

Mitochondrial Fission and Fusion: Molecular Mechanisms, Biological Functions, and Related Disorders


Abstract
1. Introduction
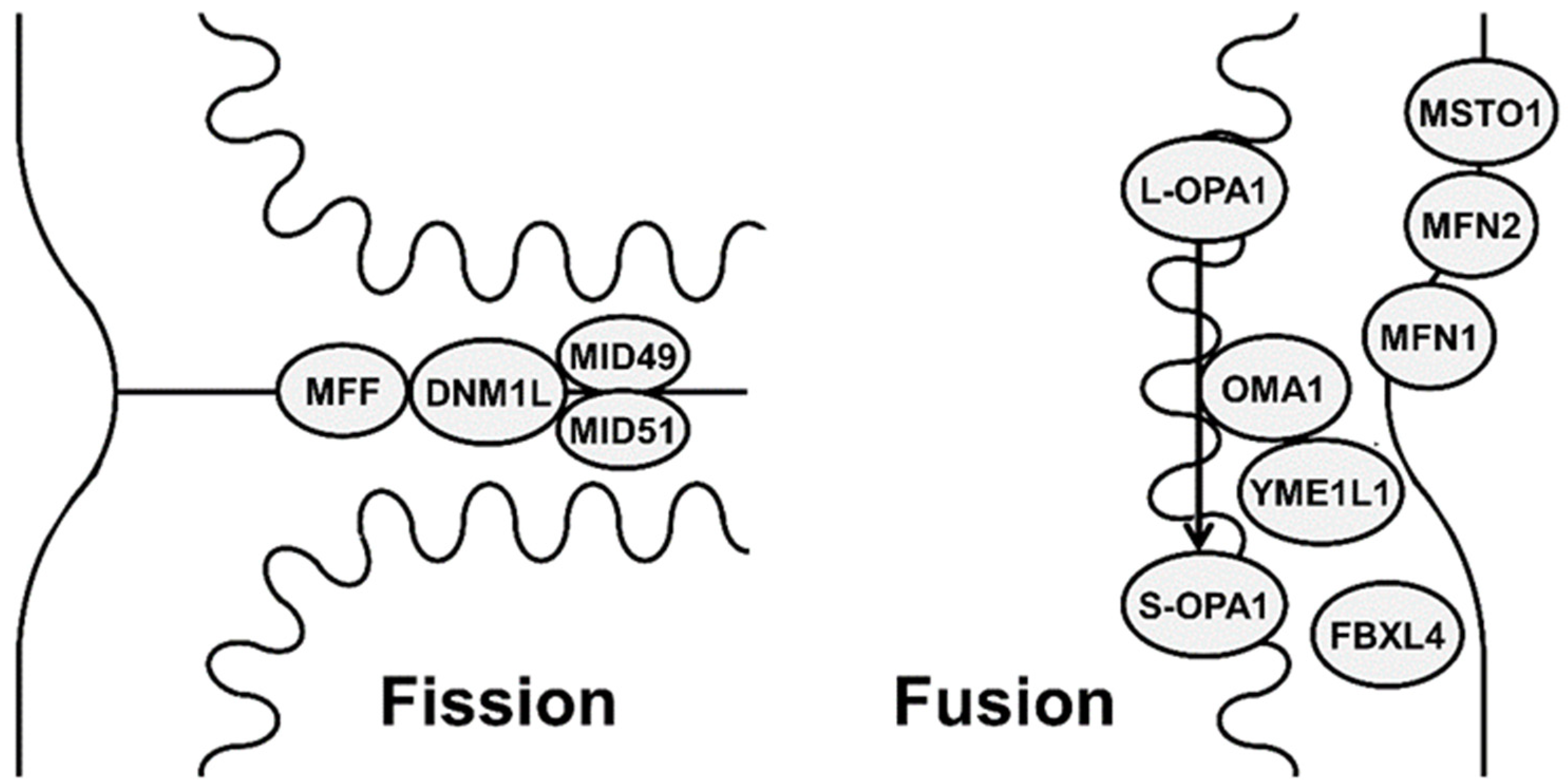
2. Mechanisms and Functions of Mitochondrial Fission and Fusion
2.1. Mitochondrial Fusion
2.2. Mitochondrial Fission
3. Disorders of Mitochondrial Fission and Fusion
3.1. MFN2-Related Disorders
3.2. MSTO1-Related Mitochondrial Myopathy and Ataxia
3.3. OPA1-Related Disorders
3.4. YME1L1-Related Optic Atrophy 11
3.5. FBXL4-Related Mitochondrial DNA Depletion Syndrome 13
3.6. DNM1L-Related Disorders
3.7. MFF-Related Encephalopathy
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- El-Hattab, A.W.; Suleiman, J.; Almannai, M.; Scaglia, F. Mitochondrial dynamics: Biological roles, molecular machinery, and related diseases. Mol. Genet. Metab. 2018, 125, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, J.; Jackson, S.; Schäfer, J.; Reichmann, H. Mitochondrial cytopathies. J. Neurol. 2003, 250, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Marshall, W.F.; Straight, A.; Murray, A.; Sedat, J.W.; Walter, P. Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol. Biol. Cell 1997, 8, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [PubMed]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- McCormick, E.M.; Zolkipli-Cunningham, Z.; Falk, M.J. Mitochondrial disease genetics update: Recent insights into the molecular diagnosis and expanding phenotype of primary mitochondrial disease. Curr Opin Pediatr. 2018, 30, 714–724. [Google Scholar] [CrossRef]
- Chen, H.C.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Gal, A.; Balicza, P.; Weaver, D.; Naghdi, S.; Joseph, S.K.; Várnai, P.; Gyuris, T.; Horváth, A.; Nagy, L.; Seifert, E.L.; et al. MSTO1 is a cytoplasmic pro-mitochondrial fusion protein, whose mutation induces myopathy and ataxia in humans. EMBO Mol. Med. 2017, 9, 967–984. [Google Scholar] [CrossRef]
- Low, H.H.; Sachse, C.; Amos, L.A.; Löwe, J. Structure of a Bacterial Dynamin-like Protein Lipid Tube Provides a Mechanism For Assembly and Membrane Curving. Cell 2009, 139, 1342–1352. [Google Scholar] [CrossRef]
- Cao, Y.-L.; Meng, S.; Chen, Y.; Feng, J.-X.; Gu, D.-D.; Yu, B.; Li, Y.-J.; Yang, J.-Y.; Liao, S.; Chan, D.C.; et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef]
- Mattie, S.; Riemer, J.; Wideman, J.G.; McBride, H.M. A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J. Cell Biol. 2017, 217, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Innao, V.; Allegra, A.G.; Musolino, C. Relationship between mitofusin 2 and cancer. Adv. Protein Chem Struct Biol. 2019, 116, 209–236. [Google Scholar] [PubMed]
