Simultaneous Monitoring of Mutation and Chimerism Using Next-Generation Sequencing in Myelodysplastic Syndrome

 , and
, and
Abstract
:1. Introduction
2. Experimental Section
2.1. Ethics Statement
2.2. Subject and DNA Isolation
2.3. STR Analysis
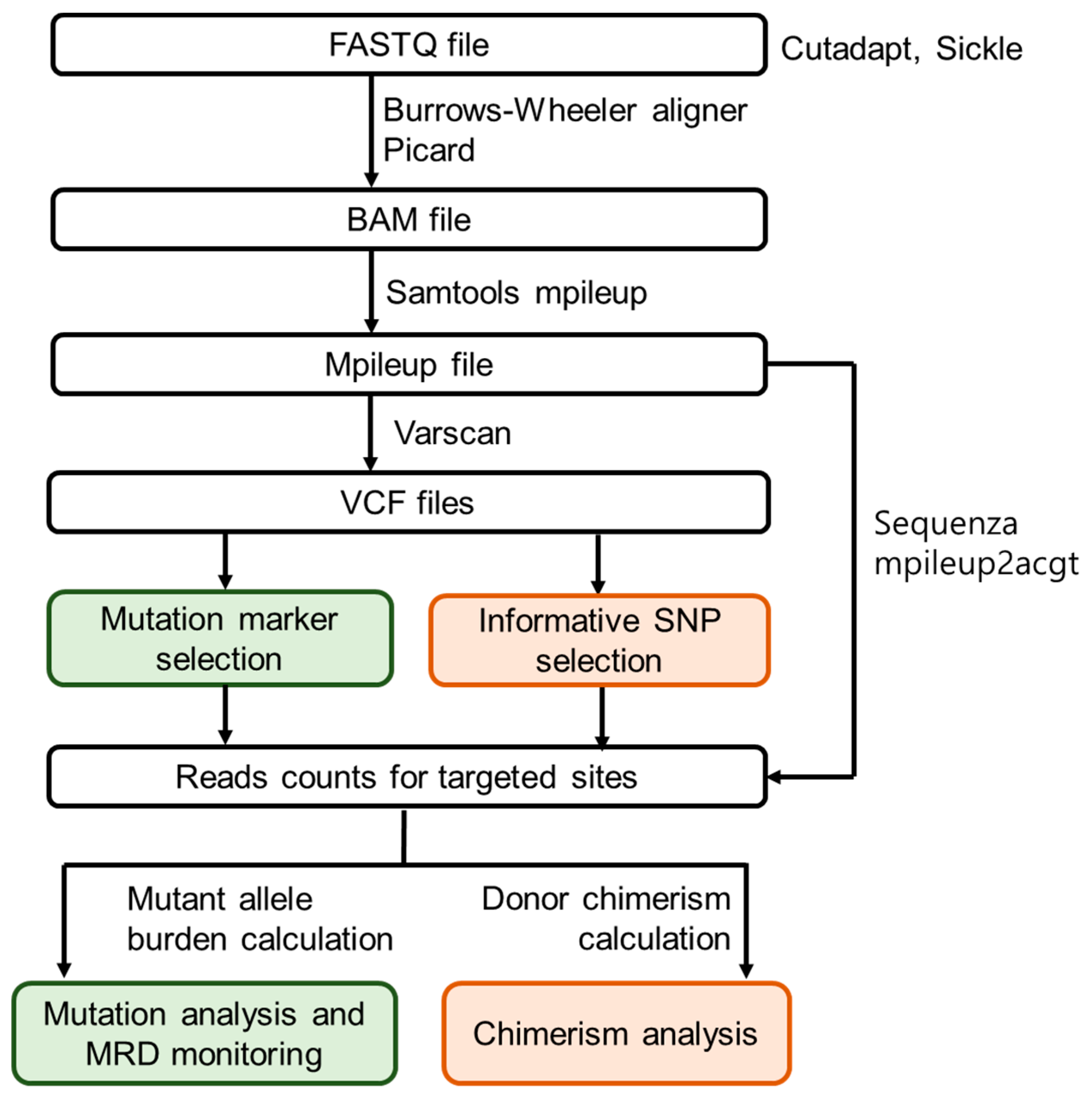
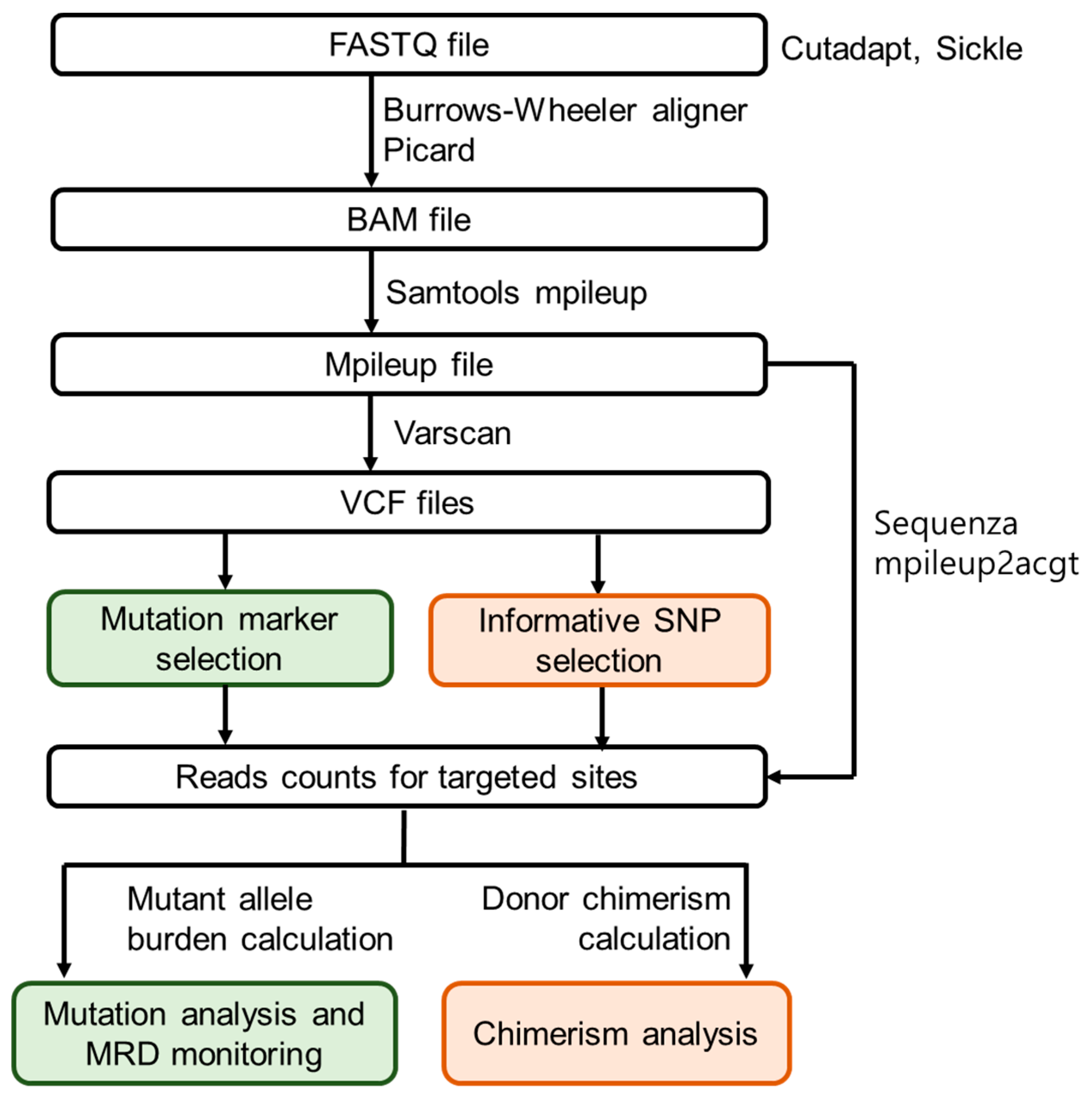
2.4. Customised NGS Panel Analysis
2.5. Minimal Residual Disease Monitoring
2.6. SNP-Based NGS Chimerism Analysis
3. Results
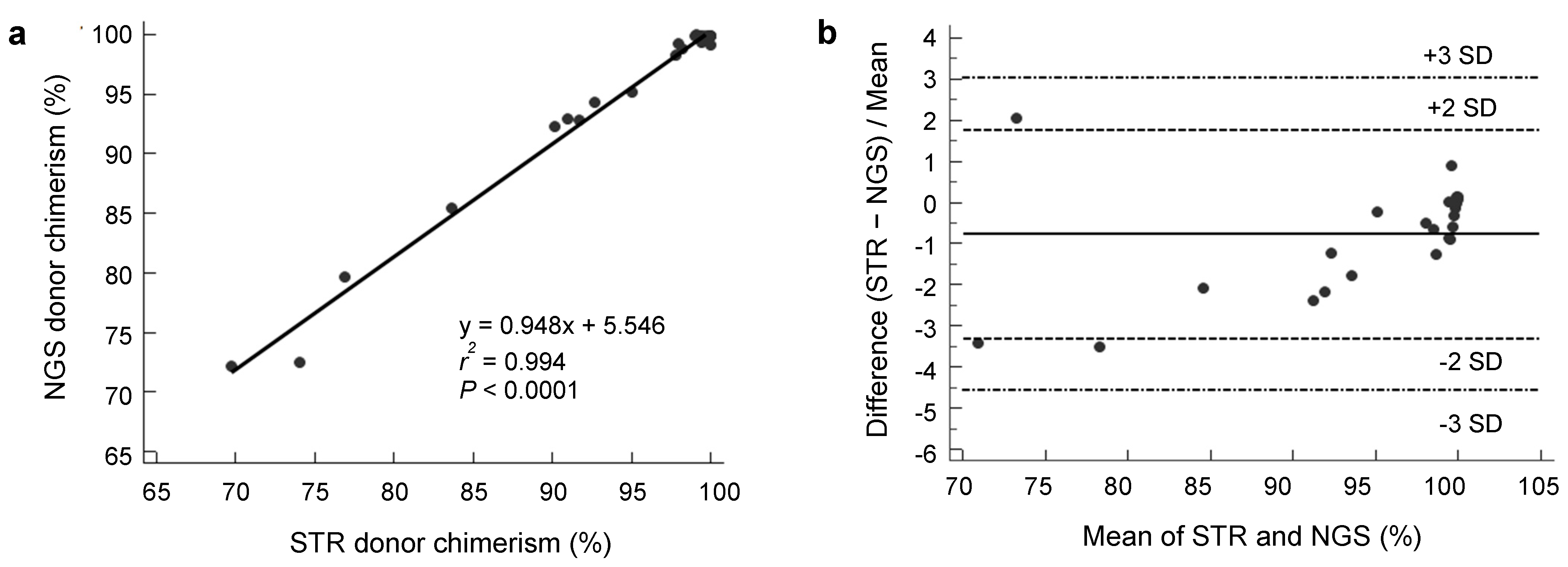
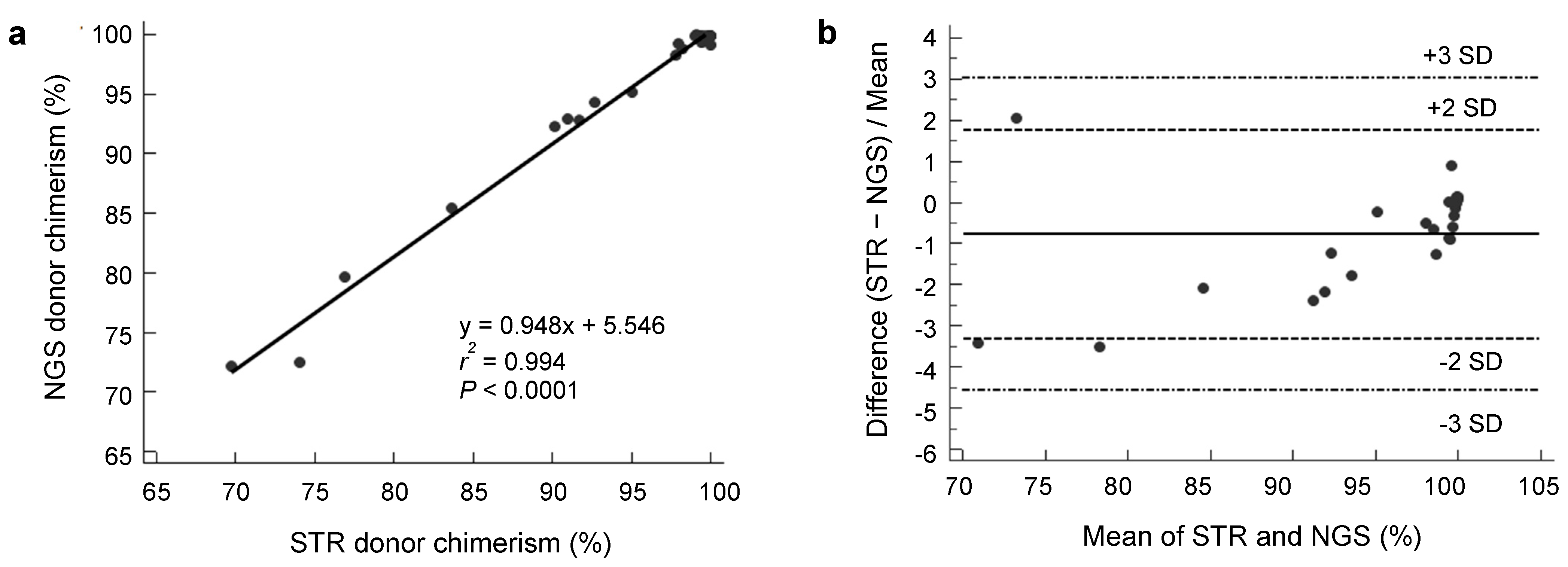
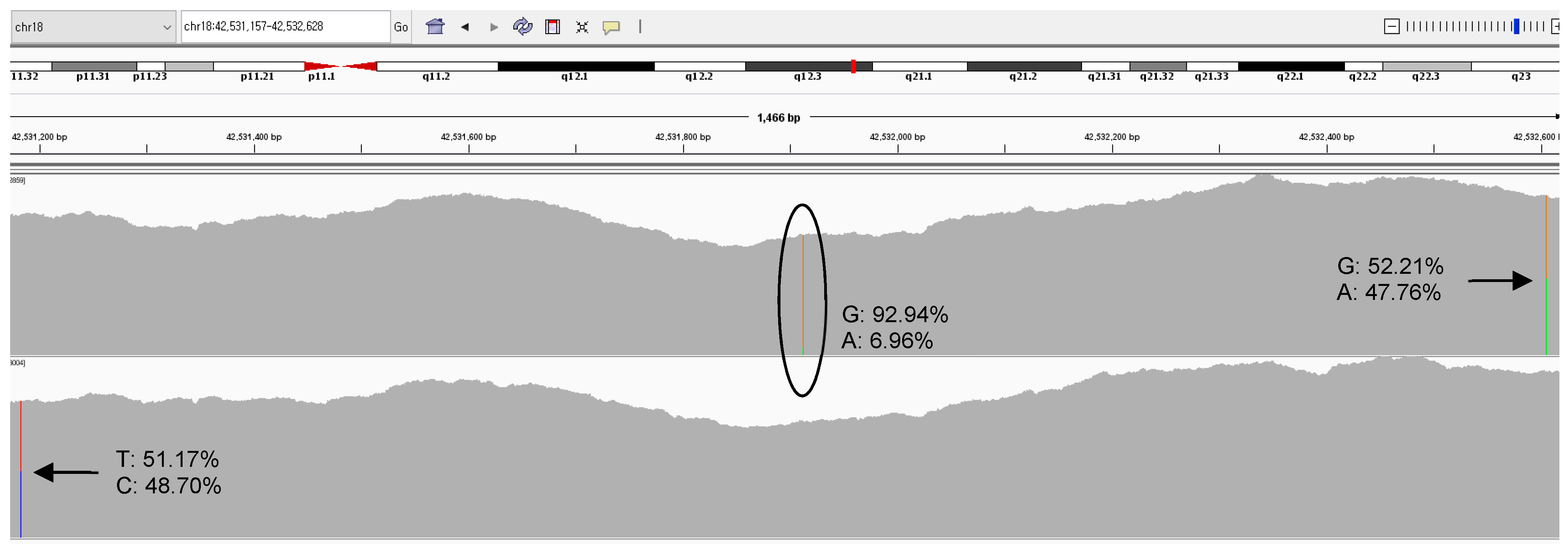
3.1. Analytical Performance of NGS Donor Chimerism
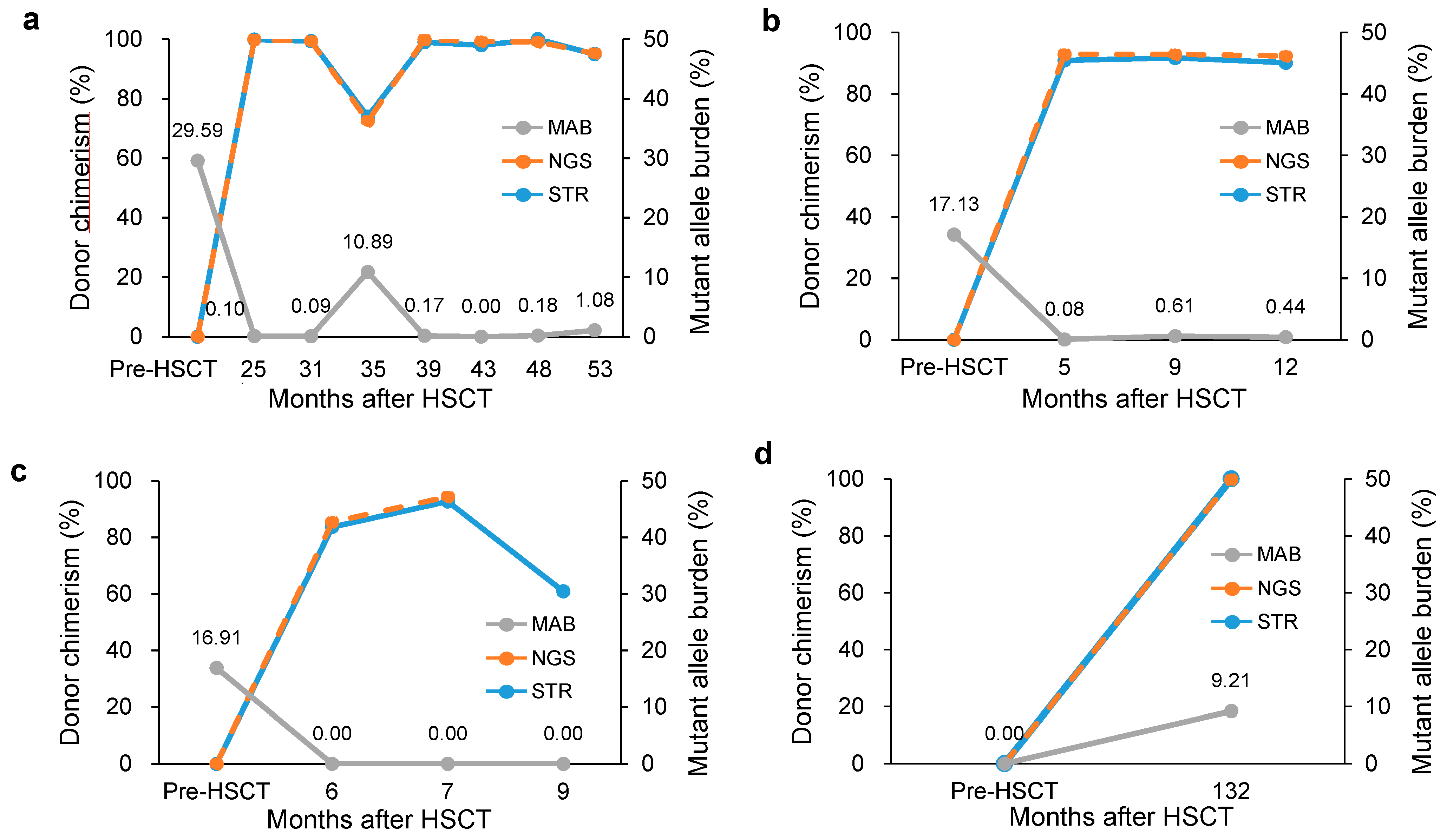
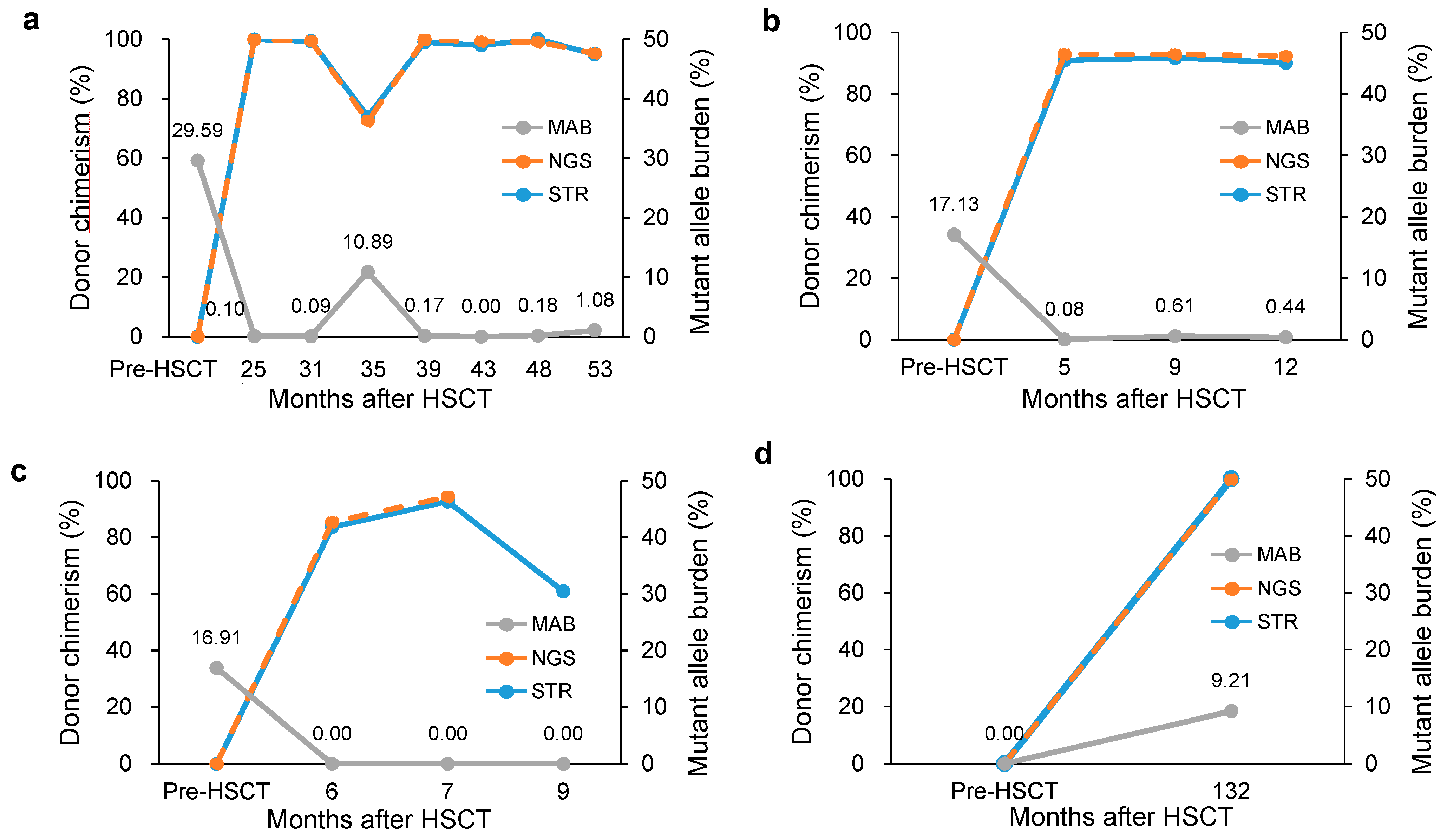
3.2. Clinical Usefulness of Simultaneous Monitoring
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Petrova, L.; Vrbacky, F.; Lanska, M.; Zavrelova, A.; Zak, P.; Hrochova, K. IDH1 and IDH2 mutations in patients with acute myeloid leukemia: Suitable targets for minimal residual disease monitoring? Clin. Biochem. 2018, 61, 34–39. [Google Scholar] [CrossRef]
- Kim, T.; Moon, J.H.; Ahn, J.S.; Kim, Y.K.; Lee, S.S.; Ahn, S.Y.; Jung, S.H.; Yang, D.H.; Lee, J.J.; Choi, S.H. Next-generation sequencing-based posttransplant monitoring of acute myeloid leukemia identifies patients at high risk of relapse. Blood 2018, 132, 1604–1613. [Google Scholar] [CrossRef]
- Morita, K.; Kantarjian, H.M.; Wang, F.; Yan, Y.; Bueso-Ramos, C.; Sasaki, K.; Issa, G.C.; Wang, S.; Jorgensen, J.; Song, X. Clearance of Somatic Mutations at Remission and the Risk of Relapse in Acute Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1788–1797. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer (IARC): Lyon, France, 2017; Volume 4, pp. 104–105. [Google Scholar]
