BRAF Mutations and Dysregulation of the MAP Kinase Pathway Associated to Sinonasal Mucosal Melanomas


 ,
,  , , ,
, , ,  , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. DNA Extraction and Next Generation Sequencing
2.3. Statistical Analysis
3. Results
3.1. Clinical Features
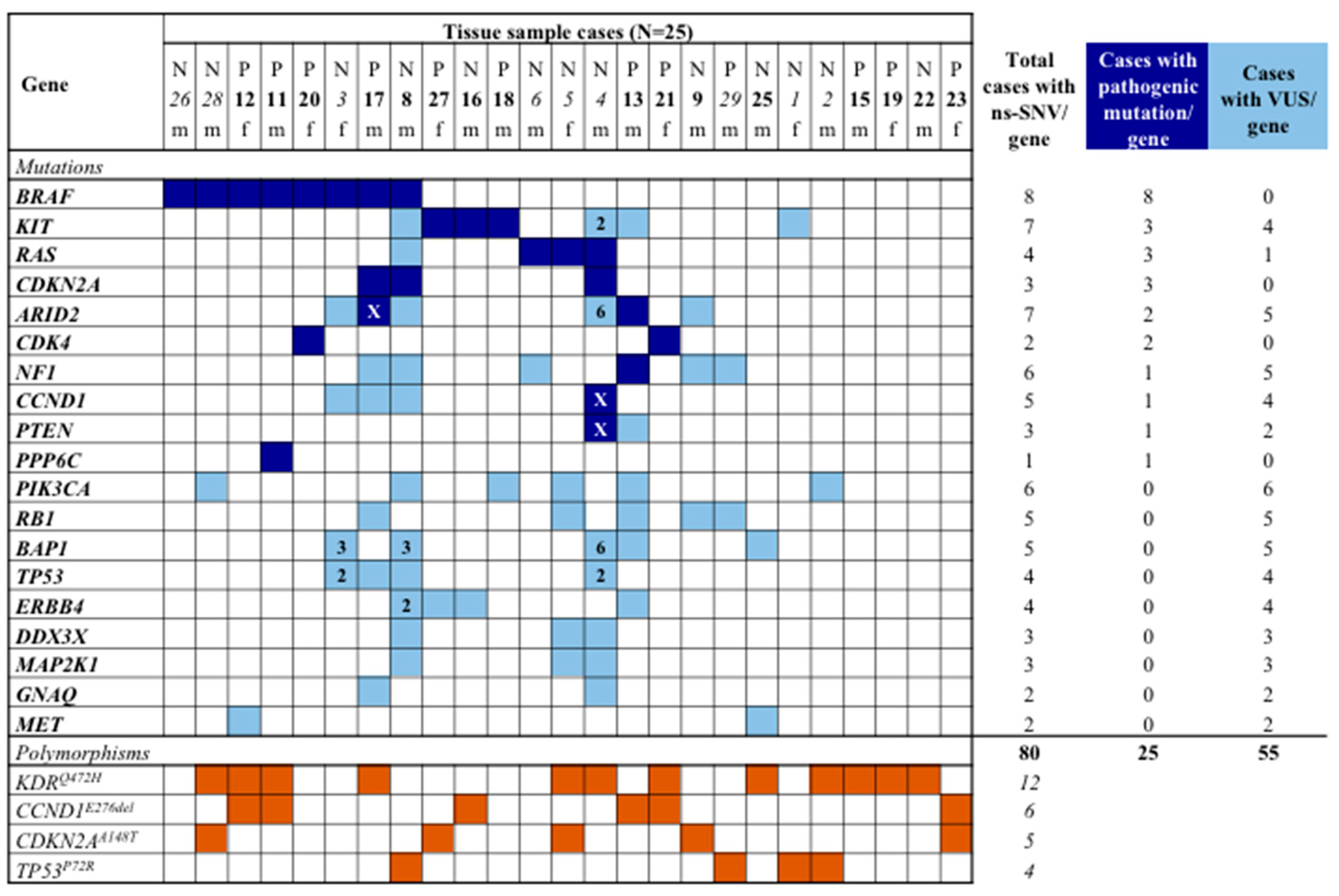
3.2. Mutation Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Cossu, A.; Casula, M.; Cesaraccio, R.; Lissia, A.; Colombino, M.; Sini, M.C.; Budroni, M.; Tanda, F.; Paliogiannis, P.; Palmieri, G. Epidemiology and genetic susceptibility of malignant melanoma in North Sardinia, Italy. Eur. J. Cancer Prev. 2017, 26, 263–267. [Google Scholar] [CrossRef]
- Lipsker, D.; Engel, F.; Cribier, B.; Velten, M.; Hedelin, G. Trends in melanoma epidemiology suggest three different types of melanoma. Br. J. Dermatol. 2007, 157, 338–343. [Google Scholar] [CrossRef]
- Postow, M.A.; Hamid, O.; Carvajal, R.D. Mucosal melanoma: Pathogenesis, clinical behavior, and management. Curr. Oncol. Rep. 2012, 14, 441–448. [Google Scholar] [CrossRef]
- Lund, V.J.; Howard, D.J.; Harding, L.; Wei, W.I. Management options and survival in malignant melanoma of the sinonasal mucosa. Laryngoscope 1999, 109, 208–211. [Google Scholar] [CrossRef]
- Mallone, S.; De Vries, E.; Guzzo, M.; Midena, E.; Verne, J.; Coebergh, J.W.; Marcos-Gragera, R.; Ardanaz, E.; Martinez, R.; Chirlaque, M.D.; et al. Descriptive epidemiology of malignant mucosal and uveal melanomas and adnexal skin carcinomas in Europe. Eur. J. Cancer 2012, 48, 1167–1175. [Google Scholar] [CrossRef]
- Clifton, N.; Harrison, L.; Bradley, P.J.; Jones, N.S. Malignant melanoma of nasal cavity and paranasal sinuses: Report of 24 patients and literature review. J. Laryngol. Otol. 2011, 125, 479–485. [Google Scholar] [CrossRef]
- Colombino, M.; Lissia, A.; Franco, R.; Botti, G.; Ascierto, P.A.; Manca, A.; Sini, M.C.; Pisano, M.; Paliogiannis, P.; Tanda, F.; et al. Unexpected distribution of KIT and BRAF mutations among southern Italian patients with sinonasal melanoma. Dermatology 2013, 226, 279–284. [Google Scholar] [CrossRef]
- McLaughlin, C.C.; Wu, X.C.; Jemal, A.; Martin, H.J.; Roche, L.M.; Chen, V.W. Incidence of noncutaneous melanomas in the U.S. Cancer 2005, 103, 1000–1007. [Google Scholar] [CrossRef]
- Prasad, M.L.; Busam, K.J.; Patel, S.G.; Hoshaw-Woodard, S.; Shah, J.P.; Huvos, A.G. Clinicopathologic differences in malignant melanoma arising in oral squamous and sinonasal respiratory mucosa of the upper aerodigestive tract. Arch. Pathol. Lab. Med. 2003, 127, 997–1002. [Google Scholar]
- Mendenhall, W.M.; Amdur, R.J.; Hinerman, R.W.; Werning, J.W.; Villaret, D.B.; Mendenhall, N.P. Head and neck mucosal melanoma. Am. J. Clin. Oncol. 2005, 28, 626–630. [Google Scholar] [CrossRef]