- Filadi, R.; Greotti, E.; Pizzo, P. Highlighting the endoplasmic reticulum-mitochondria connection: Focus on Mitofusin 2. Pharmacol. Res. 2018, 128, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Kapur, M.; Li, M.; Choi, M.-C.; Choi, S.; Kim, H.-J.; Kim, I.; Lee, E.; Taylor, J.P.; Yao, T.-P. MFN1 deacetylation activates adaptive mitochondrial fusion and protects metabolically challenged mitochondria. J. Cell Sci. 2014, 127, 4954–4963. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Cooper, J.M.; Chau, K.Y.; Rojo, M.; Schapira, A.H.V.; Taanman, J.W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet 2010, 19, 4861–4870. [Google Scholar] [CrossRef]
- Kimura, M.; Okano, Y. Human Misato regulates mitochondrial distribution and morphology. Exp. Cell Res. 2007, 313, 1393–1404. [Google Scholar] [CrossRef]
- Belenguer, P.; Pellegrini, L. The dynamin GTPase OPA1: More than mitochondria? Biochim Biophys Acta 2013, 1833, 176–183. [Google Scholar] [CrossRef]
- Olichon, A.; Emorine, L.J.; Descoins, E.; Arnauné-Pelloquin, L.; Brichese, L.; Gas, N.; Guillou, E.; Delettre, C.; Valette, A.; Hamel, C.P.; et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002, 523, 171–176. [Google Scholar] [CrossRef]
- Gilkerson, R.; De La Torre, P.; Vallier, S.S. Mitochondrial OMA1 and OPA1 as Gatekeepers of Organellar Structure/Function and Cellular Stress Response. Front. Cell Dev. Biol. 2021, 9, 527. [Google Scholar] [CrossRef]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef]
- Rainbolt, T.K.; Lebeau, J.; Puchades, C.; Wiseman, R.L. Reciprocal Degradation of YME1L and OMA1 Adapts Mitochondrial Proteolytic Activity during Stress. Cell Rep. 2016, 14, 2041–2049. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lin, L.; Wang, Y.; Wang, A.; Liu, Z.; Wu, S.; Lan, X.; Jia, J.; Zhang, Y.; Yuan, F.; et al. Novel homozygous mutation in the FBXL4 gene is associated with mitochondria DNA depletion syndrome-13. J. Neurol. Sci. 2020, 416, 116948. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Molecular Machinery of Mitochondrial Fusion and Fission. J. Biol. Chem. 2008, 283, 13501–13505. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Nakada, K.; Sato, A.; Hayashi, J.-I. Mitochondrial functional complementation in mitochondrial DNA-based diseases. Int. J. Biochem. Cell Biol. 2009, 41, 1907–1913. [Google Scholar] [CrossRef]
- Twig, G.; Shirihai, O.S. The Interplay Between Mitochondrial Dynamics and Mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Mears, J.; Lackner, L.L.; Fang, S.; Ingerman, E.; Nunnari, J.; Hinshaw, J.E. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat. Struct. Mol. Biol. 2010, 18, 20–26. [Google Scholar] [CrossRef]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA maintenance defects. Biochim. Biophys Acta Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2014, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Lawson, V.H.; Graham, B.V.; Flanigan, K.M. Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology 2005, 65, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Pipis, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Next-generation sequencing in Charcot–Marie–Tooth disease: Opportunities and challenges. Nat. Rev. Neurol. 2019, 15, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, P.J.; Rossor, A.M.; Polke, J.M.; Poh, R.; Blake, J.; Reilly, M.M. Semi-dominant mutations in MFN2 -related neuropathy and implications for genetic counselling. J. Peripher. Nerv. Syst. 2016, 21, 52–54. [Google Scholar] [CrossRef]
