
Genome-Wide Characterization of WRKY Gene Family in Camellia chekiangoleosa Identifies Potential Regulatory Components in Pigment Biosynthesis Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
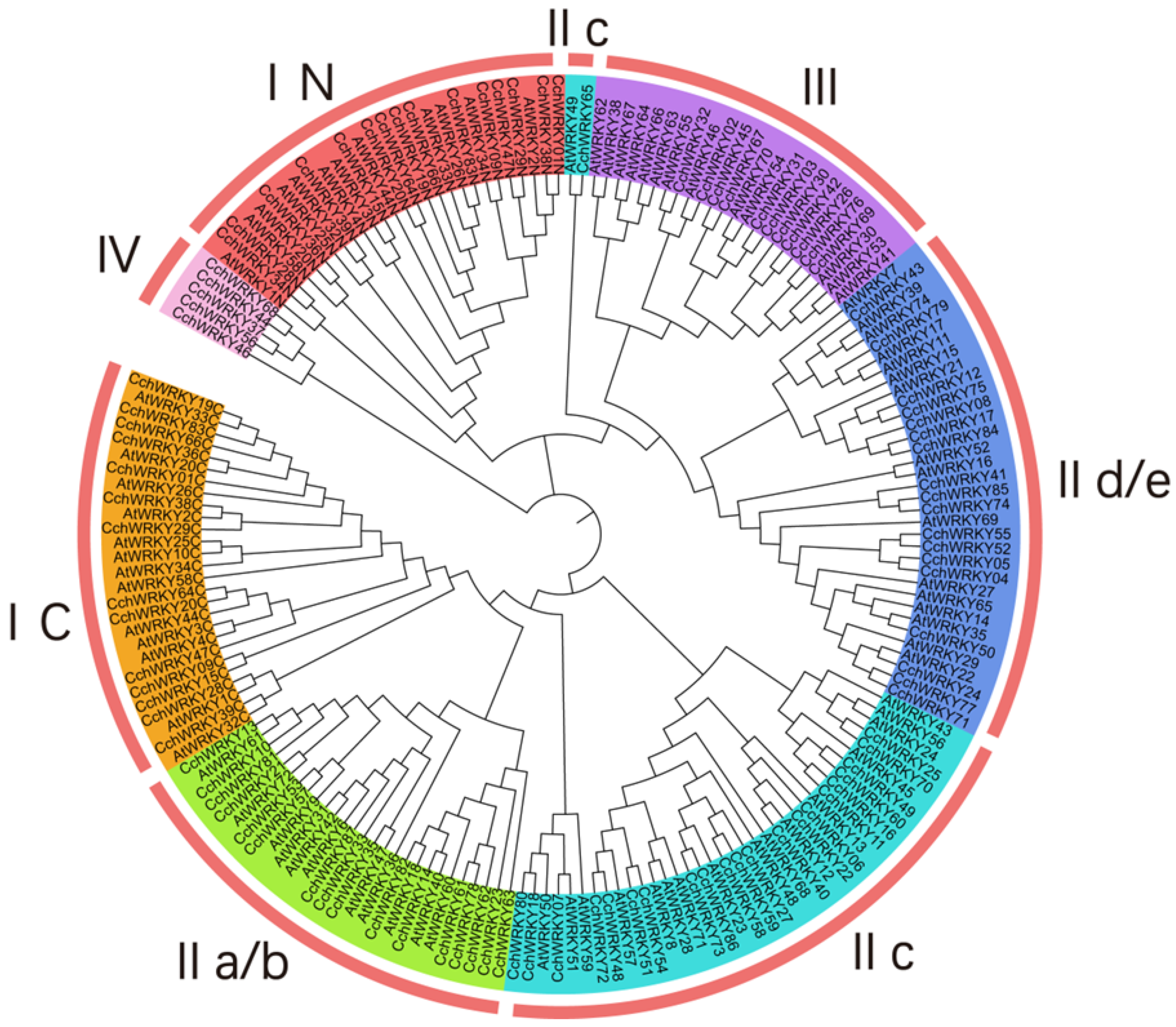
2.1. CchWRKY Protein Identification and Phylogenetic Analysis
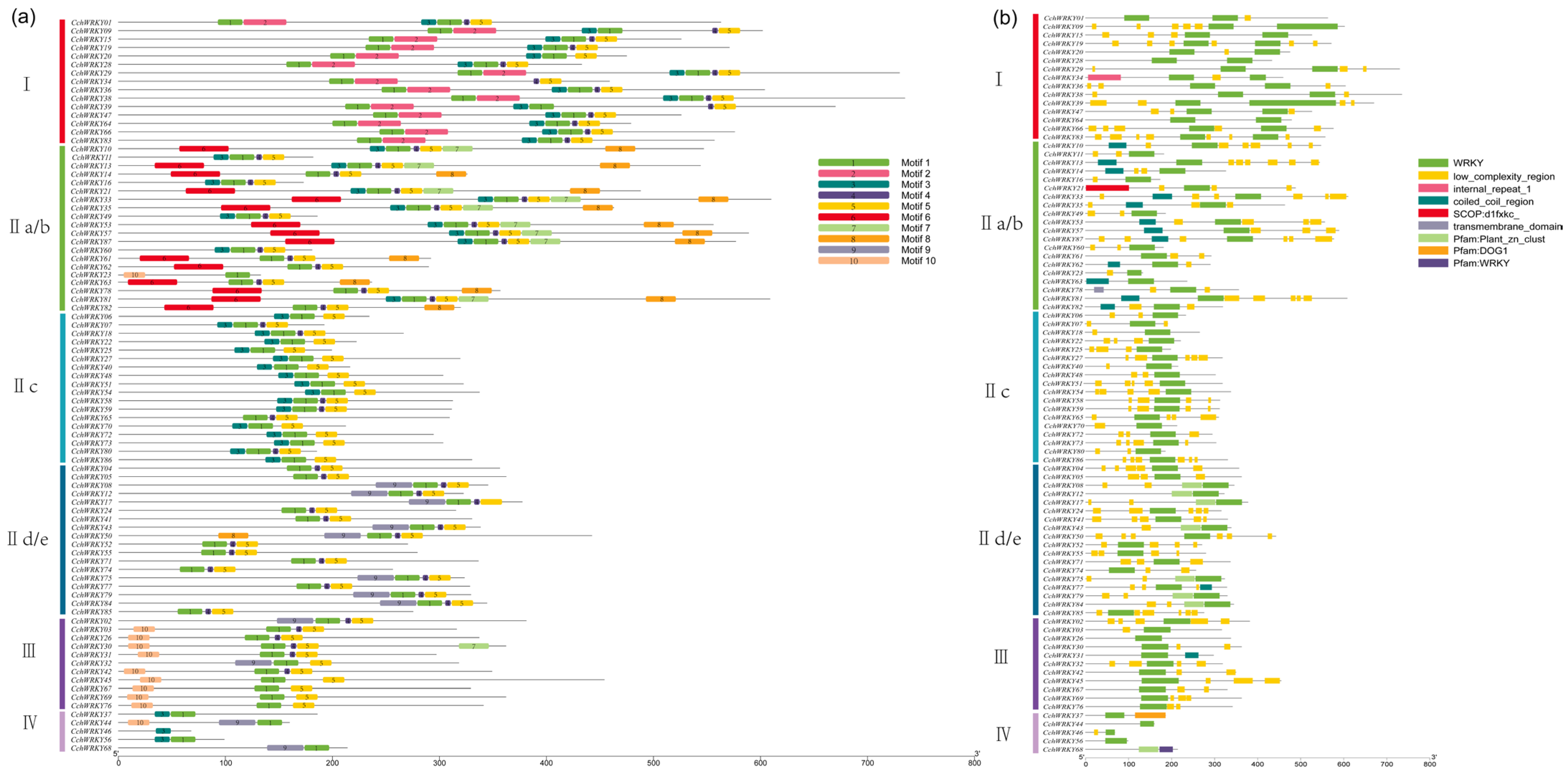
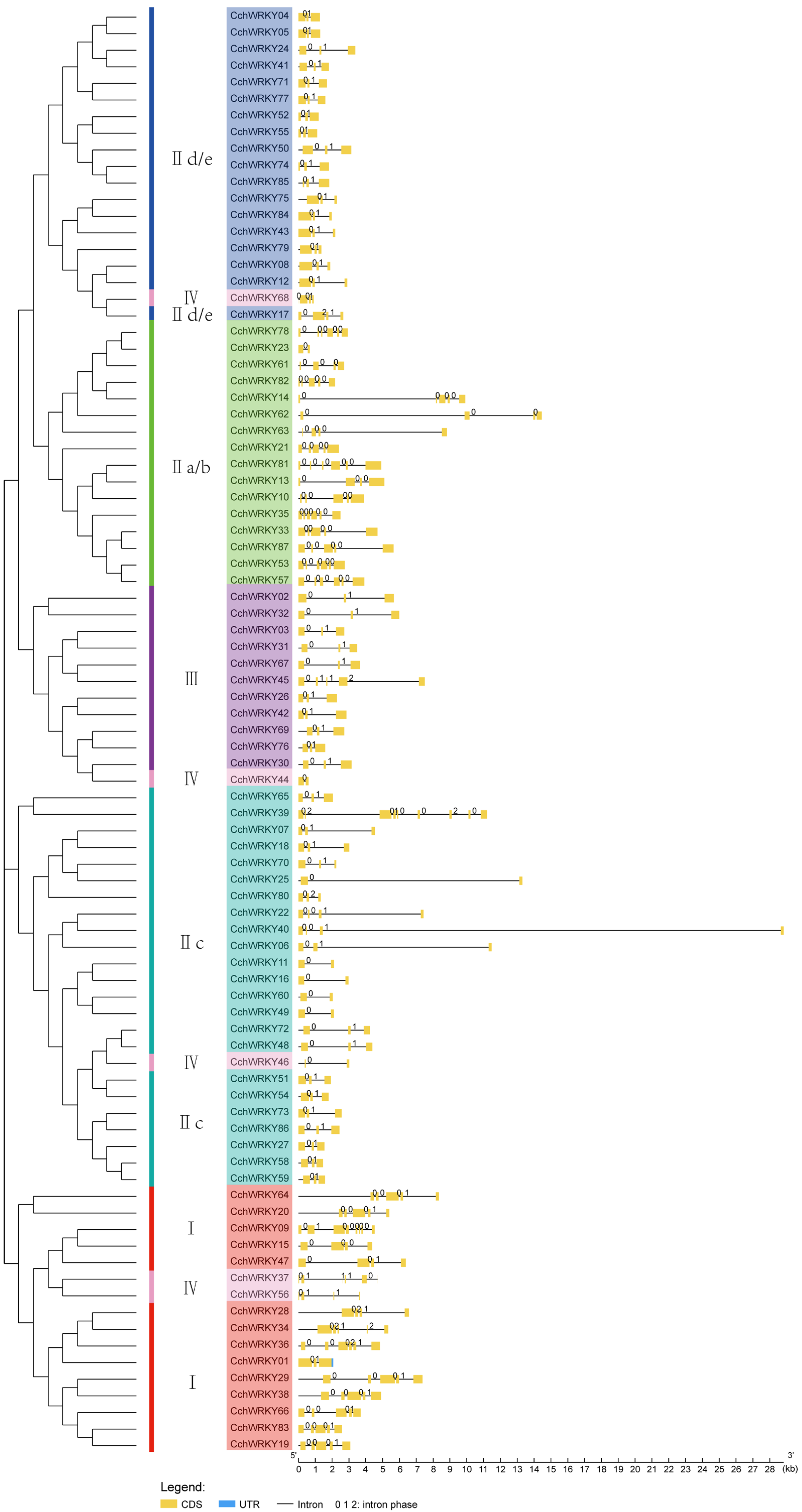
2.2. Examination of CchWRKY Gene Structures and Protein Conserved Motifs
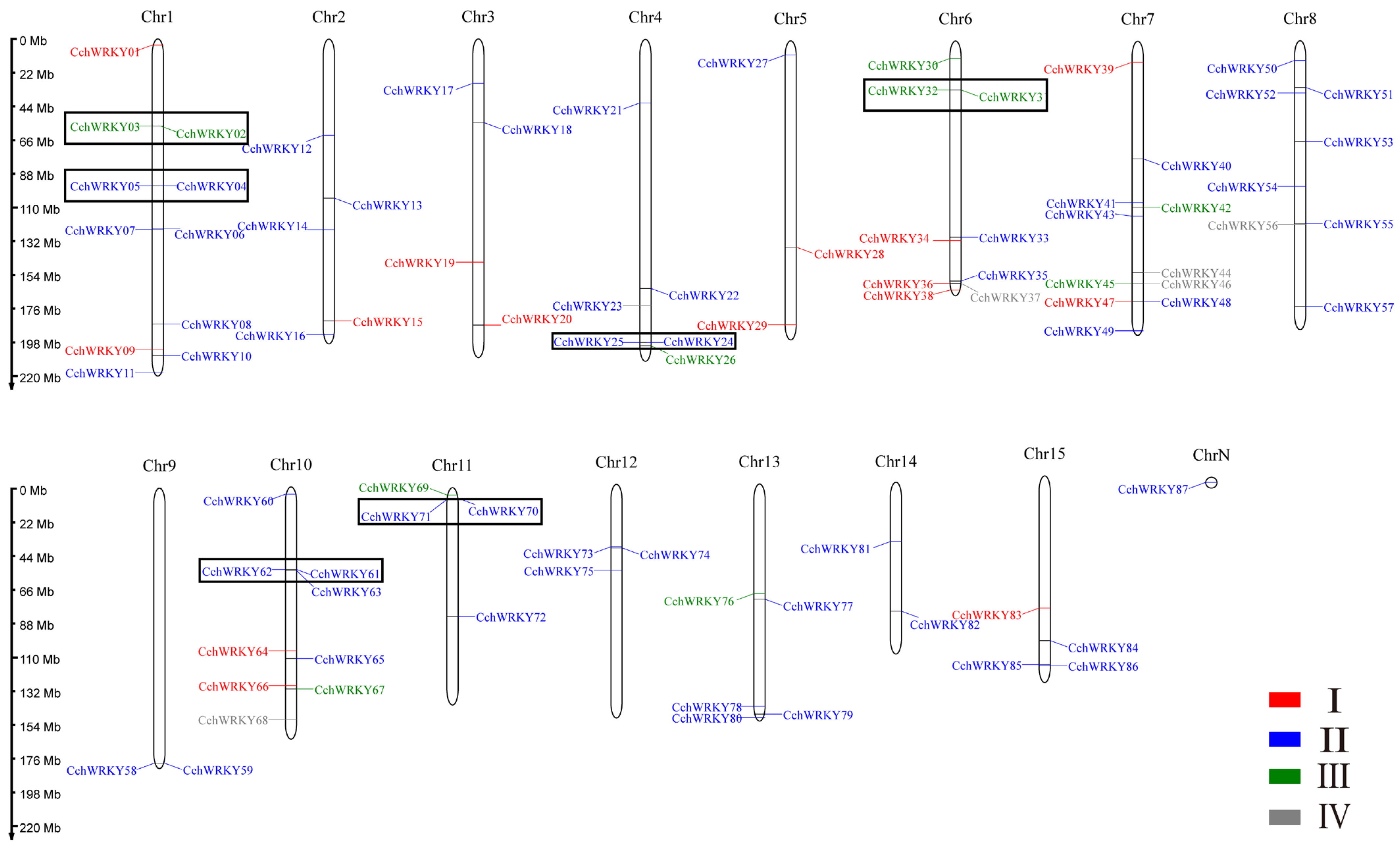
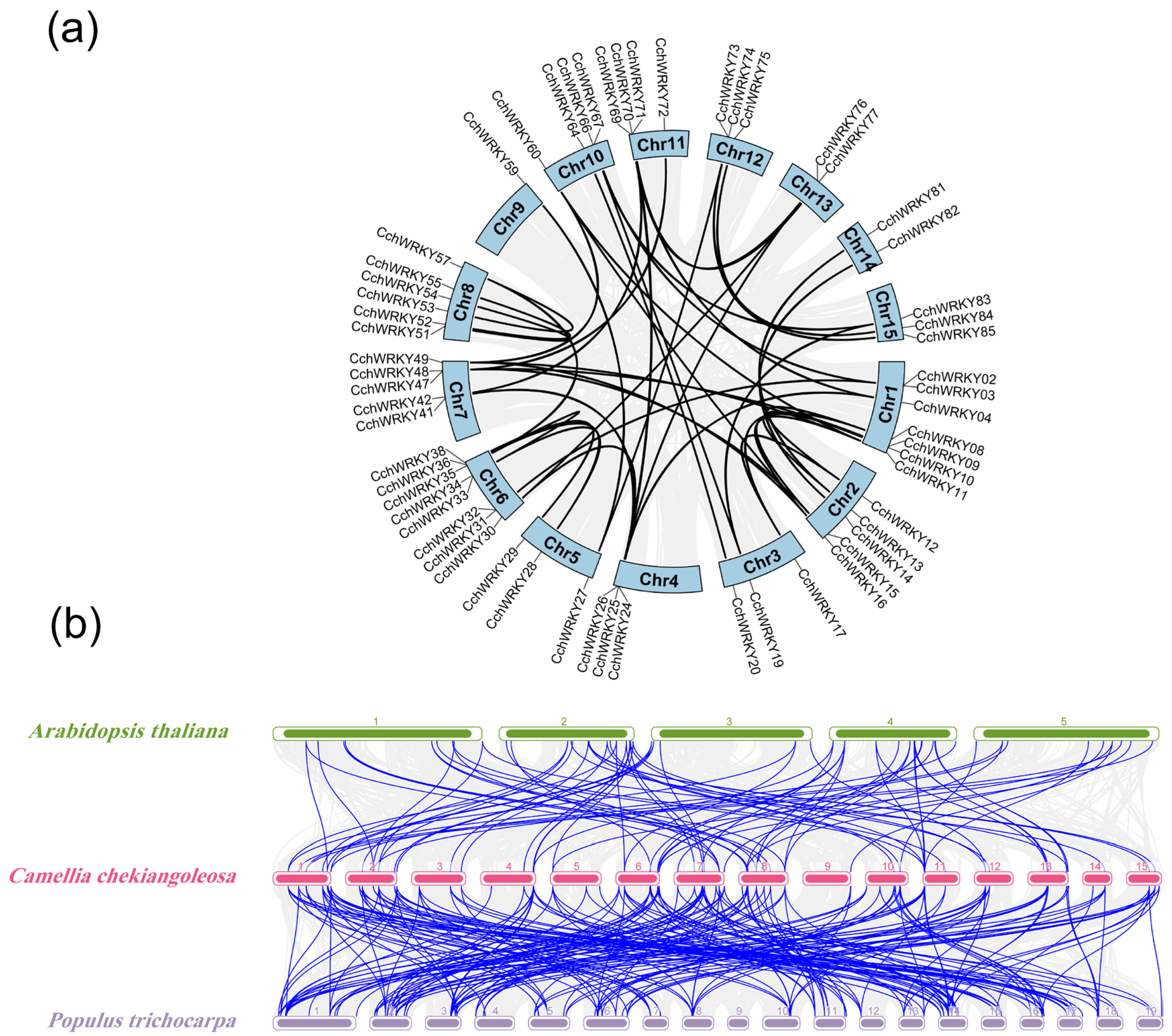
2.3. Collinearity Analysis of CchWRKYs
2.4. Examination of Cis-Acting Components in CchWRKY Promoters
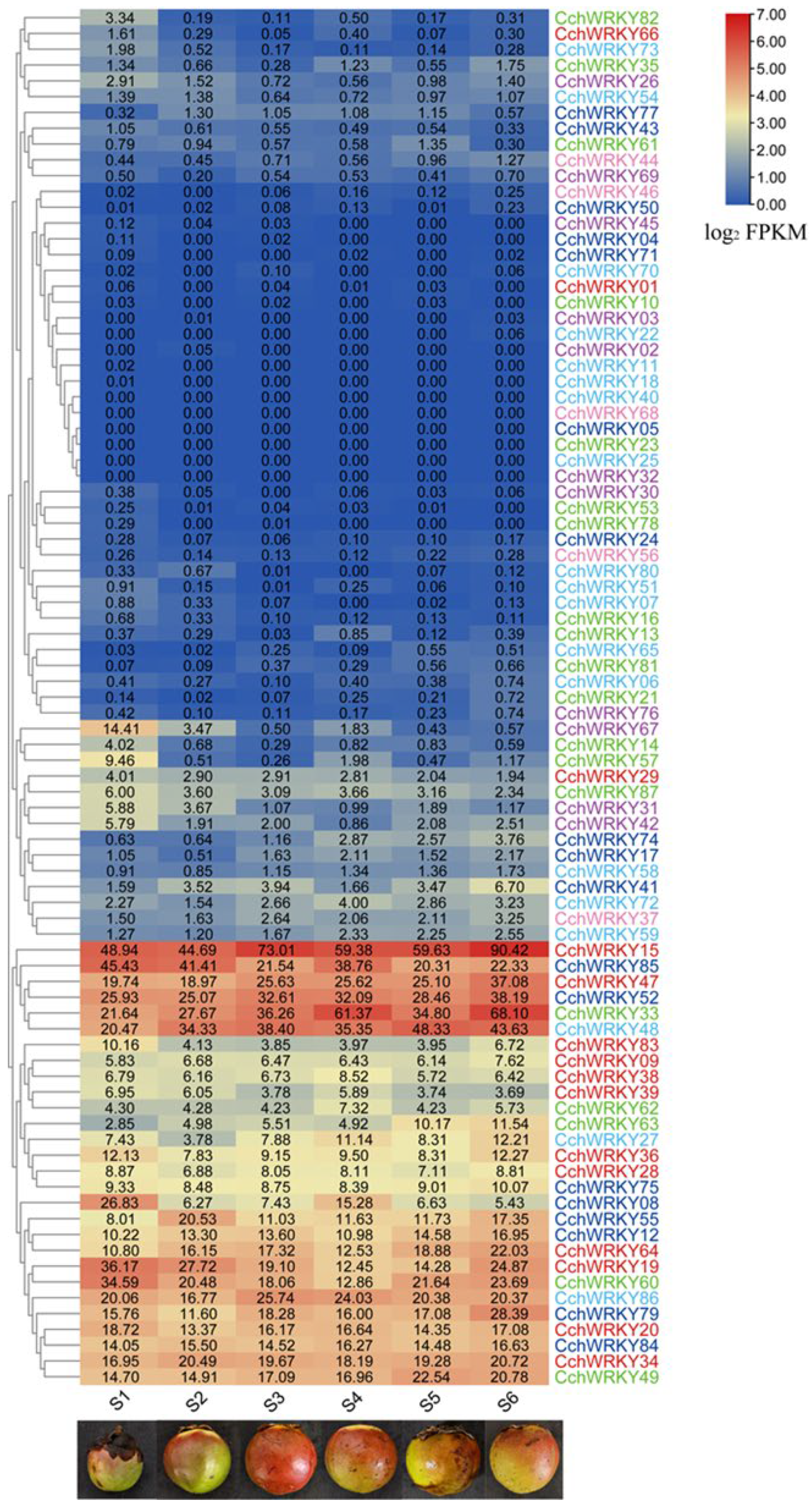
2.5. Examining and Assessing the Expression Levels of CchWRKY Genes in Camellia chekiangoleosa Pericarp at Step Development Stages
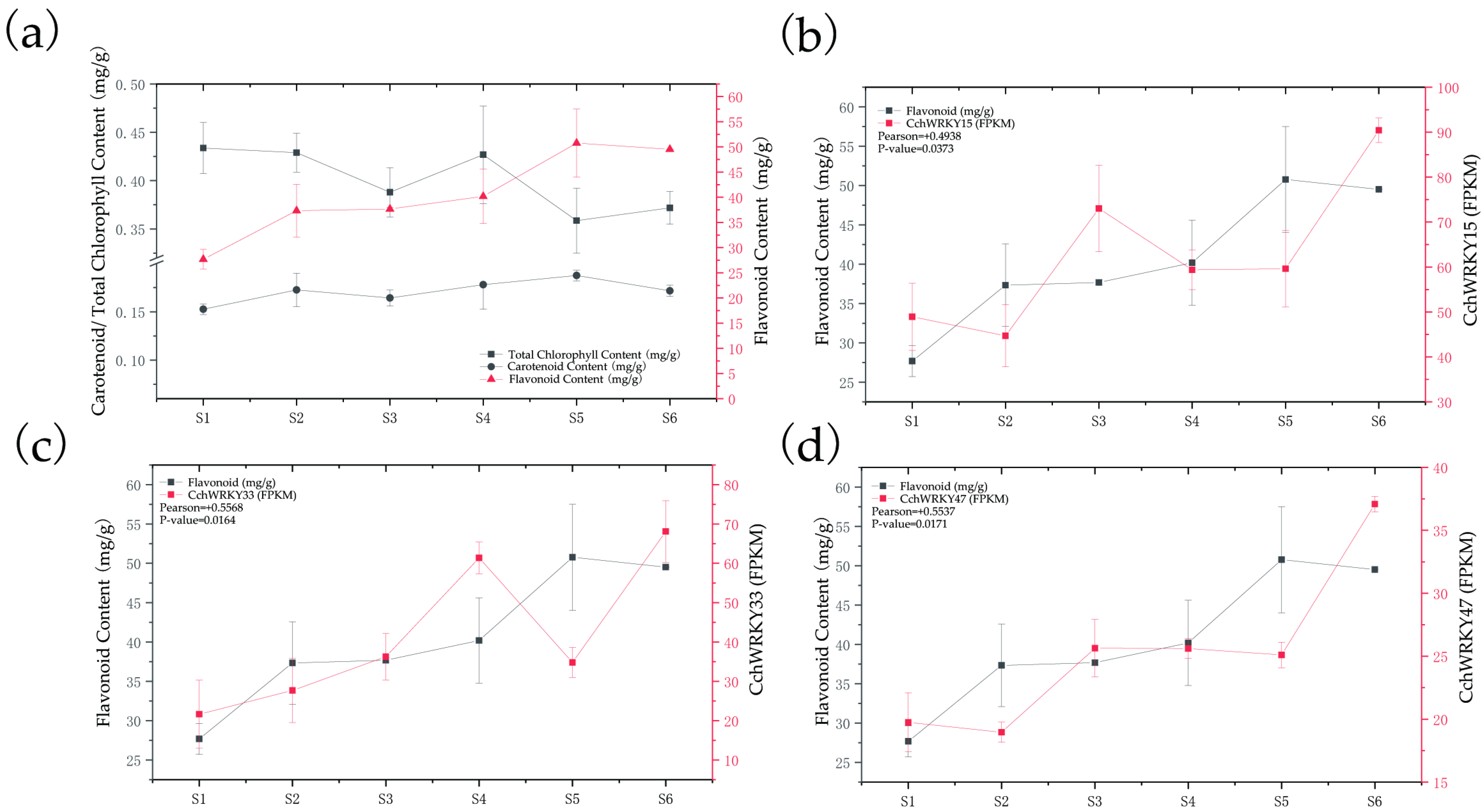
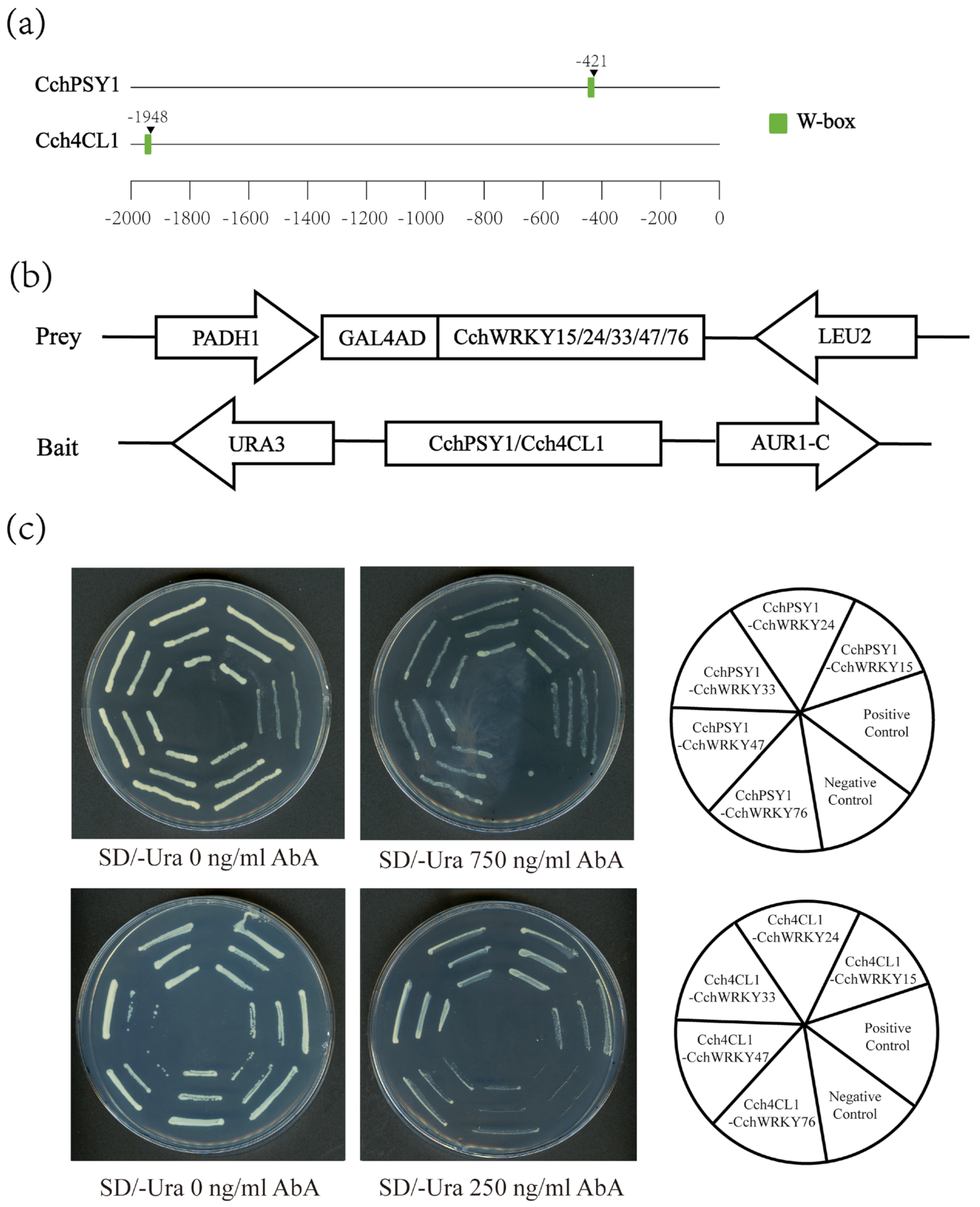
2.6. The Promoters of Pigment Biosynthetic Genes Could Be Directly Bound by CchWRKYs
3. Discussion
4. Materials and Methods
4.1. Fruit Materials of C. chekiangoleosa
4.2. Genome-Wide Identification of CchWRKYs
4.3. Chromosomal Localization, Multiple Sequence Alignment, and Phylogenetic Analysis of CchWRKYs
4.4. Analysis of Gene Structure, Conserved Motifs, and Conserved Domains of CchWRKYs
4.5. Analysis of Cis-Acting Elements in the Promoter Region and Collinearity of CchWRKYs
4.6. Analysis of CchWRKYs’ Gene Expression Profiles
4.7. DNA/RNA Extraction and Cloning of CchWRKY Members
4.8. Determination of Total Chlorophyll, Flavonoids, and Carotenoids
4.9. Yeast One-Hybrid Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jin, J.P.; Zhang, H.; Kong, L.; Gao, G.; Luo, J.C. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014, 42, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Jiang, W.J.; Zhang, Y.; Yu, H.J.; Mao, Z.C.; Gu, X.F.; Huang, S.W.; Xie, B.Y. Genome-wide analysis of WRKY gene family in Cucumis sativus. BMC Genom. 2011, 12, 471. [Google Scholar] [CrossRef]
