
Role of Flavonoids in Protecting Against Neurodegenerative Diseases—Possible Mechanisms of Action

Abstract
1. Introduction
2. Flavonoids
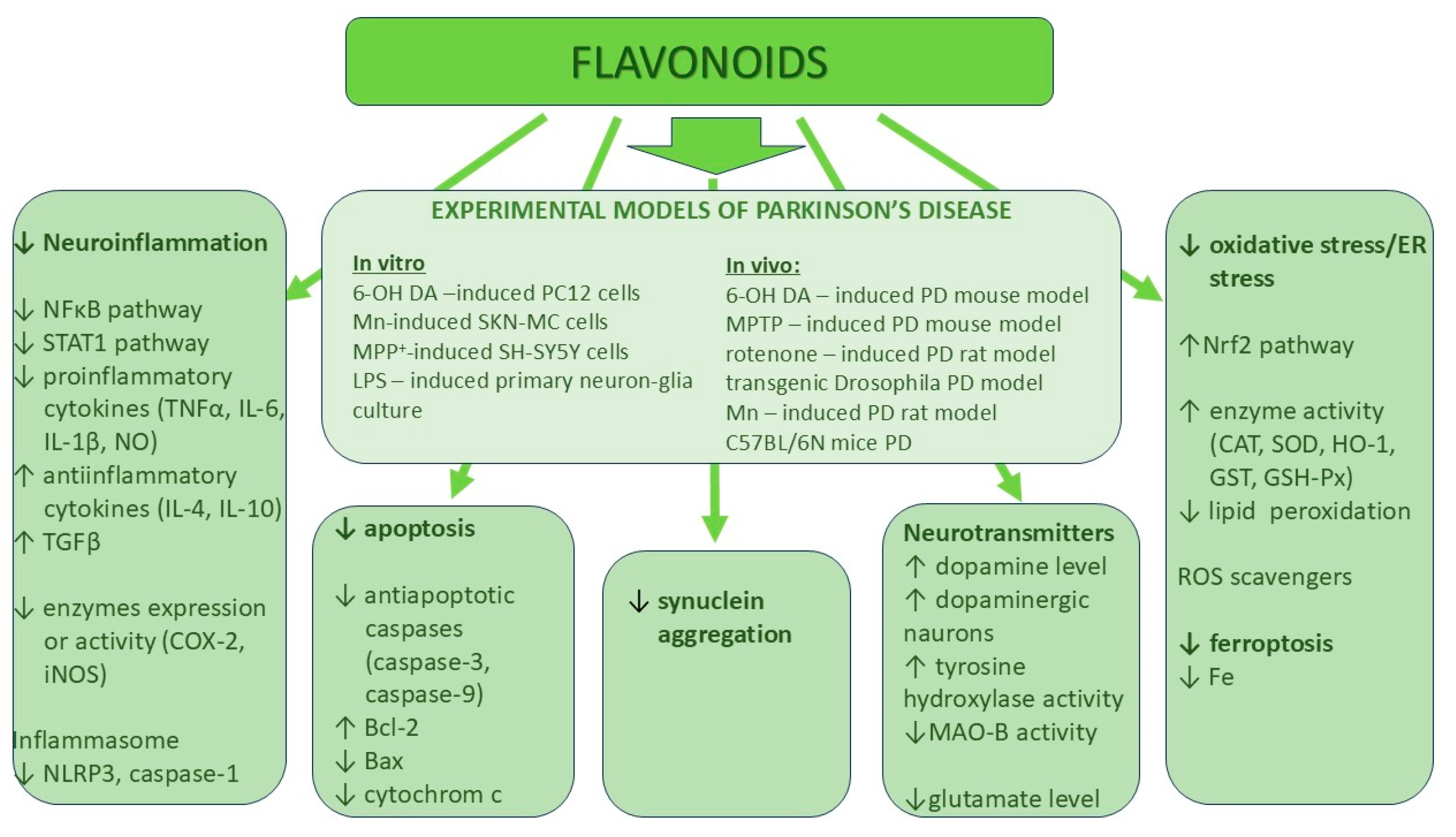
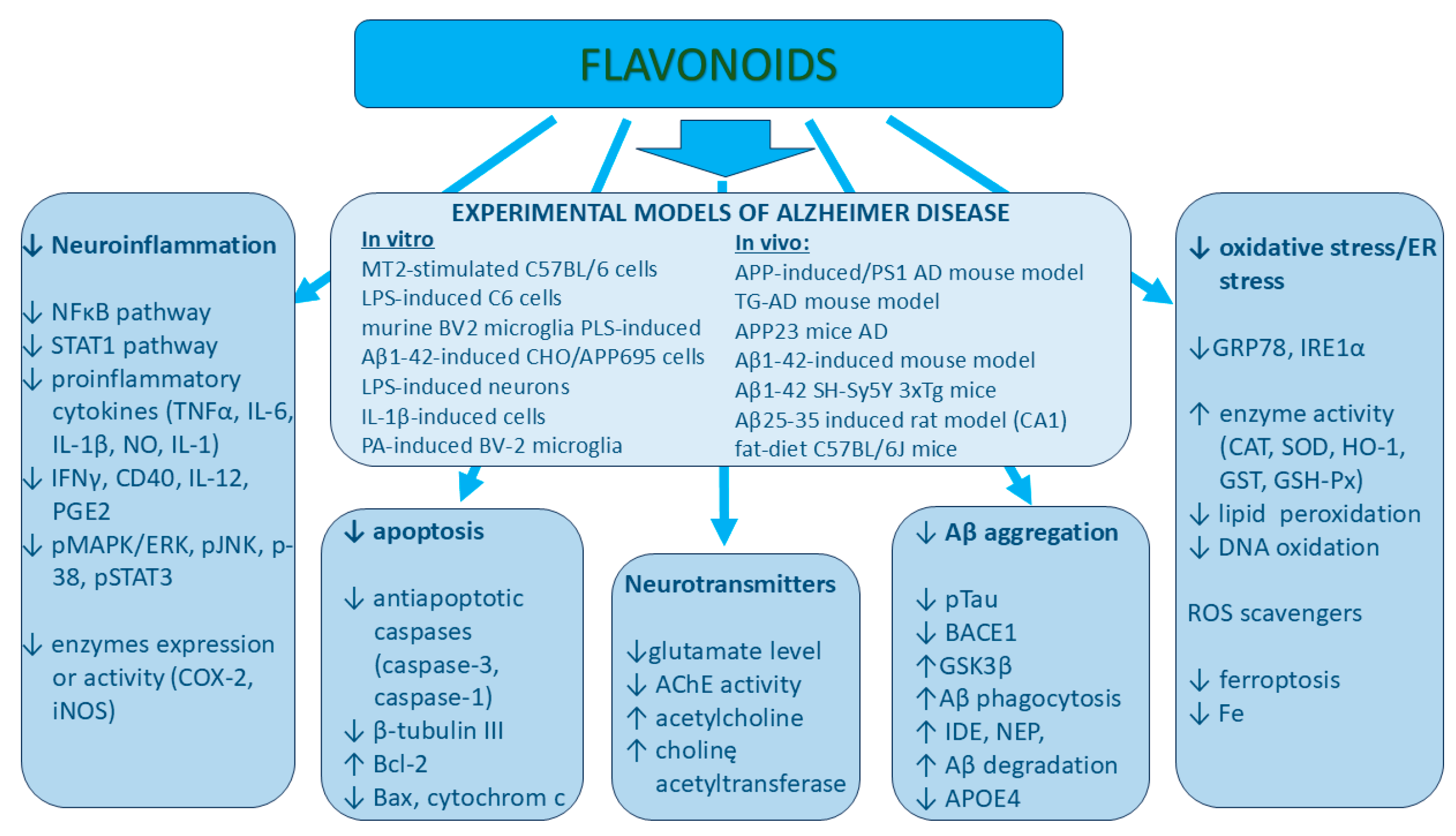
3. The Role of Various Classes of Flavonoids in Neurodegenerative Protection
3.1. Flavones
3.1.1. Chrysin (5,7-Dihydroxyflavone)
3.1.2. Luteolin (3′,4′,5,7-Tetrahydroxyflavone) (Other Names: Luteolol, Digitoflavone, Flacitran, and Luteoline)
3.1.3. Diosmetin (5,7,3′-Trihydroxy-4′-methoxyflavone)
3.1.4. Apigenin (4′,5,7-Trihydroxyflavone)
3.2. Flavonols
3.2.1. Quercetin (3,3,4,5,7-Pentahydroxyflavone)
3.2.2. Myricetin (3,3′,4′,5,5′,7-Hexahydroxyflavone)
3.3. Flavanols
Catechins (Flavan-3,3′,4′,5,7-pentol) (2R,3S)-2-(3,4-Dihydroxyphenyl)-3,4-dihydro-2H-chromene-3,5,7-triol)
3.4. Flavanone
Eriodictyol ((2S)-3′,4′,5,7-Tetrahydroxyflavan-4-one)
3.5. Isoflavone
Biochanin (5,7-Dihydroxy-40-methoxyisoflavone)
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CNS | central nervous system |
| PD | Parkinson’s disease |
| AD | Alzheimer’s disease |
| Aβ | amyloid-β |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| ROS | reactive oxygen species |
| TNF-α | tumor necrosis factor α |
| NO | nitrate oxide |
| NOS | nitrate oxide synthase |
| IL- | interleukin- |
| LPS | lipopolysaccharide |
| TRAF | TNF receptor-associated factor |
| 6-OHDA | 6-hydroxydopamine |
| COX-2 | cyclooxygenase-2 |
| CAT | catalase |
| SOD | superoxide dismutase |
| HO-1 | heme oxygenase-1 |
| Nrf2 | nuclear factor erythroid 2-like 2 |
| PPAR-γ | peroxisome proliferator-activated receptor gamma |
| BACE1 | β-site-amyloid precursor protein cleaving enzyme 1; β-secretase 1 |
| GSK3β | glycogen synthase kinase 3β |
| 38MAPK | p38 mitogen-activated protein kinase |
| PGE2 | prostaglandin E2 |
| PI3K | phosphoinositide 3-kinase |
| AKT | protein kinase B, PKB, serine/threonine-specific protein kinases |
| MAO | monoamine oxidase A or B |
| GSH | glutathione |
| GSH-Px | glutathione peroxidase |
| TGF-β | transforming growth factor β |
| STAT1 | signal transducer and activator of transcription 1 |
| AChE | acetylcholine esterase |
| ChAT | choline acetyltransferase |
| AT1R | angiotensin II type 1 receptor |
| i.p. | intraperitoneal |
| i.g. | intragastric |
References
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology. Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Sengupta, U.; Kayed, R. Amyloid β, Tau, and α-Synuclein aggregates in the pathogenesis, prognosis, cand therapeutics for neurodegenerative diseases. Prog. Neurobiol. 2022, 214, 102270. [Google Scholar] [CrossRef]
- Rostagno, A.A. Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 24, 107. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Nijveldt, R.J.; van Nood, E.; van Hoorn, D.E.C.; Boelens, P.G.; van Norren, K.; van Leeuwen, P.A.M. Flavonoids: A review of probable mechanisms of action and potential applications. Am. J. Clin. Nutr. 2001, 74, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Rebas, E.; Rzajew, J.; Radzik, T.; Zylinska, L. Neuroprotective Polyphenols: A Modulatory Action on Neurotransmitter Pathways. Curr. Neuropharmacol. 2020, 18, 431–445. [Google Scholar] [CrossRef]
- Mezzomo, T.R.; Nadal, J. Effect of nutrients and dietary substances on thyroid function and hypothyroidism. Demetra Food Nutr. Health 2016, 11, 427–443. [Google Scholar] [CrossRef]
- Youdim, K.A.; Shukitt-Hale, B.; Joseph, J.A. Flavonoids and the brain: Interactions at the blood-brain barrier and their physiological effects on the central nervous system. Free Radic. Biol. Med. 2004, 37, 1683–1693. [Google Scholar] [CrossRef]
- Melrose, J. The Potential of Flavonoids and Flavonoid Metabolites in the Treatment of Neurodegenerative Pathology in Disorders of Cognitive Decline. Antioxidants 2023, 12, 663. [Google Scholar] [CrossRef] [PubMed]