- Lund, V.J.; Chisholm, E.J.; Howard, D.J.; Wei, W.I. Sinonasal malignant melanoma: An analysis of 115 cases assessing outcomes of surgery, postoperative radiotherapy and endoscopic resection. Rhinology 2012, 50, 203–210. [Google Scholar] [CrossRef]
- Jangard, M.; Hansson, J.; Ragnarsson-Olding, B. Primary sinonasal malignant melanoma: A nationwide study of the Swedish population, 1960–2000. Rhinology 2013, 51, 22–30. [Google Scholar] [CrossRef]
- Holmstrom, M.; Lund, V.J. Malignant melanomas of the nasal cavity after occupational exposure to formaldehyde. Br. J. Ind. Med. 1991, 48, 9–11. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Palmieri, G.; Colombino, M.; Casula, M.; Budroni, M.; Manca, A.; Sini, M.C.; Lissia, A.; Stanganelli, I.; Ascierto, P.A.; Cossu, A. Epidemiological and genetic factors underlying melanoma development in Italy. Melanoma Manag. 2015, 2, 149–163. [Google Scholar] [CrossRef]
- Palmieri, G.; Ombra, M.; Colombino, M.; Casula, M.; Sini, M.; Manca, A.; Paliogiannis, P.; Ascierto, P.A.; Cossu, A. Multiple molecular pathways in melanomagenesis: Characterization of therapeutic targets. Front. Oncol. 2015, 5, 183. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Busam, K.J.; Francone, T.D.; Wong, G.C.; Guo, T.; Agaram, N.P.; Besmer, P.; Jungbluth, A.; Gimbel, M.; Chen, C.T.; et al. L576P KIT mutation in anal melanomas correlates with KIT protein expression and is sensitive to specific kinase inhibition. Int. J. Cancer 2007, 121, 257–264. [Google Scholar] [CrossRef]
- Beadling, C.; Jacobson-Dunlop, E.; Hodi, F.S.; Le, C.; Warrick, A.; Patterson, J.; Town, A.; Harlow, A.; Cruz, F., III; Azar, S.; et al. KIT gene mutations and copy number in melanoma subtypes. Clin. Cancer Res. 2008, 14, 6821–6828. [Google Scholar] [CrossRef]
- Schoenewolf, N.L.; Bull, C.; Belloni, B.; Holzmann, D.; Tonolla, S.; Lang, R.; Mihic-Probst, D.; Andres, C.; Dummer, R. Sinonasal, genital and acrolentiginous melanomas show distinct characteristics of KIT expression and mutations. Eur. J. Cancer 2012, 48, 1842–1852. [Google Scholar] [CrossRef]
- Omholt, K.; Grafström, E.; Kanter-Lewensohn, L.; Hansson, J.; Ragnarsson-Olding, B.K. KIT pathway alterations in mucosal melanomas of the vulva and other sites. Clin. Cancer Res. 2011, 17, 3933–3942. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, J.W.; Kim, Y.S. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: A meta-analysis. Br. J. Dermatol. 2011, 164, 776–784. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Ablain, J.; Xu, M.; Rothschild, H.; Jordan, R.C.; Mito, J.K.; Daniels, B.H.; Bell, C.F.; Joseph, N.M.; Wu, H.; Bastian, B.C.; et al. Human tumor genomics and zebrafish modeling identify SPRED1 loss as a driver of mucosal melanoma. Science 2018, 362, 1055–1060. [Google Scholar] [CrossRef]
- Newell, F.; Kong, Y.; Wilmott, J.S.; Johansson, P.A.; Ferguson, P.M.; Cui, C.; Li, Z.; Kazakoff, S.H.; Burke, H.; Dodds, T.J.; et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat. Commun. 2019, 10, 3163. [Google Scholar] [CrossRef]