- Ciolkowski, I.; Wanke, D.; Birkenbihl, R.P.; Somssich, I.E. Studies on DNA-binding selectivity of WRKY transcription factors lend structural clues into WRKY-domain function. Plant Mol. Biol. 2008, 68, 81–92. [Google Scholar] [CrossRef]
- Pandey, S.P.; Somssich, I.E. The Role of WRKY Transcription Factors in Plant Immunity. Plant Physiol. 2009, 150, 1648–1655. [Google Scholar] [CrossRef]
- Bjornson, M.; Pimprikar, P.; Nürnberger, T.; Zipfel, C. The transcriptional landscape of Arabidopsis thaliana pattern-triggered immunity. Nat. Plants 2021, 7, 579–586. [Google Scholar] [CrossRef]
- Guo, M.Y.; Yang, F.J.; Liu, C.X.; Zou, J.P.; Qi, Z.Y.; Fotopoulos, V.; Lu, G.; Yu, J.Q.; Zhou, J. A single-nucleotide polymorphism in WRKY33 promoter is associated with the cold sensitivity in cultivated tomato. New Phytol. 2022, 236, 989–1005. [Google Scholar] [CrossRef]
- Schluttenhofer, C.; Yuan, L. Regulation of Specialized Metabolism by WRKY Transcription Factors. Plant Physiol. 2015, 167, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Jeena, G.S.; Shikha; Kumar, R.S.; Pandey, A.; Shukla, R.K. Ocimum sanctum, OscWRKY1, regulates phenylpropanoid pathway genes and promotes resistance to pathogen infection in Arabidopsis. Plant Mol. Biol. 2022, 110, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.X.; Zhang, F.B.; Song, H.D.; Liang, K.L.; Wang, J.X.; Zhang, X.Y.; Wang, X.W.; Yang, J.G.; Han, W.H. Genome-wide analysis of the WRKY family in Nicotiana benthamiana reveals key members regulating lignin synthesis and Bemisia tabaci resistance. Ind. Crops Prod. 2024, 222, 119655. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.L.; Wang, L.; Tian, Y.N.; Jia, N.; Chen, S.Z.; Shi, N.B.; Huang, X.M.; Zhou, C.; Yu, Y.W.; et al. Regulation of ethylene-responsive SlWRKYs involved in color change during tomato fruit ripening. Sci. Rep. 2017, 7, 16674. [Google Scholar] [CrossRef]