- Roloff, G.W.; Lai, C.; Hourigan, C.S.; Dillon, L.W. Technical advances in the measurement of residual disease in acute myeloid leukemia. J. Clin. Med. 2017, 6, 87. [Google Scholar] [CrossRef]
- Bader, P.; Niethammer, D.; Willasch, A.; Kreyenberg, H.; Klingebiel, T. How and when should we monitor chimerism after allogeneic stem cell transplantation? Bone Marrow Transplant. 2005, 35, 107. [Google Scholar] [CrossRef] [PubMed]
- Ahci, M.; Stempelmann, K.; Buttkereit, U.; Crivello, P.; Trilling, M.; Heinold, A.; Steckel, N.K.; Koldehoff, M.; Horn, P.A.; Beelen, D.W. Clinical utility of quantitative PCR for chimerism and engraftment monitoring after allogeneic stem cell transplantation for hematologic malignancies. Biol. Blood Marrow Transplant. 2017, 23, 1658–1668. [Google Scholar] [CrossRef]
- Aloisio, M.; Licastro, D.; Caenazzo, L.; Torboli, V.; D’eustacchio, A.; Severini, G.M.; Athanasakis, E.A. Technical application of quantitative next generation sequencing for chimerism evaluation. Mol. Med. Rep. 2016, 14, 2967–2974. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hwang, I.S.; Shin, S.; Choi, J.R.; Lee, S.T. SNP-based next-generation sequencing reveals low-level mixed chimerism after allogeneic hematopoietic stem cell transplantation. Ann. Hematol. 2018, 97, 1731–1734. [Google Scholar] [CrossRef]
- Shapiro, R.M.; Kim, D.D.H. Next-generation sequencing-based minimal residual disease monitoring in patients receiving allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia or myelodysplastic syndrome. Curr. Opin. Hematol. 2018, 25, 425–432. [Google Scholar] [CrossRef]
