions to offspring birthweight and weight gain ininfancy. Int. J. Epidemiol. 2007, 36, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Challa, A.S.; Evagelidou, E.N.; Giapros, V.I.; Cholevas, V.I.; Andronikou, S.K. Growth factors, adiponectin, leptin and body mass index in prepubertal children born large for gestational age. J. Endocrinol. Invest. 2011, 34, 411–416. [Google Scholar] [CrossRef]
- Cetin, C.; Baş, F.; Uçar, A.; Poyrazoğlu, S.; Saka, N.; Bundak, R.; Darendeliler, F. Comparative analysis of glucoinsulinemic markers and proinflammatory cytokines in prepubertal children born large-versus appropriate-for gestational age. Endocrine 2014, 47, 816–824. [Google Scholar]
- Darendeliler, F.; Poyrazoglu, S.; Sancakli, O.; Bas, F.; Gokcay, G.; Aki, S.; Eskiyurt, N. Adiponectin is an indicator of insulin resistance in non-obese prepubertal children born large for gestational age (LGA) and is affected by birth weight. Clin. Endocrinol. 2009, 70, 710–716. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, P.; Zhou, W.; Hu, J.; Cui, L.; Chen, Z.J. Association of large for gestational age with cardiovascular metabolic risks: A systematic review and meta-analysis. Obesity 2023, 31, 1255–1269. [Google Scholar]
- Stene, L.C.; Magnus, P.; Lie, R.T.; Søvik, O.; Joner, G.; Norwegian Childhood Diabetes Study Group. Birth weight and childhood onset type 1 diabetes: Population based cohort study. BMJ 2001, 322, 889–892. [Google Scholar] [CrossRef] [PubMed]
- Flega, K.M.; Carroll, M.D.; Kit, B.K.; Ogden, C.L. Prevalence of Obesity and Trends in the Distribution of Body Mass Index Among US Adults, 1999–2010. J. Am. Med. Assoc. 2012, 307, 491–497. [Google Scholar] [CrossRef]
- Baeten, J.M.; Bukusi, E.A.; Lambe, M. Pregnancy complications and outcomes among overweight and obese nulliparous women. Am. J. Public Health. 2001, 91, 436–440. [Google Scholar]
- Pantham, P.; Aye, I.L.; Powell, T.L. Inflammation in maternal obesity and gestational diabetes mellitus. Placenta 2015, 36, 709–715. [Google Scholar] [CrossRef]
- Whitaker, R.C.; Wright, J.A.; Pepe, M.S.; Seidel, K.D.; Dietz, W.H. Predicting obesity in young adulthood from childhood and parental obesity. N. Engl. J. Med. 1997, 337, 869–873. [Google Scholar] [CrossRef]
- Gaillard, R.; Steegers, E.A.P.; Duijts, L.; Felix, J.F.; Hofman, A.; Franco, O.H.; Jaddoe, V.W. Childhood Cardiometabolic Outcomes of Maternal Obesity During Pregnancy The Generation R Study. Hypertension 2014, 63, 683–691. [Google Scholar] [CrossRef]
- Catalano, P.M.; Presley, L.; Minium, J.; Hauguel-de Mouzon, S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care 2009, 32, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Claesson, I.M.; Josefsson, A.; Olhager, E.; Oldin, C.; Sydsjö, G. Effects of a gestational weight gain restriction program for obese women: Sibling pairs’ weight development during the first five years of life. Sex Reprod. Healthc. 2018, 17, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Berglind, D.; Willmer, M.; Näslund, E.; Tynelius, P.; Sørensen, T.I.; Rasmussen, F. Differences in gestational weight gain between pregnancies before and after maternal bariatric surgery correlate with differences in birth weight but not with scores on the body mass index in early childhood. Pediatr. Obes. 2014, 9, 427–434. [Google Scholar] [CrossRef]
- ACOG Practice Bulletin No. 190. Gestational Diabetes Mellitus. Obstet. Gynecol. 2018, 131, e49–e64.