- Wang, P.J.; Yue, C.; Chen, D.; Zheng, Y.C.; Zhang, Q.; Yang, J.F.; Ye, N.X. Genome-wide identification of WRKY family genes and their response to abiotic stresses in tea plant (Camellia sinensis). Genes. Genom. 2019, 41, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Li, J.B.; Xiong, C.W.; Ruan, D.; Du, W.; Li, H.; Ruan, C.J. Identification of Camellia oleifera WRKY transcription factor genes and functional characterization of CoWRKY78. Front. Plant Sci. 2023, 14, 1110366. [Google Scholar] [CrossRef]
- Ye, H.L.; Folz, J.; Li, C.; Zhang, Y.; Hou, Z.X.; Zhang, L.Y.; Su, S.C. Response of metabolic and lipid synthesis gene expression changes in Camellia oleifera to mulched ecological mat under drought conditions. Sci. Total Environ. 2021, 795, 148856. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Yan, Y.D.; Zeng, Y.L.; Jin, S.J.; Jiang, Q.; Yu, L.; Lai, R.X. Correlation between squalene synthase promoter and WRKY transcription factor in Camellia oleifera. J. Hortic. Sci. Biotechnol. 2021, 96, 34–43. [Google Scholar] [CrossRef]
- Li, A.; Du, Q.H.; Zeng, Y.L.; Yang, R.; Ge, L.Y.; Zhu, Z.Y.; Li, C.Y.; Tan, X.F. Light Regulated CoWRKY15 Acts on CoSQS Promoter to Promote Squalene Synthesis in Camellia oleifera Seeds. Int. J. Mol. Sci. 2024, 25, 11134. [Google Scholar] [CrossRef]
- Feng, Y.; Zhao, K.K.; Li, J.Y.; Wang, M.Y.; Yin, H.F.; Fan, Z.Q.; Li, X.L.; Liu, W.X. Transcriptome and Pigment Analyzes Provide Insights into Carotenoids and Flavonoids Biosynthesis in Camellia nitidissima Stamens. Horticulturae 2024, 10, 420. [Google Scholar] [CrossRef]