- Wong, K.; van der Weyden, L.; Schott, C.R.; Foote, A.; Constantino-Casas, F.; Smith, S.; Dobson, J.M.; Murchison, E.P.; Wu, H.; Yeh, I.; et al. Cross-species genomic landscape comparison of human mucosal melanoma with canine oral and equine melanoma. Nat. Commun. 2019, 10, 353. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Sini, M.C.; Doneddu, V.; Paliogiannis, P.; Casula, M.; Colombino, M.; Manca, A.; Botti, G.; Ascierto, P.A.; Lissia, A.; Cossu, A.; et al. Genetic alterations in main candidate genes during melanoma progression. Oncotarget 2018, 9, 8531–8541. [Google Scholar] [CrossRef] [Green Version]
- Bastian, B.C. Understanding the progression of melanocytic neoplasia using genomic analysis: From fields to cancer. Oncogene 2003, 22, 3081–3086. [Google Scholar] [CrossRef]
- Smalley, K.S.; Contractor, R.; Nguyen, T.K.; Xiao, M.; Edwards, R.; Muthusamy, V.; King, A.J.; Flaherty, K.T.; Bosenberg, M.; Herlyn, M.; et al. Identification of a novel subgroup of melanomas with KIT/cyclin-dependent kinase-4 overexpression. Cancer Res. 2008, 68, 5743–5752. [Google Scholar] [CrossRef]
- Ashida, A.; Takata, M.; Murata, H.; Kido, K.; Saida, T. Pathological activation of KIT in metastatic tumors of acral and mucosal melanomas. Int. J. Cancer 2009, 124, 862–868. [Google Scholar] [CrossRef]
- Vidwans, S.J.; Flaherty, K.T.; Fisher, D.E.; Tenenbaum, J.M.; Travers, M.D.; Shrager, J. A melanoma molecular disease model. PLoS ONE 2011, 6, e18257. [Google Scholar] [CrossRef]
- Manca, A.; Paliogiannis, P.; Colombino, M.; Casula, M.; Lissia, A.; Botti, G.; Caracò, C.; Ascierto, P.A.; Sini, M.C.; Palomba, G.; et al. Mutational concordance between primary and metastatic melanoma: A next-generation sequencing approach. J. Transl. Med. 2019, 17, 289. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Landrum, M.J.; Kattman, B.L. ClinVar at five years: Delivering on the promise. Hum Mutat. 2018, 39, 1623–1630. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Palmieri, G.; Colombino, M.; Casula, M.; Manca, A.; Mandalà, M.; Cossu, A.; Italian Melanoma Intergroup (IMI). Molecular pathways in melanomagenesis: What we learned from next-generation sequencing approaches. Curr. Oncol. Rep. 2018, 20, 86. [Google Scholar] [CrossRef]
- Konuthula, N.; Khan, M.N.; Parasher, A.; Del Signore, A.; Genden, E.M.; Govindaraj, S.; Iloreta, A.M. The presentation and outcomes of mucosal melanoma in 695 patients. Int. Forum Allergy Rhinol. 2017, 7, 99–105. [Google Scholar] [CrossRef]
- Dreno, M.; Georges, M.; Espitalier, F.; Ferron, C.; Charnolé, A.; Dréno, B.; Malard, O. Sinonasal mucosal melanoma: A 44-case study and literature analysis. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2017, 134, 237–242. [Google Scholar] [CrossRef]