- Dankwa, L.; Richardson, J.; Motley, W.W.; Scavina, M.; Courel, S.; Bardakjian, T.; Züchner, S.; Scherer, S.S. A novel MFN2 mutation causes variable clinical severity in a multi-generational CMT2 family. Neuromuscul. Disord. 2018, 29, 134–137. [Google Scholar] [CrossRef]
- Del Bo, R.; Moggio, M.; Rango, M.; Bonato, S.; D’angelo, M.G.; Ghezzi, S.; Airoldi, G.; Bassi, M.T.; Guglieri, M.; Napoli, L.; et al. Mutated mitofusin 2 presents with intrafamilial variability and brain mitochondrial dysfunction. Neurology 2008, 71, 1959–1966. [Google Scholar]
- Züchner, S.; De Jonghe, P.; Jordanova, A.; Claeys, K.; Guergueltcheva, V.; Cherninkova, S.; Hamilton, S.R.; Van Stavern, G.; Krajewski, K.M.; Stajich, J.; et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann. Neurol. 2006, 59, 276–281. [Google Scholar] [CrossRef]
- Chung, K.W.; Cho, S.Y.; Hwang, S.J.; Kim, K.H.; Yoo, J.H.; Kwon, O.; Kim, S.M.; Sunwoo, I.N.; Zuchner, S.; Choi, B.O. Early-onset stroke associated with a mutation in mitofusin 2. Neurology 2008, 70, 2010–2011. [Google Scholar] [CrossRef]
- Boaretto, F.; Vettori, A.; Casarin, A.; Vazza, G.; Muglia, M.; Rossetto, M.G.; Cavallaro, T.; Rizzuto, N.; Carelli, V.; Salviati, L.; et al. Severe CMT Type 2 with fatal encephalopathy associated with a novel mfn2 splicing mutation. Neurology 2010, 74, 1919–1921. [Google Scholar] [CrossRef]
- Kitani-Morii, F.; Noto, Y. Recent Advances in Drosophila Models of Charcot-Marie-Tooth Disease. Int. J. Mol. Sci. 2020, 21, 7419. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Dreha-Kulaczewski, S.; Dechent, P.; Bönnemann, C.; Helms, G.; Kyllerman, M.; Brück, W.; Frahm, J.; Huehne, K.; Gärtner, J.; et al. Cerebral involvement in axonal Charcot-Marie-Tooth neuropathy caused by mitofusin2 mutations. J. Neurol. 2008, 255, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V.; et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [CrossRef]
- Morrow, J.M.; Evans, M.R.; Grider, T.; Sinclair, C.D.; Thedens, D.; Shah, S.; Yousry, T.A.; Hanna, M.G.; Nopoulos, P.; Thornton, J.S.; et al. Validation of MRC Centre MRI calf muscle fat fraction protocol as an outcome measure in CMT1A. Neurology 2018, 91, e1125–e1129. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef]
- Franco, A.; Dang, X.; Walton, E.K.; Ho, J.N.; Zablocka, B.; Ly, C.; Miller, T.M.; Baloh, R.H.; Shy, M.E.; Yoo, A.S.; et al. Burst mitofusin activation reverses neuromuscular dysfunction in murine CMT2A. eLife 2020, 9, e61119. [Google Scholar] [CrossRef]