- Fu, M.Y.; Yang, X.; Zheng, J.R.; Wang, L.; Yang, X.Y.; Tu, Y.; Ye, J.B.; Zhang, W.W.; Liao, Y.L.; Cheng, S.Y.; et al. Unraveling the regulatory mechanism of color diversity in Camellia japonica petals by integrative transcriptome and metabolome analysis. Front. Plant Sci. 2021, 12, 685136. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.W.; Jiang, C.; Wen, Q.; Wang, N.; Tao, Y.Y.; Xu, L.A. Deep sequencing of the Camellia chekiangoleosa transcriptome revealed candidate genes for anthocyanin biosynthesis. Genes 2014, 538, 1–7. [Google Scholar] [CrossRef]
- Cheng, G.X.; Zhu, J.X.; Si, J.J.; Wu, T.; Chen, J.Y.; Xu, X.Y.; Feng, S.L.; Chen, T.; Ding, C.B.; Zhou, L.J. Optimization of ultrasound-assisted enzymatic extraction, chemical constituents, biological activities, and stability of Camellia oleifera fruit shell brown pigments. LWT 2024, 207, 116625. [Google Scholar] [CrossRef]
- Shen, T.F.; Huang, B.; Xu, M.; Zhou, P.Y.; Ni, Z.X.; Gong, C.; Wen, Q.; Cao, F.L.; Xu, L.A. The reference genome of Camellia chekiangoleosa provides insights into Camellia evolution and tea oil biosynthesis. Hortic. Res. 2022, 9, uhab083. [Google Scholar] [CrossRef]
- Zhao, D.Q.; Tao, J. Recent advances on the development and regulation of flower color in ornamental plants. Front. Plant Sci. 2015, 261, 6. [Google Scholar] [CrossRef] [PubMed]
- Polivka, T.; Frank, H.A. Molecular factors controlling photosynthetic light harvesting by carotenoids. Acc. Chem. Res. 2010, 43, 1125–1134. [Google Scholar] [CrossRef]
- Naeem, M.; Zhao, W.H.; Ahmad, N.; Zhao, L.X. Beyond green and red: Unlocking the genetic orchestration of tomato fruit color and pigmentation. Funct. Integr. Genom. 2023, 23, 243. [Google Scholar] [CrossRef]