- Amone, F.; Spina, A.; Perri, A.; Lofaro, D.; Zaccaria, V.; Insolia, V.; Lirangi, C.; Puoci, F.; Nobile, V. Standardized Grape (Vitis vinifera L.) Extract Improves Short- and Long-Term Cognitive Performances in Healthy Older Adults: A Randomized, Double-Blind, and Placebo-Controlled Trial. Foods 2024, 13, 2999. [Google Scholar] [CrossRef]
- Parekh, P.; Serra, M.; Allaw, M.; Perra, M.; Marongiu, J.; Tolle, G.; Pinna, A.; Casu, M.A.; Manconi, M.; Caboni, P.; et al. Characterization of Nasco grape pomace-loaded nutriosomes and their neuroprotective effects in the MPTP mouse model of Parkinson’s disease. Front. Pharmacol. 2022, 13, 935784. [Google Scholar] [CrossRef] [PubMed]
- Dessì, D.; Fais, G.; Follesa, P.; Sarais, G. Neuroprotective Effects of Myrtle Berry By-Product Extracts on 6-OHDA-Induced Cytotoxicity in PC12 Cells. Antioxidants 2025, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, J.; Gu, Z.; Ouyang, T.; Gao, H.; Kan, H.; Yang, Y. Comprehensive Exploration of the Neuroprotective Mechanisms of Ginkgo biloba Leaves in Treating Neurological Disorders. Am. J. Chin. Med. 2024, 52, 1053–1086. [Google Scholar] [CrossRef]
- Dias, M.C.; Pinto, D.C.G.A.; Silva, A.M.S. Plant Flavonoids: Chemical Characteristics and Biological Activity. Molecules 2021, 26, 5377. [Google Scholar] [CrossRef]
- Li, Z.; Chu, S.; He, W.; Zhang, Z.; Liu, J.; Cui, L.; Yan, X.; Li, D.; Chen, N. A20 as a novel target for the anti-neuroinflammatory effect of chrysin via inhibition of NF-κB signaling pathway. Brain Behav. Immun. 2019, 79, 228–235. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, G.; Szeto, S.S.W.; Chong, C.M.; Quan, Q.; Huang, C.; Chu, I.K. Examining the neuroprotective effects of protocatechuic acid and chrysin on in vitro and in vivo models of Parkinson disease. Free Radic. Biol. Med. 2015, 84, 331–343. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Pyrgelis, E.S.; Piperi, C. Neuroprotective potential of chrysin in Parkinson’s disease: Molecular mechanisms and clinical implications. Neurochem. Int. 2020, 132, 104612. [Google Scholar] [CrossRef]
- Goes, A.T.R.; Jesse, C.R.; Antunes, M.S.; Lobo Ladd, F.V.; Lobo Ladd, A.A.B.; Luchese, C.; Boeira, S.P. Protective role of chrysin on 6-hydroxydopamine-induced neurodegeneration a mouse model of Parkinson’s disease: Involvement of neuroinflammation and neurotrophins. Chem. Biol. Interact. 2018, 279, 111–120. [Google Scholar] [CrossRef]
- Krishnamoorthy, A.; Sevanan, M.; Mani, S.; Balu, M.; Balaji, S.P.R. Chrysin restores MPTP induced neuroinflammation, oxidative stress and neurotrophic factors in an acute Parkinson’s disease mouse model. Neurosci. Lett. 2019, 709, 134382. [Google Scholar] [CrossRef] [PubMed]
- Zeinali, M.; Rezaee, S.A.; Hosseinzadeh, H. An overview on immunoregulatory and anti-inflammatory properties of chrysin and flavonoids substances. Biomed. Pharmacother. 2017, 92, 998–1009. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, R.; Zhou, Y.; Huang, S.; Hou, Y.; Pei, G. Dietary Flavonoid Chrysin Functions as a Dual Modulator to Attenuate Amyloid-beta and Tau Pathology in the Models of Alzheimer’s Disease. Mol. Neurobiol. 2024, 62, 4274–4291. [Google Scholar] [CrossRef]
- Bortolotto, V.C.; Araujo, S.M.; Pinheiro, F.C.; Poetini, M.R.; de Paula, M.T.; Meichtry, L.B.; de Almeida, F.P.; Musachio, E.A.S.; Guerra, G.P.; Prigol, M. Modulation of glutamate levels and Na(+),K(+)-ATPase activity contributes to the chrysin memory recovery in hypothyroidism mice. Physiol. Behav. 2020, 222, 112892. [Google Scholar] [CrossRef]
- Samarghandian, S.; Farkhondeh, T.; Azimi-Nezhad, M. Protective Effects of Chrysin Against Drugs and Toxic Agents. Dose-response 2017, 15, 1559325817711782. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Kempuraj, D.D.; Ahmed, M.E.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, S.A.; Iyer, S.S.; Govindarajan, R.; Chandrasekaran, P.N.; et al. Neuroprotective effects of flavone luteolin in neuroinflammation and neurotrauma. BioFactors 2020, 47, 190–197. [Google Scholar] [CrossRef]