- Ozturk Sari, S.; Yilmaz, İ.; Taşkin, O.Ç.; Narli, G.; Şen, F.; Çomoğlu, Ş.; Firat, P.; Bİlgİç, B.; Yilmazbayhan, D.; Özlük, Y. BRAF, NRAS, KIT, TERT, GNAQ/GNA11 mutation profile analysis of head and neck mucosal melanomas: A study of 42 cases. Pathology 2017, 49, 55–61. [Google Scholar] [CrossRef]
- Zebary, A.; Jangard, M.; Omholt, K.; Ragnarsson-Olding, B.; Hansson, J. KIT, NRAS and BRAF mutations in sinonasal mucosal melanoma: A study of 56 cases. Br. J. Cancer 2013, 109, 559–564. [Google Scholar] [CrossRef]
- Cohen, Y.; Rosenbaum, E.; Begum, S.; Goldenberg, D.; Esche, C.; Lavie, O.; Sidransky, D.; Westra, W.H. Exon 15 BRAF mutations are uncommon in melanomas arising in nonsun-exposed sites. Clin. Cancer Res. 2004, 10, 3444–3447. [Google Scholar] [CrossRef]
- Amit, M.; Tam, S.; Abdelmeguid, A.S.; Roberts, D.B.; Takahashi, Y.; Raza, S.M.; Su, S.Y.; Kupferman, M.E.; DeMonte, F.; Hanna, E.Y. Mutation status among patients with sinonasal mucosal melanoma and its impact on survival. Br. J. Cancer 2017, 116, 1564–1571. [Google Scholar] [CrossRef]
- Wroblewska, J.P.; Mull, J.; Wu, C.L.; Fujimoto, M.; Ogawa, T.; Marszalek, A.; Hoang, M.P. SF3B1, NRAS, KIT, and BRAF mutation; CD117 and cMYC Expression; and tumoral pigmentation in sinonasal melanomas: An analysis with newly found molecular alterations and some population-based molecular differences. Am. J. Surg. Pathol. 2019, 43, 168–177. [Google Scholar] [CrossRef]
- Khalili, J.S.; Liu, S.; Rodriguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immuno-suppression via induction of interleukin-1 in melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [Google Scholar] [CrossRef]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable TME in patients with metastatic melanoma. Clin. Cancer. Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef]
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y.; et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin. Cancer Res. 2013, 19, 393–403. [Google Scholar] [CrossRef]
- Konuthula, N.; Khan, M.N.; Parasher, A.; Del Signore, A.; Genden, E.M.; Govindaraj, S.; Iloreta, A.M. Targeting adenosine in BRAF-mutant melanoma reduces tumor growth and metastasis. Cancer Res. 2017, 7, 99–105. [Google Scholar]
- Ascierto, P.A.; McArthur, G.A. Checkpoint inhibitors in melanoma and early phase development in solid tumors: What’s the future? J. Transl. Med. 2017, 15, 173. [Google Scholar] [CrossRef]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef]
- Broman, K.K.; Dossett, L.A.; Sun, J.; Eroglu, Z.; Zager, J.S. Update on BRAF and MEK inhibition for treatment of melanoma in metastatic, unresectable, and adjuvant settings. Expert Opin. Drug Saf. 2019, 18, 381–392. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CHARACTERISTIC | Patients (n = 25) | % |
|---|---|---|
| Median age at diagnosis (range), years | 70 (49–91) | |
| Male/Female sex | 16/9 | 64/36 |
| Mean follow up (range), months | 36 (9–78) | |
| Anatomical site | ||