- Ma, Z.H.; Nan, X.T.; Li, W.F.; Mao, J.; Chen, B.H. Comprehensive genomic identification and expression analysis 4CL gene family in apple. Gene 2023, 30, 147197. [Google Scholar] [CrossRef]
- Su, W.B.; Zhu, C.Q.; Fan, Z.Q.; Huang, M.K.; Lin, H.; Chen, X.P.; Deng, C.J.; Cheng, Y.P.; Kou, Y.D.; Tong, Z.H.; et al. Comprehensive metabolome and transcriptome analyses demonstrate divergent anthocyanin and carotenoid accumulation in fruits of wild and cultivated loquats. Front. Plant Sci. 2023, 13, 1285456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jin, J.P.; Tang, L.; Zhao, Y.; Gu, X.C.; Gao, G.; Luo, J.C. PlantTFDB 2.0: Update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res. 2011, 39, 1114–1117. [Google Scholar] [CrossRef]
- Ishiguro, S.; Nakamura, K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5′ upstream regions of genes coding for sporamin and β-amylase from sweet potato. Molec. Gen. Genet. 1994, 244, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.X.; Chen, C.H.; Chen, Z.X. Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol. Biol. 2003, 51, 21–37. [Google Scholar] [CrossRef]
- Yang, W.; Li, W.S.; Qu, J.Z.; Li, F.H.; Du, W.L.; Weng, Z.F. Genome-Wide Characterization of the Maize (Zea mays L.) WRKY Transcription Factor Family and Their Responses to Ustilago maydis. Int. J. Mol. Sci. 2023, 24, 14916. [Google Scholar] [CrossRef]
- Yang, X.Z.; Li, H.; Yang, Y.C.; Wang, Y.Q.; Mo, Y.L.; Zhang, R.M.; Zhang, Y.; Ma, J.X.; Wei, C.H.; Zhang, X. Identification and expression analyses of WRKY genes reveal their involvement in growth and abiotic stress response in watermelon (Citrullus lanatus). PLoS ONE 2018, 13, e0191308. [Google Scholar] [CrossRef]