- Kou, J.J.; Shi, J.Z.; He, Y.Y.; Hao, J.J.; Zhang, H.Y.; Luo, D.M.; Song, J.K.; Yan, Y.; Xie, X.M.; Du, G.H.; et al. Luteolin alleviates cognitive impairment in Alzheimer’s disease mouse model via inhibiting endoplasmic reticulum stress-dependent neuroinflammation. Acta Pharmacol. Sin. 2022, 43, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.H.; Bi, W.; Qi, R.B.; Wang, H.D.; Lu, D.X. Luteolin inhibits microglial inflammation and improves neuron survival against inflammation. Int. J. Neurosci. 2011, 121, 329–336. [Google Scholar] [CrossRef]
- Nakagawa, T.T. Luteolin ameliorates depression-like behaviors by suppressing ER stress in a mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2022, 588, 168–174. [Google Scholar]
- Tao, X.; Zhang, R.; Wang, L.; Li, X.; Gong, W. Luteolin and Exercise Combination Therapy Ameliorates Amyloid-β1-42 Oligomers-Induced Cognitive Impairment in AD Mice by Mediating Neuroinflammation and Autophagy. J. Alzheimers Dis. 2023, 92, 195–208. [Google Scholar] [CrossRef]
- Chen, H.Q.; Jin, Z.Y.; Wang, X.J.; Xu, X.M.; Deng, L.; Zhao, J.W. Luteolin protects dopaminergic neurons from inflammation-induced injury through inhibition of microglial activation. Neurosci. Lett. 2008, 448, 175–179. [Google Scholar] [CrossRef]
- Akinrinde, A.S.; Adebiyi, O.E. Neuroprotection by luteolin and gallic acid against cobalt chloride-induced behavioural, morphological and neurochemical alterations in Wistar rats. Neurotoxicology 2019, 74, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Wittemer, S.M.; Ploch, M.; Windeck, T.; Muller, S.C.; Drewelow, B.; Derendorf, H.; Veit, M. Bioavailability and pharmacokinetics of caffeoylquinic acids and flavonoids after oral administration of Artichoke leaf extracts in humans. Phytomedicine 2005, 12, 28–38. [Google Scholar] [CrossRef]
- Yang, Y.; Gong, X.B.; Huang, L.G.; Wang, Z.X.; Wan, R.Z.; Zhang, P.; Zhang, Q.Y.; Chen, Z.; Zhang, B.S. Diosmetin exerts anti-oxidative, anti-inflammatory and anti-apoptotic effects to protect against endotoxin-induced acute hepatic failure in mice. Oncotarget 2017, 8, 30723–30733. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, Y.; Jiang, Y.; Lu, D. Diosmetin Suppresses Neuronal Apoptosis and Inflammation by Modulating the Phosphoinositide 3-Kinase (PI3K)/AKT/Nuclear Factor-κB (NF-κB) Signaling Pathway in a Rat Model of Pneumococcal Meningitis Diosmetin in a rat model of pneumococcal meningitis. Med. Sci. Monit. 2019, 25, 2238–2245. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Lian, D.; Zhang, Z.; Liu, Y.; Sun, J.; Li, L. Brain-derived neurotrophic factor is regulated via MyD88/NF-kB signaling in experimental Streptococcus pneumoniae meningitis. Sci. Rep. 2017, 7, 3545. [Google Scholar] [CrossRef]
- Varshney, K.K.; Gupta, J.K.; Srivastava, R. Investigating In Silico and In Vitro Therapeutic Potential of Diosmetin as the Anti-Parkinson Agent. Protein Pept. Lett. 2024, 31, 714–735. [Google Scholar] [CrossRef]
- Carradori, S.; Gidaro, M.C.; Petzer, A.; Costa, G.; Guglielmi, P.; Chimenti, P.; Alcaro, S.; Petzer, J.P. Inhibition of Human Monoamine Oxidase: Biological and Molecular Modeling Studies on Selected Natural Flavonoids. J. Agric. Food Chem. 2016, 64, 9004–9011. [Google Scholar] [CrossRef]
- Singh, A.; Sinha, S.; Singh, N.K. Dietary Natural Flavonoids: Intervention for MAO-B Against Parkinson’s Disease. Chem. Biol. Drug Des. 2024, 104, e14619. [Google Scholar] [CrossRef] [PubMed]
- Sawmiller, D.; Habib, A.; Li, S.; Darlington, D.; Hou, H.; Tian, J.; Shytle, R.D.; Smith, A.; Giunta, B.; Mori, T.; et al. Diosmin reduces cerebral Aβ levels, tau hyperphosphorylation, neuroinflammation, and cognitive impairment in the 3xTg-AD mice. J. Neuroimmunol. 2016, 299, 98–106. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Davra, V.; Benzeroual, K.E. Flavonoids and fibrate modulate apoE4-induced processing of amyloid precursor protein in neuroblastoma cells. Front. Neurosci. 2023, 17, 1245895. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Liu, W.Y.; Liou, S.-S.; Liu, I.-M. Diosmetin Targeted at Peroxisome Proliferator-Activated Receptor Gamma Alleviates Advanced Glycation End Products Induced Neuronal Injury. Nutrients 2022, 14, 2248. [Google Scholar] [CrossRef] [PubMed]