- Dang, X.; Walton, E.K.; Zablocka, B.; Baloh, R.H.; Shy, M.E.; Dorn, G.W. Mitochondrial Phenotypes in Genetically Diverse Neurodegenerative Diseases and Their Response to Mitofusin Activation. Cells 2022, 11, 1053. [Google Scholar] [CrossRef]
- Zhou, Y.; Carmona, S.; Muhammad, A.K.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W.; et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J. Clin. Investig. 2019, 129, 1756–1771. [Google Scholar] [CrossRef]
- Di Nottia, M.; Verrigni, D.; Torraco, A.; Rizza, T.; Bertini, E.; Carrozzo, R. Mitochondrial Dynamics: Molecular Mechanisms, Related Primary Mitochondrial Disorders and Therapeutic Approaches. Genes 2021, 12, 247. [Google Scholar] [CrossRef]
- Nasca, A.; Scotton, C.; Zaharieva, I.; Neri, M.; Selvatici, R.; Magnusson, O.T.; Gal, A.; Weaver, D.; Rossi, R.; Armaroli, A.; et al. Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia. Hum. Mutat. 2017, 38, 970–977. [Google Scholar] [CrossRef]
- Ardicli, D.; Sarkozy, A.; Zaharieva, I.; Deshpande, C.; Bodi, I.; Siddiqui, A.; U-King-Im, J.M.; Selfe, A.; Phadke, R.; Jungbluth, H.; et al. A novel case of MSTO1 gene related congenital muscular dystrophy with progressive neurological involvement. Neuromuscul. Disord. 2019, 29, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Stewart, J.D.; Hudson, G.; Andrews, R.M.; Griffiths, P.G.; Birch, M.K.; Chinnery, P.F. OPA1 increases the risk of normal but not high tension glaucoma. J. Med. Genet. 2009, 47, 120–125. [Google Scholar] [CrossRef]
- Aung, T.; Ocaka, L.; Ebenezer, N.D.; Morris, A.G.; Krawczak, M.; Thiselton, D.L.; Alexander, C.; Votruba, M.; Brice, G.; Child, A.H.; et al. A major marker for normal tension glaucoma: Association with polymorphisms in the OPA1 gene. Qual. Life Res. 2001, 110, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, C.P.; Blazo, M.; Lewis, R.A.; Tonini, R.E.; Takei, H.; Wang, J.; Wong, L.-J.; Scaglia, F. Early-onset severe neuromuscular phenotype associated with compound heterozygosity for OPA1 mutations. Mol. Genet. Metab. 2011, 103, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Toomes, C.; Marchbank, N.J.; Mackey, D.A.; Craig, J.; Newbury-Ecob, R.A.; Bennett, C.P.; Vize, C.J.; Desai, S.P.; Black, G.C.; Patel, N.; et al. Spectrum, frequency and penetrance of OPA1 mutations in dominant optic atrophy. Hum. Mol. Genet. 2001, 10, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Lenaers, G.; Hamel, C.; Delettre, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Reynier, P.; Milea, D. Dominant optic atrophy. Orphanet J. Rare Dis. 2012, 7, 46. [Google Scholar] [CrossRef]
- Eiberg, H.; Kjer, B.; Kjer, P.; Rosenberg, T. Dominant optic atrophy (OPA1) mapped to chromosome 3q region. I. Linkage analysis. Hum. Mol. Genet. 1994, 3, 977–980. [Google Scholar] [CrossRef]