- Han, E.; Kim, M.; Kim, Y.; Han, K.; Lim, J.; Kang, D.; Lee, G.D.; Kim, J.R.; Lee, J.W.; Chung, N.G. Practical informativeness of short tandem repeat loci for chimerism analysis in hematopoietic stem cell transplantation. Clin. Chim. Acta 2017, 468, 51–59. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Favero, F.; Joshi, T.; Marquard, A.M.; Birkbak, N.J.; Krzystanek, M.; Li, Q.; Szallasi, Z.; Eklund, A.C. Sequenza: Allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. 2015, 26, 64–70. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Thol, F.; Gabdoulline, R.; Liebich, A.; Klement, P.; Schiller, J.; Kandziora, C.; Hambach, L.; Stadler, M.; Koenecke, C.; Flintrop, M. Measurable residual disease monitoring by NGS before allogeneic hematopoietic cell transplantation in AML. Blood 2018, 132, 1703–1713. [Google Scholar] [CrossRef]
- Kristt, D.; Klein, T. Reliability of quantitative chimerism results: Assessment of sample performance using novel parameters. Leukemia 2006, 20, 1169. [Google Scholar] [CrossRef]
- Frick, M.; Chan, W.; Arends, C.M.; Hablesreiter, R.; Halik, A.; Heuser, M.; Michonneau, D.; Blau, O.; Hoyer, K.; Christen, F. Role of donor clonal hematopoiesis in allogeneic hematopoietic stem-cell transplantation. J. Clin. Oncol. 2018, 37, 375–385. [Google Scholar] [CrossRef]
- Scharf, S.J.; Smith, A.G.; Hansen, J.A.; McFarland, C.; Erlich, H.A. Quantitative determination of bone marrow transplant engraftment using fluorescent polymerase chain reaction primers for human identity markers. Blood 1995, 85, 1954–1963. [Google Scholar] [CrossRef]
- Dubovsky, J.; Daxberger, H.; Fritsch, G.; Printz, D.; Peters, C.; Matthes, S.; Gadner, H.; Lion, T.; Muller-Berat, N. Kinetics of chimerism during the early post-transplant period in pediatric patients with malignant and non-malignant hematologic disorders: Implications for timely detection of engraftment, graft failure and rejection. Leukemia 1999, 13, 2060–2069. [Google Scholar] [CrossRef]