- Dourado, N.S.; Souza, C.S.; de Almeida, M.M.A.; Bispo da Silva, A.; dos Santos, B.L.; Silva, V.D.A.; De Assis, A.M.; da Silva, J.S.; Souza, D.O.; Costa, M.F.D.; et al. Neuroimmunomodulatory and Neuroprotective Effects of the Flavonoid Apigenin in in vitro Models of Neuroinflammation Associated With Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 119. [Google Scholar] [CrossRef]
- Anusha, C.; Sumathi, T.; Joseph, L.D. Protective role of apigenin on rotenone induced rat model of Parkinson’s disease: Suppression of neuroinflammation and oxidative stress mediated apoptosis. Chem. Biol. Interact. 2017, 269, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Yarim, G.F.; Kazak, F.; Yarim, M.; Sozmen, M.; Genc, B.; Ertekin, A.; Gokceoglu, A. Apigenin alleviates neuroinflammation in a mouse model of Parkinson’s disease. Int. J. Neurosci. 2022, 26, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; An, J.; Zhang, J.; Zhao, J.; Sun, P.; He, Z. Network pharmacology combined with experimental validation show that apigenin as the active ingredient of Campsis grandiflora flower against Parkinson’s disease by inhibiting the PI3K/AKT/NF-κB pathway. PLoS ONE 2024, 19, e0311824. [Google Scholar] [CrossRef]
- Siddique, Y.H.; Jyoti, S. Alteration in biochemical parameters in the brain of transgenic Drosophila melanogaster model of Parkinson’s disease exposed to apigenin. Integr. Med. Res. 2017, 6, 245–253. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Andres, S.; Pevny, S.; Ziegenhagen, R.; Bakhiya, N.; Schafer, B.; Hirsch-Ernst, K.I.; Lampen, A. Safety Aspects of the Use of Quercetin as a Dietary Supplement. Mol. Nutr. Food Res. 2018, 62, 170047. [Google Scholar] [CrossRef]
- Deepika; Maurya, P.K. Health Benefits of Quercetin in Age-Related Diseases. Molecules 2022, 27, 2498. [Google Scholar] [CrossRef]
- Alizadeh, S.R.; Ebrahimzadeh, M.A. Quercetin derivatives: Drug design, development, and biological activities, a review. Eur. J. Med. Chem. 2022, 229, 114068. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Du, G.; Wu, H.; Gao, X.; Yang, Z.; Liu, B.; Cui, S. Protective effects of quercetin on traumatic brain injury induced inflammation and oxidative stress in cortex through activating Nrf2/HO-1 pathway. Restor. Neurol. Neurosci. 2021, 39, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lu, C.; Wang, T.; Qiao, C.; Lu, L.; Wu, D.; Lu, M.; Chen, R.; Fan, L.; Tang, J. Hyperoside suppresses NLRP3 inflammasome in Parkinson’s disease via Pituitary Adenylate Cyclase-Activating Polypeptide. Neurochem. Int. 2022, 152, 105254. [Google Scholar] [CrossRef] [PubMed]
- Jain, J.; Hasan, W.; Biswas, P.; Yadav, R.S.; Jat, D. Neuroprotective effect of quercetin against rotenone-induced neuroinflammation and alterations in mice behavior. J. Biochem. Mol. Toxicol. 2022, 36, e23165. [Google Scholar] [CrossRef] [PubMed]
- Underwood, E.J. Trace metals in human and animal health. J. Hum. Nutr. 1981, 35, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, Y.; Zhang, N.; Fitsanakis, V.A.; Avison, M.J.; Gore, J.C.; Aschner, M. Differential deposition of manganese in the rat brain following subchronic exposure to manganese: A T1-weighted magnetic resonance imaging study. Isr. Med. Assoc. J. 2008, 10, 793–798. [Google Scholar] [PubMed]