- Votruba, M.; Fitzke, F.W.; Holder, G.E.; Carter, A.; Bhattacharya, S.S.; Moore, A.T. Clinical Features in Affected Individuals from 21 Pedigrees with Dominant Optic Atrophy. Arch. Ophthalmol. 1998, 116, 351–358. [Google Scholar] [CrossRef]
- Barboni, P.; Savini, G.; Parisi, V.; Carbonelli, M.; LA Morgia, C.; Maresca, A.; Sadun, F.; De Negri, A.M.; Carta, A.; Sadun, A.A.; et al. Retinal Nerve Fiber Layer Thickness in Dominant Optic Atrophy: Measurements by Optical Coherence Tomography and Correlation with Age. Ophthalmology 2011, 118, 2076–2080. [Google Scholar] [CrossRef]
- Johnston, P.B.; Gaster, R.N.; Smith, V.C.; Tripathi, R.C. A Clinicopathologic Study of Autosomal Dominant Optic Atrophy. Am. J. Ophthalmol. 1979, 88, 868–875. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain A J. Neurol. 2010, 133, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, D.; Colin, E.; Oca, F.; Ferré, M.; Chevrollier, A.; Guéguen, N.; Desquiret-Dumas, V.; N’Guyen, S.; Barth, M.; Zanlonghi, X.; et al. Early-onset Behr syndrome due to compound heterozygous mutations in OPA1. Brain 2014, 137, e301. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Saada, A.; Flannery, P.J.; Burté, F.; Soiferman, D.; Khayat, M.; Eisner, V.; Vladovski, E.; Taylor, R.W.; Bindoff, L.A.; et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J. Med. Genet 2016, 53, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Jüschke, C.; Klopstock, T.; Catarino, C.B.; Owczarek-Lipska, M.; Wissinger, B.; Neidhardt, J. Autosomal dominant optic atrophy: A novel treatment for OPA1 splice defects using U1 snRNA adaption. Mol. Ther. Nucleic Acids 2021, 26, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Yarosh, W.; Monserrate, J.; Tong, J.J.; Tse, S.; Le, P.K.; Nguyen, K.; Brachmann, C.B.; Wallace, D.C.; Huang, T. The Molecular Mechanisms of OPA1-Mediated Optic Atrophy in Drosophila Model and Prospects for Antioxidant Treatment. PLoS Genet. 2008, 4, e6. [Google Scholar] [CrossRef]
- Barboni, P.; Valentino, M.L.; LA Morgia, C.; Carbonelli, M.; Savini, G.; De Negri, A.; Simonelli, F.; Sadun, F.; Caporali, L.; Maresca, A.; et al. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain 2013, 136, e231. [Google Scholar] [CrossRef]
- Aleo, S.J.; Del Dotto, V.; Fogazza, M.; Maresca, A.; Lodi, T.; Goffrini, P.; Ghelli, A.; Rugolo, M.; Carelli, V.; Baruffini, E.; et al. Drug repositioning as a therapeutic strategy for neurodegenerations associated with OPA1 mutations. Hum. Mol. Genet. 2020, 29, 3631–3645. [Google Scholar] [CrossRef]
- Zhao, L.; Hu, C.; Zhang, P.; Jiang, H.; Chen, J. Mesenchymal stem cell therapy targeting mitochondrial dysfunction in acute kidney injury. J. Transl. Med. 2019, 17, 142. [Google Scholar] [CrossRef]
- Modeling Autosomal Dominant Optic Atrophy Using Induced Pluripotent Stem Cells and Identifying Potential Therapeutic Targets. Available online: https://pubmed.ncbi.nlm.nih.gov/26738566/ (accessed on 28 May 2022).