- Guo, C.L.; Guo, R.R.; Xu, X.Z.; Gao, M.; Li, X.Q.; Song, J.Y.; Zheng, Y.; Wang, X.P. Evolution and expression analysis of the grape (Vitis vinifera L.) WRKY gene family. J. Exp. Bot. 2014, 65, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.J.; Li, X.H.; Liu, Z.W.; Li, H.; Wang, Y.X.; Zhuang, J. Transcriptome-wide identification of Camellia sinensis WRKY transcription factors in response to temperature stress. Mol. Genet. Genom. 2016, 291, 255–269. [Google Scholar] [CrossRef]
- Yang, X.; Zhou, Z.C.; Fu, M.Y.; Han, M.X.; Liu, Z.B.; Zhu, C.Y.; Wang, L.; Zheng, J.R.; Liao, Y.L.; Zhang, W.W.; et al. Transcriptome-wide identification of WRKY family genes and their expression profiling toward salicylic acid in Camellia japonica. Plant Signal Behav. 2021, 16, 1844508. [Google Scholar] [CrossRef] [PubMed]
- Aken, O.V.; Zhang, B.T.; Law, S.; Narsai, R.; Whelan, J. AtWRKY40 and AtWRKY63 modulate the expression of stress-responsive nuclear genes encoding mitochondrial and chloroplast proteins. Plant Physiol. 2013, 162, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Mahadani, P.; Hazra, A. Expression and splicing dynamics of WRKY family genes along physiological exigencies of tea plant (Camellia sinensis). Biologia 2021, 76, 2491–2499. [Google Scholar] [CrossRef]
- Su, W.J.; Zhou, Z.L.; Zeng, J.; Cao, R.L.; Zhang, Y.Y.; Hu, D.N.; Liu, J. Genome-wide identification of the WRKY gene family in Camellia oleifera and expression analysis under phosphorus deficiency. Front. Plant Sci. 2023, 14, 1082496. [Google Scholar] [CrossRef]
- Li, W.X.; Xiao, N.; Wang, Y.W.; Liu, X.M.; Chen, Z.Y.; Gu, X.Y.; Chen, Y.D. Genome-Wide Identification, Evolutionary and Functional Analyses of WRKY Family Members in Ginkgo biloba. Genes 2023, 4, 343. [Google Scholar] [CrossRef]
- Zhao, N.N.; He, M.J.; Li, L.; Cui, S.L.; Hou, M.Y.; Wang, L.; Mu, G.J.; Liu, L.F.; Yang, X.L. Identification and expression analysis of WRKY gene family under drought stress in peanut (Arachis hypogaea L.). PLoS ONE 2020, 15, e0231396. [Google Scholar] [CrossRef]