- Bahar, E.; Lee, G.H.; Bhattarai, K.R.; Lee, H.Y.; Choi, M.K.; Rashid, H.O.; Kim, J.Y.; Chae, H.J.; Yoon, H. Polyphenolic extract of euphorbia supina attenuates manganese-induced neurotoxicity by enhancing antioxidant activity through regulation of ER stress and ER stress-mediated apoptosis. Int. J. Mol. Sci. 2017, 18, 300. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, J.Y.; Yoon, H. Quercetin Attenuates Manganese-Induced Neuroinflammation by Alleviating Oxidative Stress through Regulation of Apoptosis, iNOS/NF-κB and HO-1/Nrf2 Pathways. Int. J. Mol. Sci. 2017, 18, 1989. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, G.Y.; Chen, Z.; Jasmer, K.J.; Chuang, D.Y.; Gu, Z.; Hannink, M.; Simonyi, A. Quercetin Attenuates Inflammatory Responses in BV-2 Microglial Cells: Role of MAPKs on the Nrf2 Pathway and Induction of Heme Oxygenase-1. PLoS ONE 2015, 10, e0141509. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bayazid, A.B.; Lim, B.O. Quercetin Is An Active Agent in Berries against Neurodegenerative Diseases Progression through Modulation of Nrf2/HO1. Nutrients 2022, 14, 5132. [Google Scholar] [CrossRef]
- Lu, J.; Wu, D.M.; Zheng, Y.L.; Hu, B.; Zhang, Z.F.; Shan, Q.; Zheng, Z.H.; Liu, C.M.; Wang, Y.J. Quercetin activates AMP-activated protein kinase by reducing PP2C expression protecting old mouse brain against high cholesterol-induced neurotoxicity. J. Pathol. 2010, 222, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Ono, K.; Murase, A.; Yamada, M. Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-beta aggregation pathway. Am. J. Pathol. 2009, 175, 2557–2565. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Joshi, V.; Mishra, R.; Upadhyay, A.; Amanullah, A.; Poluri, K.M.; Singh, S.; Kumar, A.; Mishra, A. Polyphenolic flavonoid (Myricetin) upregulated proteasomal degradation mechanisms: Eliminates neurodegenerative proteins aggregation. J. Cell Physiol. 2019, 234, 20900–20914. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, Z.; Li, B.; Peng, Y.; Yang, X.; Xiao, Y.; Ni, R.; Qi, X.L. Myricetin ameliorates cognitive impairment in 3×Tg Alzheimer’s disease mice by regulating oxidative stress and tau hyperphosphorylation. Biomed. Pharmacother. 2024, 177, 116963. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2′ -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 120–139. [Google Scholar] [CrossRef] [PubMed]
- Shoeb, M.; Ansari, N.H.; Srivastava, S.K.; Ramana, K.V. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 2014, 21, 230–237. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jang, J.H.; Lee, S.H.; Jung, K.; Yoo, H.; Park, G. Inhibitory Effects of Myricetin on Lipopolysaccharide-Induced Neuroinflammation. Brain Sci. 2020, 10, 32. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Boriero, D.; Carcereri de Prati, A.; Antonini, L.; Ragno, R.; Sohji, K.; Mariotto, S.; Butturini, E. The anti-STAT1 polyphenol myricetin inhibits M1 microglia activation and counteracts neuronal death. FEBS J. 2021, 288, 2347–2359. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.C.; Xie, Z.G.; Gu, M.J.; Wang, C.D.; Xu, L.M.; Gao, C.; Yuan, X.L.; Wu, Y.; Hu, Y.Q.; Cao, Y.; et al. Myricetin mitigates motor disturbance and decreases neuronal ferroptosis in a rat model of Parkinson’s disease. Sci. Rep. 2024, 14, 15107. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chang, Y.; Chang, C.Y.; Wang, S.J.; Huang, S.K. Myricetin inhibits the release of glutamate in rat cerebrocortical nerve terminals. J. Med. Food 2015, 18, 516–523. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Musial, C.; Kuban-Jankowska, A.; Gorska-Ponikowska, M. Beneficial Properties of Green Tea Catechins. Int. J. Mol. Sci. 2020, 21, 1744. [Google Scholar] [CrossRef]
- Mao, L.; Hochstetter, D.; Yao, L.; Zhao, Y.; Zhou, J.; Wang, Y.; Xu, P. Green Tea Polyphenol (−)-Epigallocatechin Gallate (EGCG) Attenuates Neuroinflammation in Palmitic Acid-Stimulated BV-2 Microglia and High-Fat Diet-Induced Obese Mice. Int. J. Mol. Sci. 2019, 20, 5081. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yang, C.; Si, N.; Chu, T.; Yu, J.; Yuan, X.; Chen, X.T. Epigallocatechin-3-gallate Inhibits LPS/AβO-induced Neuroinflammation in BV2 Cells through Regulating the ROS/TXNIP/NLRP3 Pathway. J. Neuroimmune Pharmacol. 2024, 19, 31. [Google Scholar] [CrossRef] [PubMed]