- Antin, J.H.; Childs, R.; Filipovich, A.H.; Giralt, S.; Mackinnon, S.; Spitzer, T.; Weisdorf, D. Establishment of complete and mixed donor chimerism after allogeneic lymphohematopoietic transplantation: Recommendations from a workshop at the 2001 Tandem Meetings of the International Bone Marrow Transplant Registry and the American Society of Blood and Marrow Transplantation. Biol. Blood Marrow Transplant. 2001, 7, 473–485. [Google Scholar] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506. [Google Scholar] [CrossRef] [PubMed]
- Rautenberg, C.; Germing, U.; Haas, R.; Kobbe, G.; Schroeder, T. Relapse of acute myeloid leukemia after allogeneic stem cell transplantation: Prevention, detection, and treatment. Int. J. Mol. Sci. 2019, 20, 228. [Google Scholar] [CrossRef]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef]
- Hamilton, B.K.; Rybicki, L.; Hirsch, C.; Przychodzen, B.; Nazha, A.; Gerds, A.T.; Hanna, R.; Kalaycio, M.; Sekeres, M.A.; Sobecks, R. Mutation clonal burden and allogeneic hematopoietic cell transplantation outcomes in acute myeloid leukemia and myelodysplastic syndromes. Bone Marrow Transplant. 2019, 54, 1281–1286. [Google Scholar] [CrossRef]
- Schaap, N.; Schattenberg, A.; Mensink, E.; Preijers, F.; Hillegers, M.; Knops, R.; Pennings, A.; Boezeman, J.; Geurts van Kessel, A.; de Pauw, B.; et al. Long-term follow-up of persisting mixed chimerism after partially T cell-depleted allogeneic stem cell transplantation. Leukemia 2002, 16, 13–21. [Google Scholar] [CrossRef]