| Nasal cavity | 13 | 52 |
| Paranasal sinuses | 12 | 48 |
| Histological variables | ||
| Mitosis (<1 / >1) | 14/11 | 56/44 |
| Necrosis (present/absent) | 10/15 | 40/60 |
| Ulceration (present/absent) | 14/11 | 56/44 |
| Status at end of follow-up | ||
| Death from disease | 17 | 68 |
| Death from uncertain cause | 5 | 20 |
| Alive with disease | 2 | 8 |
| Alive with no evidence of disease | 1 | 4 |
| Disease’s stage at diagnosis | ||
| pT2 | 3 | 12 |
| pT3 | 9 | 36 |
| pT4 | 8 | 32 |
| N0/N+ | 21/4 | 84/16 |
| M0/M1 | 24/1 | 96/4 |
| Unknown | 5 | 20 |
| CHARACTERISTIC | Cases with Pathogenetic Gene Mutations | Cases with Mutations in BRAF + RAS Genes | ||||
|---|---|---|---|---|---|---|
| No. | % | p | No. | % | p | |
| Total cases (n = 25) | 16 | 64.0 | 11 | 44.0 | ||
| Sex | ||||||
| Female (n = 9) | 6 | 66.7 | 1.000 | 4 | 44.4 | 1.000 |
| Male (n = 16) | 10 | 62.5 | 7 | 43.8 | ||
| Population origin | ||||||
| Sardinian (n = 9) | 6 | 66.7 | 1.000 | 6 | 66.7 | 0.115 |
| Non-Sardinian (n = 16) | 10 | 62.5 | 5 | 31.3 | ||
| Anatomical site | ||||||
| Nasal cavity (n = 13) | 8 | 61.5 | 0.881 | 7 | 53.8 | 0.453 |
| Paranasal sinuses (n = 12) | 8 | 66.7 | 4 | 33.3 | ||
| Mitosis | ||||||
| < 1 (n = 14) | 7 | 50.0 | 0.208 | 5 | 35.7 | 0.592 |
| ≥1 (n = 11) | 9 | 81.8 | 6 | 54.5 | ||
| Necrosis | ||||||
| Present (n = 10) | 6 | 60.0 | 1.000 | 4 | 40.0 | 1.000 |
| Absent (n = 15) | 10 | 66.7 | 7 | 46.7 | ||
| Ulceration | ||||||
| Present (n = 14) | 9 | 64.3 | 1.000 | 6 | 42.9 | 0.783 |
| Absent (n = 11) | 7 | 63.6 | 5 | 45.5 | ||
| Disease stage at diagnosis | ||||||
| pT2-3N0 (n = 8) | 5 | 62.5 | 0.970 | 3 | 37.5 | 0.875 |
| pT4N0 (n = 7) | 5 | 71.4 | 4 | 57.1 | ||
| pTanyN+/M1 (n = 5) | 3 | 60.0 | 2 | 40.0 | ||
| Unknown (n = 5) | 3 | 60.0 | 2 | 40.0 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colombino, M.; Paliogiannis, P.; Cossu, A.; De Re, V.; Miolo, G.; Botti, G.; Scognamiglio, G.; Ascierto, P.A.; Santeufemia, D.A.; Fraggetta, F.; et al. BRAF Mutations and Dysregulation of the MAP Kinase Pathway Associated to Sinonasal Mucosal Melanomas. J. Clin. Med. 2019, 8, 1577. https://doi.org/10.3390/jcm8101577
Colombino M, Paliogiannis P, Cossu A, De Re V, Miolo G, Botti G, Scognamiglio G, Ascierto PA, Santeufemia DA, Fraggetta F, et al. BRAF Mutations and Dysregulation of the MAP Kinase Pathway Associated to Sinonasal Mucosal Melanomas. Journal of Clinical Medicine. 2019; 8(10):1577. https://doi.org/10.3390/jcm8101577
Chicago/Turabian StyleColombino, Maria, Panagiotis Paliogiannis, Antonio Cossu, Valli De Re, Gianmaria Miolo, Gerardo Botti, Giosuè Scognamiglio, Paolo Antonio Ascierto, Davide Adriano Santeufemia, Filippo Fraggetta, and et al. 2019. "BRAF Mutations and Dysregulation of the MAP Kinase Pathway Associated to Sinonasal Mucosal Melanomas" Journal of Clinical Medicine 8, no. 10: 1577. https://doi.org/10.3390/jcm8101577