- Nan, S.; Wang, P.; Zhang, Y.; Fan, J. Epigallocatechin-3-Gallate Provides Protection Against Alzheimer’s Disease-Induced Learning and Memory Impairments in Rats. Drug Des. Devel Ther. 2021, 15, 2013–2024. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shen, J.; Xie, J.; Ye, L.; Mao, J.; Sun, S.; Chen, W.; Wei, S.; Ruan, S.; Wang, L.; Hu, H.; et al. Neuroprotective effect of green tea extract (-)-epigallocatechin-3-gallate in a preformed fibril-induced mouse model of Parkinson’s disease. Neuroreport 2024, 35, 421–430. [Google Scholar] [CrossRef]
- Özduran, G.; Becer, E.; Vatansever, H.S.; Yücecan, S. Neuroprotective effects of catechins in an experimental Parkinson’s disease model and SK-N-AS cells: Evaluation of cell viability, anti-inflammatory and anti-apoptotic effects. Neurol. Res. 2022, 44, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Bawono, L.C.; Khairinisa, M.A.; Jiranusornkul, S.; Levita, J. The role of catechins of Camellia sinensis leaves in modulating antioxidant enzymes: A review and case study. J. Appl. Pharm. Sci. 2023, 13, 052–065. [Google Scholar] [CrossRef]
- Li, S.; Wang, Z.; Liu, G.; Chen, M. Neurodegenerative diseases and catechins: (−)-epigallocatechin-3-gallate is a modulator of chronic neuroinflammation and oxidative stress. Front. Nutr. 2024, 11, 1425839. [Google Scholar] [CrossRef]
- Islam, A.; Islam, M.S.; Rahman, M.K.; Uddin, M.N.; Akanda, M.R. The pharmacological and biological roles of eriodictyol. Arch. Pharm. Res. 2020, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Yan, S.; Wen, X.; Zhang, S.; Liu, Z.; Liu, X.; Xiao, C. Eriodictyol alleviates lipopolysaccharide-triggered oxidative stress and synaptic dysfunctions in BV-2 microglial cells and mouse brain. J. Cell Biochem. 2019, 120, 14756–14770. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Yan, S.; Zheng, J.; Gao, Y.; Zhang, S.; Liu, Z.; Liu, X.; Xiao, C. Eriodictyol Attenuates LPS-Induced Neuroinflammation, Amyloidogenesis, and Cognitive Impairments via the Inhibition of NF-κB in Male C57BL/6J Mice and BV2 Microglial Cells. J. Agric. Food Chem. 2018, 66, 10205–10214. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Shi, H.; Zhu, X.; Wei, X.; Ren, M.; Han, M.; Ren, D.; Lou, H. Eriodictyol Attenuates β-Amyloid 25-35 Peptide-Induced Oxidative Cell Death in Primary Cultured Neurons by Activation of Nrf2. Neurochem. Res. 2015, 40, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Zeng, M.; Wang, S.; Cao, B.; Liu, M.; Zhang, Y.; Jia, J.; Zhang, Q.; Zhang, B.; Wang, R.; et al. Eriodictyol and Homoeriodictyol Improve Memory Impairment in Aβ25–35-Induced Mice by Inhibiting the NLRP3 Inflammasome. Molecules 2022, 27, 2488. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, W.J.; Zheng, X.R.; Liu, Q.L.; Du, Q.; Lai, Y.J.; Liu, S.Q. Eriodictyol ameliorates cognitive dysfunction in APP/PS1 mice by inhibiting ferroptosis via vitamin D receptor-mediated Nrf2 activation. Mol. Med. 2022, 28, 11. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Křížová, L.; Dadáková, K.; Kašparovská, J.; Kašparovský, T. Isoflavones. Molecules 2019, 24, 1076. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sarfraz, A.; Javeed, M.; Shah, M.A.; Hussain, G.; Shafiq, N.; Sarfraz, I.; Riaz, A.; Sadiqa, A.; Zara, R.; Zafar, S.; et al. Biochanin A: A novel bioactive multifunctional compound from nature. Sci. Total Environ. 2020, 722, 137907. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.Y.; Wu, Y.Y.; Huang, H.; He, C.; Li, W.Z.; Wang, H.L.; Chen, H.Q.; Yin, Y.Y. Biochanin A attenuates LPS-induced pro-inflammatory responses and inhibits the activation of the MAPK pathway in BV2 microglial cells. Int. J. Mol. Med. 2015, 35, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Q.; Jin, Z.Y.; Li, G.H. Biochanin A protects dopaminergic neurons against lipopolysaccharide-induced damage through inhibition of microglia activation and proinflammatory factors generation. Neurosci. Lett. 2007, 417, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, C.; Wu, W.Y.; Chen, F.; Wu, Y.Y.; Li, W.Z.; Chen, H.Q.; Yin, Y.Y. Biochanin A protects dopaminergic neurons against lipopolysaccharide-induced damage and oxidative stress in a rat model of Parkinson’s disease. Pharmacol. Biochem. Behav. 2015, 138, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, W.Y.; Huang, H.; Li, W.Z.; Chen, H.Q.; Yin, Y.Y. Biochanin A Protects Against Lipopolysaccharide-Induced Damage of Dopaminergic Neurons Both In Vivo and In Vitro via Inhibition of Microglial Activation. Neurotox. Res. 2016, 30, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.X.; Kong, H.; Yu, Y.G.; Zhou, J.W.; Chen, H.Q.; Yin, Y.Y. Biochanin A protects against angiotensin II-induced damage of dopaminergic neurons in rats associated with the increased endophilin A2 expression. Behav. Pharmacol. 2019, 30, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhao, W.; Yu, H.; Zhang, F.; Zhang, H.T.; Zhou, Y. Biochanin A alleviates cognitive impairment and hippocampal mitochondrial damage in ovariectomized APP/PS1 mice. Phytomedicine 2022, 100, 154056. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, B.; Yu, H.; Zhao, W.; Song, X.; Liu, Y.; Wang, K.; Peacher, N.; Zhao, X.; Zhang, H.T. Biochanin A Attenuates Ovariectomy-Induced Cognition Deficit via Antioxidant Effects in Female Rats. Front. Pharmacol. 2021, 12, 603316. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Won, J.P.; Yoon, H.J.; Lee, H.G.; Seo, H.G. Biochanin A inhibits excitotoxicity-triggered ferroptosis in hippocampal neurons. Eur. J. Pharmacol. 2024, 985, 177104. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.W.; Tham, C.L.; Israf, D.A.; Lee, S.H.; Kim, M.K. Neuroprotective effects of biochanin A against glutamate-induced cytotoxicity in PC12 cells via apoptosis inhibition. Neurochem. Res. 2013, 38, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.W.; Kim, M.K. Neuroprotective Effects of Biochanin A against β-Amyloid-Induced Neurotoxicity in PC12 Cells via a Mitochondrial-Dependent Apoptosis Pathway. Molecules 2016, 21, 548. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zarmouh, N.O.; Eyunni, S.K.; Soliman, K.F. The Benzopyrone Biochanin-A as a reversible, competitive, and selective monoamine oxidase B inhibitor. BMC Complement. Altern. Med. 2017, 17, 34. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Youn, K.; Park, J.H.; Lee, J.; Jeong, W.S.; Ho, C.T.; Jun, M. The Identification of Biochanin A as a Potent and Selective β-Site App-Cleaving Enzyme 1 (Bace1) Inhibitor. Nutrients 2016, 8, 637. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Manach, C.; Scalbert, A.; Moran, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef]
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 29, 162750. [Google Scholar] [CrossRef]
- Hu, L.; Luo, Y.; Yang, J.; Cheng, C. Botanical Flavonoids: Efficacy, Absorption, Metabolism and Advanced Pharmaceutical Technology for Improving Bioavailability. Molecules 2025, 30, 1184. [Google Scholar] [CrossRef] [PubMed]
- Açar, Y.; Ağagündüz, D.; De Cicco, P.; Capasso, R. Flavonoids: Their putative neurologic roles, epigenetic changes, and gut microbiota alterations in Parkinson’s disease. Biomed. Pharmacother. 2023, 168, 115788. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Class of Flavonoids | Name of Compound | Sources | Structure | Range of Effective Concentration/Dose | Affected Components Involved in Neuroprotection |
|---|---|---|---|---|---|
| Flavones | Chrysin | Honey, propolis, passion flowers, carrots, and chamomile | 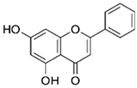 | In vitro: 5–10 μM In vivo: Orally: 25 and 50 mg/kg for 4 days i.p. 25 mg/kg/day | ↓NF-κB, ↓NO, ↓IL-6, ↓TNF-α, TRL-4, ↓COX-2, ↓iNOS, ↓MPO ↑IL-4, ↑IL-10 ↑Nrf2, ↑SOD, ↑CAT, HO-1 ↓Aβ, ↓p-Tau, ↓BACE-1, ↓GSK3β, ↓glutamate, ↓AChE |