- Bouvier, A.; Ribourtout, B.; François, S.; Orvain, C.; Paz, D.L.; Beucher, A.; Guérard, A.; Guardiola, P.; Ugo, V.; Blanchet, O. Donor cell-derived acute promyelocytic leukemia after allogeneic hematopoietic stem cell transplantation. Eur. J. Haematol. 2018, 101, 570–574. [Google Scholar] [CrossRef]
- Kobayashi, S.; Kobayashi, A.; Osawa, Y.; Nagao, S.; Takano, K.; Okada, Y.; Tachi, N.; Teramoto, M.; Kawamura, T.; Horiuchi, T. Donor cell leukemia arising from preleukemic clones with a novel germline DDX41 mutation after allogenic hematopoietic stem cell transplantation. Leukemia 2017, 31, 1020. [Google Scholar] [CrossRef]
- Clark, J.R.; Scott, S.D.; Jack, A.L.; Lee, H.; Mason, J.; Carter, G.I.; Pearce, L.; Jackson, T.; Clouston, H.; Sproul, A. Monitoring of chimerism following allogeneic haematopoietic stem cell transplantation (HSCT): Technical recommendations for the use of short tandem repeat (STR) based techniques, on behalf of the United Kingdom National External Quality Assessment Service for Leucocyte Immunophenotyping Chimerism Working Group. Br. J. Haematol. 2015, 168, 26–37. [Google Scholar]
- Hiatt, J.B.; Pritchard, C.C.; Salipante, S.J.; O’Roak, B.J.; Shendure, J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013, 23, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Waalkes, A.; Penewit, K.; Wood, B.L.; Wu, D.; Salipante, S.J. Ultrasensitive detection of acute myeloid leukemia minimal residual disease using single molecule molecular inversion probes. Haematologica 2017, 102, 1549–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Location | Read Depth | %BE | %SD |
|---|---|---|---|---|
| NRAS:c.35G>A | Chr1:115258747 | 2048.7 ± 416.2 | 0.026 | 0.041 |
| SF3B1:c.2098A>G | Chr2:198266834 | 1930.5 ± 365.2 | 0.018 | 0.031 |