- Pedersen, J. Diabetes and pregnancy; blood sugar of newborn infants during fasting and glucose administration. Ugeskr. Laeger. 1952, 114, 685. [Google Scholar]
- HAPO Study Cooperative Research Group; Metzger, B.E.; Lowe, L.P.; Dyer, A.R.; Trimble, E.R.; Chaovarindr, U.; Coustan, D.R.; Hadden, D.R.; McCance, D.R.; Hod, M.; et al. Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar]
- Leth-Møller, M.; Hulman, A.; Kampmann, U.; Hede, S.; Ovesen, P.G.; Knorr, S. Effect of gestational diabetes on fetal growth rate and later overweight in the offspring. J. Clin. Endocrinol. Metab. 2024, dgae428. [Google Scholar] [CrossRef]
- Schaefer-Graf, U.M.; Kjos, S.L.; Bühling, K.J.; Henrich, W.; Brauer, M.; Heinze, T.; Dudenhausen, J.W.; Vetter, K. Amniotic fluid insulin levels and fetal abdominal circumference at time of amniocentesis in pregnancies with diabetes. Diabet. Med. 2003, 20, 349–354. [Google Scholar] [CrossRef]
- Villar, J.; Ochieng, R.; Gunier, R.B.; Papageorghiou, A.T.; Rauch, S.; McGready, R.; Gauglitz, J.M.; Barros, F.C.; Vatish, M.; Fernandes, M.; et al. Association between fetal abdominal growth trajectories, maternal metabolite signatures early in pregnancy, and childhood growth and adiposity: Prospective observational multinational INTERBIO-21st fetal study. Lancet Diabetes Endocrinol. 2022, 10, 710–719. [Google Scholar] [CrossRef]
- Pathirana, M.M.; Lassi, Z.S.; Ali, A.; Arstall, M.A.; Roberts, C.T.; Andraweera, P.H. Association between metabolic syndrome and gestational diabetes mellitus in women and their children: A systematic review and meta-analysis. Endocrine 2021, 71, 310–320. [Google Scholar]
- Gillman, M.W.; Oakey, H.; Baghurst, P.A.; Volkmer, R.E.; Robinson, J.S.; Crowther, C.A. Effect of treatment of gestational diabetes mellitus on obesity in the next generation. Diabetes Care 2010, 33, 964–968. [Google Scholar] [CrossRef]
- Scholtens, D.M.; Kuang, A.; Lowe, L.P.; Hamilton, J.; Lawrence, J.M.; Lebenthal, Y.; Brickman, W.J.; Clayton, P.; Ma, R.C.; McCance, D.; et al. Hyperglycemia and Adverse Pregnancy Outcome Follow-up Study (HAPO FUS): Maternal Glycemia and Childhood Glucose Metabolism. Diabetes Care 2019, 42, 381–392. [Google Scholar]
- Bendor, C.D.; Bardugo, A.; Rotem, R.S.; Derazne, E.; Gerstein, H.C.; Tzur, D.; Pinhas-Hamiel, O.; Tsur, A.M.; Cukierman-Yaffe, T.; Lebenthal, Y.; et al. Glucose Intolerance in Pregnancy and Offspring Obesity in Late Adolescence. Diabetes Care 2022, 45, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Chen, H. Established maternal obesity in the rat reprograms hypothalamic appetite regulators and leptin signaling at birth. Int. J. Obes. 2009, 33, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Barbour, L.A.; Hernandez, T.L. Maternal Lipids and Fetal Overgrowth: Making Fat from Fat. Clin Ther. 2018, 40, 1638–1647. [Google Scholar] [CrossRef]
- Page, K.A.; Luo, S.; Wang, X.; Chow, T.; Alves, J.; Buchanan, T.A.; Xiang, A.H. Children Exposed to Maternal Obesity or Gestational Diabetes Mellitus During Early Fetal Development Have Hypothalamic Alterations That Predict Future Weight Gain. Diabetes Care 2019, 42, 1473–1480. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Ann. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, J.P.; Hauguel-De Mouzon, S.; Lepercq, J.; Challier, J.C.; Huston-Presley, L.; Friedman, J.E.; Kalhan, S.C.; Catalano, P.M. TNF-alpha is a predictor of insulin resistance in human pregnancy. Diabetes 2002, 51, 2207–2213. [Google Scholar] [CrossRef]