| Luteolin | Celery, broccoli, artichoke, green pepper, parsley, thyme, dandelion, perilla, chamomile tea, carrots, olive oil, peppermint, rosemary, navel oranges, and oregano | 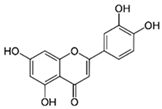 | In vitro: 1–25 μM In vivo: i.p. 20, 100 mg/kg/d i.p. 20 mg/kg/d Orally: 30–100 mg/kg/day | ↓NF-κB, ↓NO, ↓IL-6, ↓IL-β1, ↓TNF-α, ↓COX-2, ↓iNOS, ↓GRP78, ↓IRE1, ↓GFAP ↑Nrf2, ↑SOD, ↑GST ↓Aβ, ↓p-Tau, ↑dopamine, ↑tyrosine hydroxylase | |
| Diosmetin | Caucasian vetch |  | In vitro: 2.5–50 μM Orally: 100–200 mg/kg/day | ↓PI3K/AKT pathway ↓NF-κB, ↓IL-6, ↓TNF-α, ↓IL-12 ↑Nrf2, ↑SOD, ↑CAT, ↑GSH-Px ↓Aβ, ↓p-Tau, ↑GSF-3β ↑Aβ phagocytosis, ↓ApoE4, ↓IFNγ, ↓CD40 ↓MAO-B, Trkβ ↑IDE, ↑NEP, ↑Aβ degradation | |
| Apigenin | Parsley, celery, celeriac, and chamomile tea |  | In vitro: 10–80 μM, In vivo: i.p. 10–50 mg/kg/day, Orally: 20 mg/kg/day for 30 days | PI3K/AKT ↓NF-κB, ↓IL-6, ↓TNF-α, ↓IL-1β, ↓iNOS, ↑BDNF, ↑GDNF ↑GSH, ↓lipid oxidation ROS scavenger ↑tyrosine hydroxylase ↑dopamine, ↓MAO-B ↓α-synuclein aggregation ↑caspase-3, ↑caspase-9 | |
| Flavonols | Quercetin | Capers, radish, red onion, radicchio, lovage, dock, and honey | 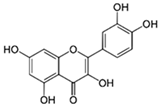 | In vitro: 10–30 μg/mL In vivo: i.p. 10–100 mg/kg/day Orally: 30–50 mg/kg/day 60 days | MAPK/ERK1/2 ↓NF-κB, ↓IL-6, ↓TNF-α, ↓IL-1β, ↓COX-2, ↓iNOS, ↑Nrf2/HO-1, ↑SOD, ↑CAT, ↑GSH-Px, ↑GSH ROS scavenger ↓Aβ, ↓p-Tau, ↓BACE-1, ↓NLRP3, ↓caspase-3, ↓caspase-1, ↓Bax, ↑Bcl-2, ↓cytochrome c |
| Myricetin | Vegetables, tea, nuts fruits, berries, red wine, and medical plants | 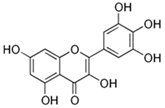 | In vitro: 10–50 μM In vivo: i.p. 10–100 mg/kg/day i.g. 25 mg/kg/day | ↓pMAPK/ERK1/2, ↓pJNK, ↓pp38, ↓IBA-1 ↓NF-κB/STAT1 ↓IL-6, ↓TNF-α, ↓IL-1β, ↓COX-2, ↓iNOS, ↓PGE2 ↑Nrf2, ↑GSH-Px, ↑GSH ↓Fe2+, ↓ferroptosis ↓Aβ, ↓GSK-3β, ↓α-synuclein, ↑tyrosine hydrolase, ↓glutamate ↓DNA oxidation ↓lipid oxidation | |
| Flavanol | Catechins | Tea, cacao, pome fruits, and wine | 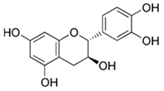 | In vitro: 5–20 μM | ↓NF-κB/STAT1, ↓pSTAT3, ↓pJak2 ↓IL-6, ↓TNF-α, ↓IL-1β, ↑Nrf2, ↑SOD, ↑CAT, ↑GSH-Px, ↑GSH ↓Aβ, ↓p-Tau, ↓BACE-1, ↓AChE, ↓caspase-3, ↑IL-10, ↑IL-4, ↑TGF-β |
| Flavanone | Eriodictyol | Yerba santa, citrus fruits, and peanuts |  | In vitro: 50 μM In vivo: i.g. 25–100 mg/kg/day | ↓NF-κB/MAPKs, ↓pERK, ↓pJak2, ↓p-p38, ↓IL-6, ↓TNF-α, ↓IL-1β, ↓COX-2, ↓iNOS ↑Nrf2/HO-1, ↑SOD, ↑CAT, ↑GSH-Px, ↑GSH, ↑GCS ↓ferroptosis ↓Aβ, ↓p-Tau, ↓BACE-1, ↓APP, ↓TRL4 ↓AChE, ↑ChAT ↓caspase-1, ↓IL-1, ↓IL-18 |
| Isoflavone | Biochanin A | Soya, red clover, alfalfa, chickpeas, fruits, vegetables, and nuts | 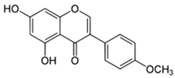 | In vitro: 1.25–5 μM | ↓NF-κB/MAPKs, ↓pERK, ↓pJNK, ↓p-p38, ↓IL-6, ↓TNF-α, ↓IL-1β, ↓COX-2, ↓iNOS ↑Nrf2, ↑SOD, ↑CAT, ↑GSH-Px, ↑GSH, ↓Fe2+, ↓ferroptosis ↓Aβ, ↓BACE-1, ↓MAO-B ↓NLRP3, ↓caspase-1, ↓caspase-3, caspase-8, caspase-9, ↓Bax, ↑Bcl-2 ↓cytochrome ↑EPA2 and ↓AT1R |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rębas, E. Role of Flavonoids in Protecting Against Neurodegenerative Diseases—Possible Mechanisms of Action. Int. J. Mol. Sci. 2025, 26, 4763. https://doi.org/10.3390/ijms26104763
Rębas E. Role of Flavonoids in Protecting Against Neurodegenerative Diseases—Possible Mechanisms of Action. International Journal of Molecular Sciences. 2025; 26(10):4763. https://doi.org/10.3390/ijms26104763
Chicago/Turabian StyleRębas, Elżbieta. 2025. "Role of Flavonoids in Protecting Against Neurodegenerative Diseases—Possible Mechanisms of Action" International Journal of Molecular Sciences 26, no. 10: 4763. https://doi.org/10.3390/ijms26104763
APA StyleRębas, E. (2025). Role of Flavonoids in Protecting Against Neurodegenerative Diseases—Possible Mechanisms of Action. International Journal of Molecular Sciences, 26(10), 4763. https://doi.org/10.3390/ijms26104763