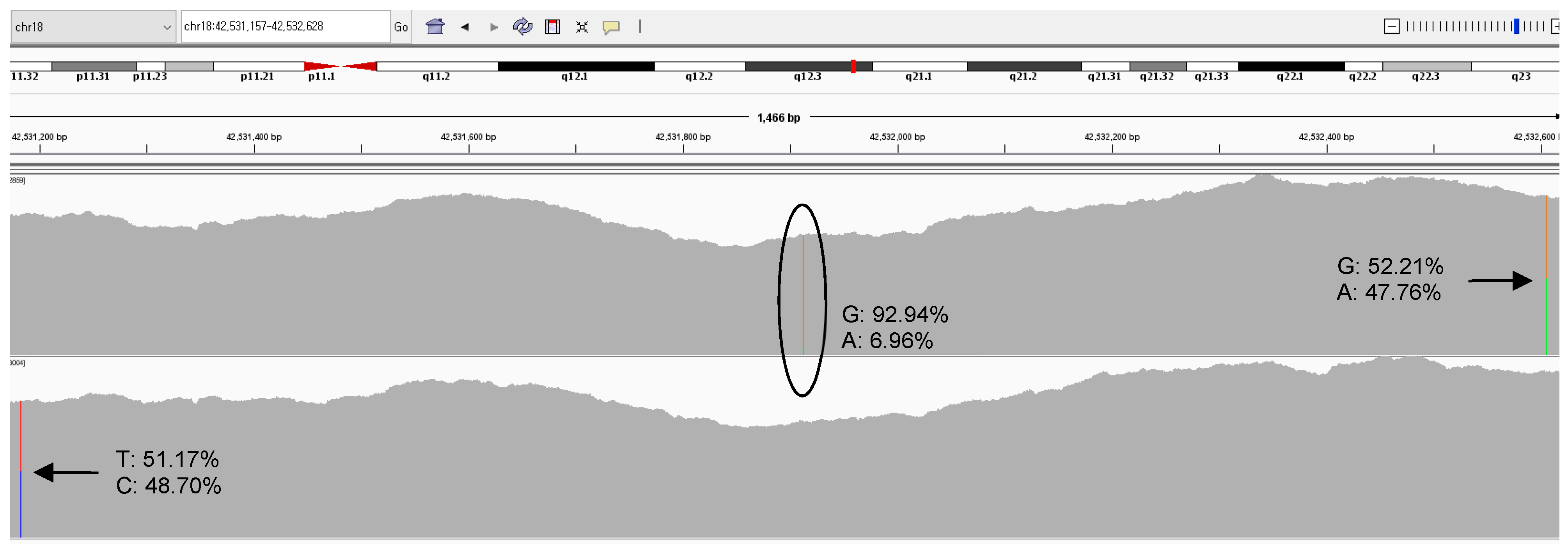
| SETBP1:c.2608G>A | Chr18:42531913 | 1598.5 ± 313.4 | 0.041 | 0.064 |
| U2AF1:c.101C>A,T | Ch21:44524456 | 1410.7 ± 351.4 | 0.026 | 0.055 |
| PHF6:c.820C>T | ChrX:133549136 | 1184.0 ± 455.8 | 0.023 | 0.052 |
| Case Type | No. | Gender/Age | Type of HSCT | Status | Sample | F/U Months | BM Findings Cellularity (Blasts) | Chromosome Analysis | %Donor STR | No. IRA STR | %Donor NGS | No. IRA NGS | MAB (%) † | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Complete donor chimerism | 1 | M/61 | Unrelated matched - PBSCT | Pre | PB | RCMD * | 46,XY[20] * | 22.40 | U2AF1:c.101C>T | |||||
| 6.60 | SETBP1:c.2608G>A | |||||||||||||
| Post 1 | PB | 30 | NA | NA | 99.4 | 14 | 99.94 | 41 | 0.00 | U2AF1:c.101C>T | ||||
| 0.00 | SETBP1:c.2608G>A | |||||||||||||
| 2 | F/39 | Family mismatched -PBSCT | Pre | PB | Hypo-MDS * | 46,XX,t(2;3)(p23;q29)[14]/ 46,XX[6] * | 10.33 | SF3B1:c.2098A>G | ||||||
| Post 1 | BM | 25 | 30% (1%) | 46,XX[20] | 100 | 8 | 99.87 | 24 | 0.00 | |||||
| 3 | M/29 | Family mismatched -PBSCT | Pre | PB | MDS EB-1 * | 46,XY[20] * | 31.78 | U2AF1:c.101C>T | ||||||
| Post 1 | PB | 46 | NA | NA | 99.1 | 11 | 99.97 | 31 | 0.00 | |||||
| 4 | F/29 | Sibling matched -PBSCT | Pre | PB | RCMD * | 47,XX,+8,del(11)(q21)[8]/ 46,XX[12] * | 37.64 | U2AF1:c.101C>A | ||||||
| Post 1 | PB | 54 | NA | NA | 99.9 | 5 | 99.91 | 17 | 0.15 | |||||
| 5 | M/45 | Sibling matched -PBSCT | Pre | PB | MDS EB-2 * | 46,XY[15] * | 26.22 | U2AF1:c.101C>T | ||||||
| Post 1 | PB | 37 | NA | NA | 100 | 6 | 99.93 | 27 | 0.00 | |||||
| 6 | F/26 | Sibling matched -PBSCT | Pre | PB | RCMD * | 47,XX,+8[3]/46,XX[17] * | 6.88 | U2AF1:c.101C>A | ||||||