- Hartmann, B.; Wai, T.; Hu, H.; MacVicar, T.; Musante, L.; Fischer-Zirnsak, B.; Stenzel, W.; Gräf, R.; Heuvel, L.V.D.; Ropers, H.-H.; et al. Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation. eLife 2016, 5, e16078. [Google Scholar] [CrossRef]
- Bonnen, P.E.; Yarham, J.W.; Besse, A.; Wu, P.; Faqeih, E.A.; Al-Asmari, A.M.; Saleh, M.A.; Eyaid, W.; Hadeel, A.; He, L.; et al. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am. J. Hum. Genet 2013, 93, 471–481. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Dai, H.; Almannai, M.; Wang, J.; Faqeih, E.A.; Al Asmari, A.; Saleh, M.A.M.; Elamin, M.A.O.; Alfadhel, M.; Alkuraya, F.S.; et al. Molecular and clinical spectra of FBXL4 deficiency. Hum. Mutat. 2017, 38, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Gai, X.; Ghezzi, D.; Johnson, M.A.; Biagosch, C.A.; Shamseldin, H.E.; Haack, T.B.; Reyes, A.; Tsukikawa, M.; Sheldon, C.A.; Srinivasan, S.; et al. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 2013, 93, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Kimura, N.; Oda, N.; Shimomura, H.; Kumada, T.; Miyajima, T.; Murayama, K.; Tanaka, M.; Fujii, T. Pyruvate therapy for mitochondrial DNA depletion syndrome. Biochim. Et Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Montero, R.; Sánchez-Alcázar, J.A.; Briones, P.; Navarro-Sastre, A.; Gallardo, E.; Bornstein, B.; Herrero-Martín, D.; Rivera, H.; Martin, M.A.; Marti, R.; et al. Coenzyme Q10 deficiency associated with a mitochondrial DNA depletion syndrome: A case report. Clin. Biochem. 2009, 42, 742–745. [Google Scholar] [CrossRef]
- Montero, R.; Grazina, M.; López-Gallardo, E.; Montoya, J.; Briones, P.; Navarro-Sastre, A.; Land, J.M.; Hargreaves, I.P.; Artuch, R.; O’Callaghan, M.D.M.; et al. Coenzyme Q10 deficiency in mitochondrial DNA depletion syndromes. Mitochondrion 2013, 13, 337–341. [Google Scholar] [CrossRef]
- Sheffer, R.; Douiev, L.; Edvardson, S.; Shaag, A.; Tamimi, K.; Soiferman, D.; Meiner, V.; Saada, A. Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. Am. J. Med. Genet A 2016, 170, 1603–1607. [Google Scholar] [CrossRef]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A Lethal Defect of Mitochondrial and Peroxisomal Fission. New Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Yoon, G.; Malam, Z.; Paton, T.; Marshall, C.R.; Hyatt, E.; Ivakine, Z.; Scherer, S.W.; Lee, K.-S.; Hawkins, C.; Cohn, R.D.; et al. Lethal Disorder of Mitochondrial Fission Caused by Mutations in DNM1L. J. Pediatrics 2016, 171, 313–316.e2. [Google Scholar] [CrossRef]
- Fahrner, J.A.; Liu, R.; Perry, M.S.; Klein, J.; Chan, D.C. A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am. J. Med. Genet A 2016, 170, 2002–2011. [Google Scholar] [CrossRef]
- Chao, Y.H.; Robak, L.A.; Xia, F.; Koenig, M.K.; Adesina, A.; Bacino, C.A.; Scaglia, F.; Bellen, H.J.; Wangler, M.F. Missense variants in the middle domain of DNM1L in cases of infantile encephalopathy alter peroxisomes and mitochondria when assayed in Drosophila. Hum. Mol. Genet 2016, 25, 1846–1856. [Google Scholar] [CrossRef]
- Vandeleur, D.; Chen, C.V.; Huang, E.J.; Connolly, A.J.; Sanchez, H.; Moon-Grady, A.J. Novel and lethal case of cardiac involvement in DNM1L mitochondrial encephalopathy. Am. J. Med. Genet. Part A 2019, 179, 2486–2489. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.; Charif, M.; Chevrollier, A.; Chaumette, T.; Angebault, C.; Kane, M.S.; Paris, A.; Alban, J.; Quiles, M.; Delettre, C.; et al. Mutations in DNM1L, as in OPA1, result in dominant optic atrophy despite opposite effects on mitochondrial fusion and fission. Brain 2017, 140, 2586–2596. [Google Scholar] [CrossRef] [PubMed]