- Yan, X.; Zhu, M.J.; Xu, W.; Tong, J.F.; Ford, S.P.; Nathanielsz, P.W.; Du, M. Up-regulation of Toll-like receptor 4/nuclear factor-kappaB signaling is associated with enhanced adipogenesis and insulin resistance in fetal skeletal muscle of obese sheep at late gestation. Endocrinology 2010, 151, 380–387. [Google Scholar] [CrossRef]
- Shang, Y.C.; Zhang, C.; Wang, S.H.; Xiong, F.; Zhao, C.P.; Peng, F.N.; Feng, S.W.; Yu, M.J.; Li, M.S.; Zhang, Y.N. Activated β-catenin induces myogenesis and inhibits adipogenesis in BM-derived mesenchymal stromal cells. Cytotherapy 2007, 9, 667–681. [Google Scholar] [CrossRef]
- Artaza, J.N.; Bhasin, S.; Magee, T.R.; Reisz-Porszasz, S.; Shen, R.; Groome, N.P.; Meerasahib, M.F.; Fareez, M.M.; Gonzalez-Cadavid, N.F. Myostatin inhibits myogenesis and promotes adipogenesis in C3H 10T(1/2) mesenchymal multipotent cells. Endocrinology 2005, 146, 3547–3557. [Google Scholar] [CrossRef]
- Atègbo, J.M.; Grissa, O.; Yessoufou, A.; Hichami, A.; Dramane, K.L.; Moutairou, K.; Miled, A.; Grissa, A.; Jerbi, M.; Tabka, Z.; et al. Modulation of adipokines and cytokines in gestational diabetes and macrosomia. J. Clin. Endocrinol. Metab. 2006, 91, 4137–4143. [Google Scholar] [CrossRef]
- Hauguel-de Mouzon, S.; Guerre-Millo, M. The placenta cytokine network and inflammatory signals. Placenta 2006, 27, 794–798. [Google Scholar] [CrossRef]
- Challier, J.C.; Basu, S.; Bintein, T.; Minium, J.; Hotmire, K.; Catalano, P.M.; Hauguel-de Mouzon, S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 2008, 29, 274–281. [Google Scholar] [CrossRef]
- Saben, J.; Lindsey, F.; Zhong, Y.; Thakali, K.; Badger, T.M.; Andres, A.; Gomez-Acevedo, H.; Shankar, K. Maternal obesity is associated with a lipotoxic placental environment. Placenta. 2014, 35, 171–177. [Google Scholar] [CrossRef]
- Kuzmicki, M.; Telejko, B.; Wawrusiewicz-Kurylonek, N.; Citko, A.; Lipinska, D.; Pliszka, J.; Wilk, J.; Kalejta, K.; Lemancewicz, A.; Grabiec, M.; et al. The expression of suppressor of cytokine signaling 1 and 3 in fat and placental tissue from women with gestational diabetes. Gynecol. Endocrinol. 2012, 28, 841–844. [Google Scholar] [CrossRef]
- Keats, E.C.; Oh, C.; Chau, T.; Khalifa, D.S.; Imdad, A.; Bhutta, Z.A. Effects of vitamin and mineral supplementation during pregnancy on maternal, birth, child health and development outcomes in low- and middle-income countries: A systematic review. Campbell Syst. Rev. 2021, 17, e1127. [Google Scholar] [CrossRef]
- Bieswal, F.; Ahn, M.T.; Reusens, B.; Holvoet, P.; Raes, M.; Rees, W.D.; Remacle, C. The importance of catch-up growth after early malnutrition for the programming of obesity in male rat. Obesity 2006, 14, 1330–1343. [Google Scholar]
- Desai, M.; Babu, J.; Ross, M.G. Programmed metabolic syndrome: Prenatal undernutrition and postweaning overnutrition. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R2306–R2314. [Google Scholar] [CrossRef]
- Brenseke, B.; Prater, M.R.; Bahamonde, J.; Gutierrez, J.C. Current thoughts on maternal nutrition and fetal programming of the metabolic syndrome. J. Pregnancy. 2013, 2013, 368461. [Google Scholar]
- Agote, M.; Goya, L.; Ramos, S.; Alvarez, C.; Gavete, M.L.; Pascual-Leone, A.M.; Escrivá, F. Glucose uptake and glucose transporter proteins in skeletal muscle from undernourished rats. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E1101–E1109. [Google Scholar]