| Post 1 | PB | 32 | NA | NA | 97.8 | 14 | 98.28 | 30 | 0.10 | |||||
| 7 | M/58 | Family mismatched -PBSCT | Pre | PB | RCMD * | 46,XY[20] * | 46.74 | U2AF1:c.101C>T | ||||||
| 3.69 | NRAS:c.35G>A | |||||||||||||
| Post 1 | BM | 10 | 50% (<1%) | 46,XY[20] | 98.2 | 13 | 98.81 | 28 | 0.00 | U2AF1:c.101C>T | ||||
| 0.00 | NRAS:c.35G>A | |||||||||||||
| 8 | M/24 | Family mismatched -PBSCT | Pre | PB | RCMD * | 47,XY,+8[20] * | 7.10 | U2AF1:c.101C>A | ||||||
| Post 1 | PB | 37 | NA | NA | 99.6 | 7 | 99.87 | 11 | 0.08 | |||||
| 9 | F/31 | Sibling matched -PBSCT | Pre | PB | MDS EB-1 * | 46,XX[20] * | - | None | ||||||
| Post 1 | PB | 30 | NA | NA | 99.7 | 10 | 99.86 | 20 | - | |||||
| Mixed chimerism | 10 | M/3 | Sibling matched - BMT | Pre | PB | RCC * | 46,XY[20] * | - | None | |||||
| Post 1 | PB | 1 | 20% (1%) * | 46,XY[2]//46,XX[28] * | 76.9 | 9 | 80.56 | 9 | - | |||||
| Post 2 | PB | 3 | 80% (<1%) * | 46,XY[8]//46,XX[12] * | 69.7 | 6 | 72.00 | 10 | - | |||||
| 11 | F/59 | Unrelated matched - PBSCT | Pre | PB | RCMD * | 46,XX[20] * | 29.59 | U2AF1:c.101C>T | ||||||
| Post 1 | PB | 25 | NA | NA | 99.9 | 14 | 99.88 | 37 | 0.10 | |||||
| Post 2 | PB | 31 | NA | NA | 99.4 | 14 | 99.39 | 37 | 0.09 | |||||
| Post 3 | BM | 35 | 70% (18%) | 47,XX,+21[9]//46,XY[11] | 74.0 | 14 | 72.49 | 28 | 10.89 | |||||
| Post 4 | PB | 39 | NA | NA | 99.0 | 14 | 99.85 | 37 | 0.17 | |||||
| Post 5 | BM | 43 | 30% (5%) | //46,XY[30] | 98.0 | 14 | 99.23 | 37 | 0.00 | |||||
| Post 6 | PB | 48 | NA | NA | 100 | 14 | 99.09 | 37 | 0.18 | |||||
| Post 7 | PB | 53 | 5% (18%) * | 47,XX,+21[10]//46,XY[10] * | 95.0 | 14 | 95.23 | 37 | 1.08 | |||||
| 12 | M/54 | Sibling matched - BMT | Pre | PB | RARS * | 46,XY[12] * | 17.13 | SF3B1:c.2098A>G | ||||||