- Zou, X.H.; Dong, C.; Liu, H.T.; Gao, Q.H. Genome-wide characterization and expression analysis of WRKY family genes during development and resistance to Colletotrichum fructicola in cultivated strawberry (Fragariaxananassa Duch.). J. Integr. Agric. 2022, 21, 1658–1672. [Google Scholar]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Bi, C.W.; Xu, Y.Q.; Ye, Q.L.; Yin, T.M.; Ye, N. Genome-wide identification and characterization of WRKY gene family in Salix suchowensis. PeerJ 2016, 4, e2437. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.Q.; Yang, C.K.; Zheng, B.; Ni, J.B.; Zhou, K.B.; Qian, M.J.; Wu, H.X. Genome-Wide Identification and Expression Analysis of GST Genes during Light-Induced Anthocyanin Biosynthesis in Mango (Mangifera indica L.). Plants 2024, 13, 2726. [Google Scholar] [CrossRef]
- Wu, K.L.; Guo, Z.J.; Wang, H.H.; Li, J. The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Res. 2005, 12, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhang, Z.L.; Zou, X.L.; Huang, J.; Ruas, P.; Thompson, D.; Shen, Q.J. Annotations and functional analyses of the rice WRKY gene superfamily reveal positive and negative regulators of abscisic acid signaling in aleurone cells. Plant Physiol. 2005, 137, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef]
- Meng, D.; Li, Y.Y.; Bai, Y.; Li, M.J.; Cheng, L.L. Genome-wide identification and characterization of WRKY transcriptional factor family in apple and analysis of their responses to waterlogging and drought stress. Plant Physiol. Biochem. 2016, 103, 71–83. [Google Scholar] [CrossRef]