- Douiev, L.; Sheffer, R.; Horvath, G.; Saada, A. Bezafibrate Improves Mitochondrial Fission and Function in DNM1L-Deficient Patient Cells. Cells 2020, 9, 301. [Google Scholar] [CrossRef] [PubMed]
- Djouadi, F.; Bastin, J. Mitochondrial Genetic Disorders: Cell Signaling and Pharmacological Therapies. Cells 2019, 8, 289. [Google Scholar] [CrossRef]
- Panda, I.; Ahmad, I.; Sagar, S.; Zahra, S.; Shamim, U.; Sharma, S.; Faruq, M.; Msc, I.A.; Msc, S.S.; Msc, S.Z.; et al. Encephalopathy due to defective mitochondrial and peroxisomal fission 2 caused by a novel MFF gene mutation in a young child. Clin. Genet. 2020, 97, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.; Feichtinger, R.G.; Freisinger, P.; Pies, M.; Schrödl, F.; Iuso, A.; Sperl, W.; Mayr, J.A.; Prokisch, H.; Haack, T.B. Disturbed mitochondrial and peroxisomal dynamics due to loss of MFF causes Leigh-like encephalopathy, optic atrophy and peripheral neuropathy. J. Med. Genet. 2016, 53, 270–278. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]


{kind=link}
| Diseases | Gene | Main Clinical Manifestations | |
|---|---|---|---|
| Mitochondrial fusion disorders | Charcot–Marie–Tooth neuropathy 2A | MFN2 | Axonal sensorimotor neuropathy |
| Hereditary motor and sensory neuropathy VIA with optic atrophy disease | MFN2 | Axonal sensorimotor neuropathy and optic atrophy | |
| Mitochondrial myopathy and ataxia | MSTO1 | Cognitive impairment, myopathy, and ataxia | |
| Optic atrophy 1 | OPA1 | Optic atrophy | |
| Optic atrophy plus syndrome | OPA1 | Optic atrophy, hearing impairment, ataxia, neuropathy, and myopathy | |
| Behr syndrome | OPA1 | Optic atrophy, ataxia, and pyramidal signs | |
| Mitochondrial DNA depletion syndrome 14 | OPA1 | Profound developmental delay, hypotonia, and hypertrophic cardiomyopathy | |
| Optic atrophy 11 | YME1L1 | Optic atrophy, developmental delay, and leukoencephalopathy | |
| Mitochondrial DNA depletion syndrome 13 | FBXL4 | Developmental delay, hypotonia, seizures, and lactic acidosis | |
| Mitochondrial fission disorders | Encephalopathy due to defective mitochondrial and peroxisomal fission 1 | DNM1L | Developmental delay, regression, hypotonia, and seizures |
| Optic atrophy 5 | DNM1L | Optic atrophy | |
| Encephalopathy due to defective mitochondrial and peroxisomal fission 2 | MFF | Developmental delay, hypotonia, and microcephaly | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Ojaimi, M.; Salah, A.; El-Hattab, A.W. Mitochondrial Fission and Fusion: Molecular Mechanisms, Biological Functions, and Related Disorders. Membranes 2022, 12, 893. https://doi.org/10.3390/membranes12090893
Al Ojaimi M, Salah A, El-Hattab AW. Mitochondrial Fission and Fusion: Molecular Mechanisms, Biological Functions, and Related Disorders. Membranes. 2022; 12(9):893. https://doi.org/10.3390/membranes12090893
Chicago/Turabian StyleAl Ojaimi, Mode, Azza Salah, and Ayman W. El-Hattab. 2022. "Mitochondrial Fission and Fusion: Molecular Mechanisms, Biological Functions, and Related Disorders" Membranes 12, no. 9: 893. https://doi.org/10.3390/membranes12090893
APA StyleAl Ojaimi, M., Salah, A., & El-Hattab, A. W. (2022). Mitochondrial Fission and Fusion: Molecular Mechanisms, Biological Functions, and Related Disorders. Membranes, 12(9), 893. https://doi.org/10.3390/membranes12090893