- Gavete, M.L.; Martín, M.A.; Alvarez, C.; Escrivá, F. Maternal food restriction enhances insulin-induced GLUT-4 translocation and insulin signaling pathway in skeletal muscle from suckling rats. Endocrinology 2005, 146, 3368–3378. [Google Scholar]
- Chango, A.; Pogribny, I.P. Considering maternal dietary modulators for epigenetic regulation and programming of the fetal epigenome. Nutrients 2015, 7, 2748–2770. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S. Impact of Maternal Diet on the Epigenome during In Utero Life and the Developmental Programming of Diseases in Childhood and Adulthood. Nutrients 2015, 7, 9492–9507. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Narang, M.; Banerjee, B.D.; Basu, S. Oxidative stress in term small for gestational age neonates born to undernourished mothers: A case control study. BMC Pediatr. 2004, 20, 14. [Google Scholar]
- Baydas, G.; Karatas, F.; Gursu, M.F.; Bozkurt, H.A.; Ilhan, N.; Yasar, A.; Canatan, H. Antioxidant vitamin levels in term and preterm infants and their relation to maternal vitamin status. Arch. Med. Res. 2002, 33, 276–280. [Google Scholar]
- Zheng, J.; Liu, X.; Zheng, B.; Zheng, Z.; Zhang, H.; Zheng, J.; Sun, C.; Chen, H.; Yang, J.; Wang, Z.; et al. Maternal 25-Hydroxyvitamin D Deficiency Promoted Metabolic Syndrome and Downregulated Nrf2/CBR1 Pathway in Offspring. Front. Pharmacol. 2020, 11, 97. [Google Scholar] [CrossRef]
- Miliku, K.; Felix, J.F.; Voortman, T.; Tiemeier, H.; Eyles, D.W.; Burne, T.H.; McGrath, J.J.; Jaddoe, V.W.V. Associations of maternal and fetal vitamin D status with childhood body composition and cardiovascular risk factors. Matern. Child. Nutr. 2019, 15, e12672. [Google Scholar] [CrossRef]
- Pilz, S.; Kienreich, K.; Rutters, F.; de Jongh, R.; van Ballegooijen, A.J.; Grübler, M.; Tomaschitz, A.; Dekker, J.M. Role of vitamin D in the development of insulin resistance and type 2 diabetes. Curr. Diab. Rep. 2013, 13, 261–270. [Google Scholar]
- Rush, E.C.; Katre, P.; Yajnik, C.S. Vitamin B12: One carbon metabolism, fetal growth and programming for chronic disease. Eur. J. Clin. Nutr. 2014, 68, 2–7. [Google Scholar]
- Behere, R.V.; Deshmukh, A.S.; Otiv, S.; Gupte, M.D.; Yajnik, C.S. Maternal Vitamin B12 Status During Pregnancy and Its Association With Outcomes of Pregnancy and Health of the Offspring: A Systematic Review and Implications for Policy in India. Front. Endocrinol. 2021, 12, 619176. [Google Scholar]
- Yajnik, C.S.; Deshpande, S.S.; Jackson, A.A.; Refsum, H.; Rao, S.; Fisher, D.J.; Bhat, D.S.; Naik, S.S.; Coyaji, K.J.; Joglekar, C.V.; et al. Vitamin B12 and folate concentrations during pregnancy and insulin resistance in the offspring: The Pune Maternal Nutrition Study. Diabetologia 2008, 51, 29–38. [Google Scholar]
- Stewart, C.P.; Christian, P.; Schulze, K.J.; Arguello, M.; LeClerq, S.C.; Khatry, S.K.; West, K.P., Jr. Low Maternal Vitamin B-12 Status Is Associated with Offspring Insulin Resistance Regardless of Antenatal Micronutrient Supplementation in Rural Nepal. J. Nutr. 2011, 141, 1912–1917. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.F.; Lazdam, M.; Lewandowski, A.J.; Worton, S.A.; Kelly, B.; Kenworthy, Y.; Adwani, S.; Wilkinson, A.R.; McCormick, K.; Sargent, I.; et al. Cardiovascular risk factors in children and young adults born to preeclamptic pregnancies: A systematic review. Pediatrics 2012, 129, e1552–e1561. [Google Scholar]