| Post 1 | PB | 5 | 15% (<1%) * | 46,XY[20] * | 90.9 | 7 | 92.91 | 19 | 0.08 | |||||
| Post 2 | PB | 9 | 30% (<1%) * | 46,XY[20] * | 91.7 | 8 | 92.84 | 19 | 0.61 | |||||
| Post 3 | PB | 12 | 10% (3%) * | 46,XY[10] * | 90.2 | 8 | 92.35 | 19 | 0.44 | |||||
| 13 | M/52 | Sibling matched - BMT | Pre | PB | MDS EB-2 * | 46,XY[20] * | 16.91 | NRAS:c.35G>A | ||||||
| Post 1 | PB | 6 | 30% (<1%) * | 46,XY[20] * | 83.7 | 9 | 85.44 | 9 | 0.00 | |||||
| Post 2 | BM | 7 | 15% (1%) | 46,XY[20] | 92.7 | 8 | 94.37 | 9 | 0.00 | |||||
| Post 3 | BM | 9 | 40% (5%) | 46,XY,t(1;21)(p36.3;q11.2)[19]/ 46,XY[1] | 60.9 | 9 | NA | NA | 0.00 | |||||
| Donor-cell derived MDS | 14 | F/59 | Sibling matched - BMT | Pre | PB | MDS EB-1 * | NA * | 0.00 | PHF6:c.820C>T | |||||
| Post 1 | BM | 132 | 15% (5%) | //46,XY,+1,der(1;7)(q10;q10)[5]/46,XY[5] | 100 | 5 | 99.80 | 13 | 9.21 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-M.; Kim, Y.-J.; Park, S.-S.; Han, E.; Kim, M.; Kim, Y. Simultaneous Monitoring of Mutation and Chimerism Using Next-Generation Sequencing in Myelodysplastic Syndrome. J. Clin. Med. 2019, 8, 2077. https://doi.org/10.3390/jcm8122077
Lee J-M, Kim Y-J, Park S-S, Han E, Kim M, Kim Y. Simultaneous Monitoring of Mutation and Chimerism Using Next-Generation Sequencing in Myelodysplastic Syndrome. Journal of Clinical Medicine. 2019; 8(12):2077. https://doi.org/10.3390/jcm8122077
Chicago/Turabian StyleLee, Jong-Mi, Yoo-Jin Kim, Sung-Soo Park, Eunhee Han, Myungshin Kim, and Yonggoo Kim. 2019. "Simultaneous Monitoring of Mutation and Chimerism Using Next-Generation Sequencing in Myelodysplastic Syndrome" Journal of Clinical Medicine 8, no. 12: 2077. https://doi.org/10.3390/jcm8122077