- Miettola, S.; Hartikainen, A.L.; Vääräsmäki, M.; Bloigu, A.; Ruokonen, A.; Järvelin, M.R.; Pouta, A. Offspring’s blood pressure and metabolic phenotype after exposure to gestational hypertension in utero. Eur. J. Epidemiol. 2013, 28, 87–98. [Google Scholar] [PubMed]
- Huxley, R.R.; Shiell, A.W.; Law, C.M. The role of size at birth and postnatal catch-up growth in determining systolic blood pressure: A systematic review of the literature. J. Hypertens. 2000, 18, 815–831. [Google Scholar] [CrossRef]
- Hutcheon, J.A.; Lisonkova, S.; Joseph, K.S. Epidemiology of pre-eclampsia and the other hypertensive disorders of pregnancy. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Jonker, S.S.; Louey, S. Endocrine and other physiologic modulators of perinatal cardiomyocyte endowment. J. Endocrinol. 2016, 228, R1–R18. [Google Scholar] [CrossRef]
- Balli, S.; Kibar, A.E.; Ece, I.; Oflaz, M.B.; Yilmaz, O. Assessment of Fetal Cardiac Function in Mild Preeclampsia. Pediatr. Cardiol. 2013, 34, 1674–1679. [Google Scholar]
- Yesil, G.D.; Gishti, O.; Felix, J.F.; Reiss, I.; Ikram, M.K.; Steegers, E.A.; Hofman, A.; Jaddoe, V.W.; Gaillard, R. Influence of Maternal Gestational Hypertensive Disorders on Microvasculature in School-Age Children: The Generation R Study. Am. J. Epidemiol. 2016, 184, 605–615. [Google Scholar] [PubMed]
- Kvehaugen, A.S.; Dechend, R.; Ramstad, H.B.; Troisi, R.; Fugelseth, D.; Staff, A.C. Endothelial function and circulating biomarkers are disturbed in women and children after preeclampsia. Hypertension 2011, 58, 63–69. [Google Scholar]
- Koulouraki, S.; Paschos, V.; Pervanidou, P.; Christopoulos, P.; Gerede, A.; Eleftheriades, M. Short- and Long-Term Outcomes of Preeclampsia in Offspring: Review of the Literature. Children 2023, 10, 826. [Google Scholar] [CrossRef]
- Jansen, M.A.; Pluymen, L.P.; Dalmeijer, G.W.; Groenhof, T.K.J.; Uiterwaal, C.S.; Smit, H.A.; van Rossem, L. Hypertensive disorders of pregnancy and cardiometabolic outcomes in childhood: A systematic review. Eur. J. Prev. Cardiol. 2019, 26, 1718–1747. [Google Scholar]
- Bi, S.; Zhang, L.; Huang, L.; Li, Y.; Liang, Y.; Huang, M.; Huang, B.; Liang, J.; Gu, S.; Chen, J.; et al. Long-term effects of preeclampsia on metabolic and biochemical outcomes in offspring: What can be expected from a meta-analysis? Obes. Rev. 2022, 23, e13411. [Google Scholar] [CrossRef]
- Indrio, F.; Martini, S.; Francavilla, R.; Corvaglia, L.; Cristofori, F.; Mastrolia, S.A.; Neu, J.; Rautava, S.; Russo Spena, G.; Raimondi, F.; et al. Epigenetic Matters: The Link between Early Nutrition, Microbiome, and Long-term Health Development. Front. Pediatr. 2017, 5, 178. [Google Scholar]
- Franzago, M.; Fraticelli, F.; Stuppia, L.; Vitacolonna, E. Nutrigenetics, epigenetics and gestational diabetes: Consequences in mother and child. Epigenetics 2019, 14, 215–235. [Google Scholar] [PubMed]
- Engel, S.M.; Joubert, B.R.; Wu, M.C.; Olshan, A.F.; Håberg, S.E.; Ueland, P.M.; Nystad, W.; Nilsen, R.M.; Vollset, S.E.; Peddada, S.D.; et al. Neonatal genome-wide methylation patterns in relation to birth weight in the Norwegian Mother and Child Cohort. Am. J. Epidemiol. 2014, 179, 834–842. [Google Scholar]
- Williams, L.; Seki, Y.; Delahaye, F.; Cheng, A.; Fuloria, M.; Hughes Einstein, F.; Charron, M.J. DNA hypermethylation of CD3(+) T cells from cord blood of infants exposed to intrauterine growth restriction. Diabetologia 2016, 59, 1714–1723. [Google Scholar]
- Ortiz-Dosal, A.; Arellanes-Licea, E.D.C.; Rodil-García, P.; Salazar-Olivo, L.A. Circulating microRNAs overexpressed in macrosomia: An experimental and bioinformatic approach. J. Dev. Orig. Health Dis. 2020, 11, 464–472. [Google Scholar]
- Shen, Z.; Tang, Y.; Song, Y.; Shen, W.; Zou, C. Differences of DNA methylation patterns in the placenta of large for gestational age infant. Medicine 2020, 99, e22389. [Google Scholar] [PubMed]
- Meyrueix, L.P.; Gharaibeh, R.; Xue, J.; Brouwer, C.; Jones, C.; Adair, L.; Norris, S.A.; Ideraabdullah, F. Gestational diabetes mellitus placentas exhibit epimutations at placental development genes. Epigenetics 2022, 17, 2157–2177. [Google Scholar]
- Bhushan, R.; Rani, A.; Gupta, D.; Ali, A.; Dubey, P.K. MicroRNA-7 Regulates Insulin Signaling Pathway by Targeting IRS1, IRS2, and RAF1 Genes in Gestational Diabetes Mellitus. Microrna 2022, 11, 57–72. [Google Scholar]
- Zhang, L.; Yu, X.; Wu, Y.; Fu, H.; Xu, P.; Zheng, Y.; Wen, L.; Yang, X.; Zhang, F.; Hu, M.; et al. Gestational Diabetes Mellitus-Associated Hyperglycemia Impairs Glucose Transporter 3 Trafficking in Trophoblasts Through the Downregulation of AMP-Activated Protein Kinase. Front. Cell Dev. Biol. 2021, 9, 722024. [Google Scholar]
- Harary, D.; Akinyemi, A.; Charron, M.J.; Fuloria, M. Fetal Growth and Intrauterine Epigenetic Programming of Obesity and Cardiometabolic Disease. Neoreviews 2022, 23, e363–e372. [Google Scholar]

{kind=link}
| Cook et al. [12] | De Ferranti et al. [13] | Weiss et al. [14] | Cruz et al. [15] | |
|---|---|---|---|---|
| Abdominal obesity | WC > 90th p | WC > 75 p | BMI ≥ 2 SDS | WC ≥ 90th p (NHANES III) |
| HDL-c | ≤40 mg/dL | <50 mg/dL | ≤5 th p (NGHS) | ≤10th p (NHANES III) |
| Triglycerides | ≥110 mg/dL | ≥100 mg/dL | ≥95th p (NGHS) | ≥90th p (NHANES III) |
| Blood pressure | ≥90th p | >90 p | ≥95th p | ≥90th p |
| Hyperglycemia | FPG ≥ 110 mg/dL | FPG ≥ 110 mg/dL | GI (ADA criteria) # | GI (ADA criteria) |
| MetS definition | ≥3 criteria | ≥3 criteria | ≥3 criteria | ≥3 criteria |
| Age * (Years) | Abdominal Obesity (WC) | HDL-c | Triglycerides | Blood Pressure | FPG |
|---|---|---|---|---|---|
| 6–<10 | ≥90th p | - | - | - | - |
| 10–<16 | ≥90th p or adult cut-off | <40 mg/dL | ≥150 mg/dL | SBP ≥ 130 or DBP ≥ 85 mmHg | ≥100 mg/dL or known T2DM |
| 16+ | ≥94 cm for males and >80 cm for females ** | <40 mg/dL in males and < 50 mg/dL in females or treatment for low HDL | ≥150 mg/dL or specific treatment for high TG | SBP ≥ 130 or DBP ≥ 85 mmHg or receiving hypertension treatment | ≥100 mg/dL or known T2DM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kardelen, A.D.; Darendeliler, F. The Role of the Intrauterine Environment in Shaping Childhood and Adolescence Metabolic Outcomes. Metabolites 2025, 15, 252. https://doi.org/10.3390/metabo15040252
Kardelen AD, Darendeliler F. The Role of the Intrauterine Environment in Shaping Childhood and Adolescence Metabolic Outcomes. Metabolites. 2025; 15(4):252. https://doi.org/10.3390/metabo15040252
Chicago/Turabian StyleKardelen, Asli Derya, and Feyza Darendeliler. 2025. "The Role of the Intrauterine Environment in Shaping Childhood and Adolescence Metabolic Outcomes" Metabolites 15, no. 4: 252. https://doi.org/10.3390/metabo15040252
APA StyleKardelen, A. D., & Darendeliler, F. (2025). The Role of the Intrauterine Environment in Shaping Childhood and Adolescence Metabolic Outcomes. Metabolites, 15(4), 252. https://doi.org/10.3390/metabo15040252