- Brand, L.H.; Fischer, N.M.; Harter, K.; Kohlbacher, O.; Wanke, D. Elucidating the evolutionary conserved DNA-binding specificities of WRKY transcription factors by molecular dynamics and in vitro binding assays. Nucleic Acids Res. 2013, 41, 9764–9778. [Google Scholar] [CrossRef]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef]
- Takahashi, T.; Hasegawa, H. Influence of environmental factors on pigment synthesis in plants. J. Plant Physiol. 2017, 213, 21–30. [Google Scholar]
- Wang, A.H.; Ma, H.Y.; Zhang, X.T.; Zhang, B.H.; Li, F. Transcriptomic analysis reveals the mechanism underlying the anthocyanin changes in Fragaria nilgerrensis Schlecht. and its interspecific hybrids. BMC Plant Biol. 2023, 23, 356. [Google Scholar] [CrossRef]
- Bao, Y.R.; Nie, T.K.; Wang, D.D.; Chen, Q. Anthocyanin regulatory networks in Solanum tuberosum L. leaves elucidated via integrated metabolomics, transcriptomics, and StAN1 overexpression. BMC Plant Biol. 2022, 22, 228. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.W.; Su, W.Q.; Cai, Z.Y.; Dong, L.; Li, C.B.; Xin, M.; Fang, W.K.; Liu, Y.Q.; Wang, X.M.; Huang, Z.B.; et al. Combined Analysis of Transcriptome and Metabolome Reveals the Potential Mechanism of Coloration and Fruit Quality in Yellow and Purple Passiflora edulis Sims. J. Agric. Food Chem. 2020, 68, 12096–12106. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, J.X.; Nageswaran, D.; Li, L. Carotenoid metabolism and regulation in horticultural crops. Hortic. Res. 2015, 2, 15036. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.R.; Kim, H.T.; Shanmugam, A.; Nath, U.K.; Goswami, G.; Song, J.Y.; Park, J.I.; Nou, I.S. Expression Profiling of Regulatory and Biosynthetic Genes in Contrastingly Anthocyanin Rich Strawberry (Fragaria x ananassa) Cultivars Reveals Key Genetic Determinants of Fruit Color. Int. J. Mol. Sci. 2018, 19, 656. [Google Scholar] [CrossRef]
- Li, H.; Yang, Z.; Zeng, Q.W.; Wang, S.B.; Luo, Y.W.; Huang, Y.; Xin, Y.C.; He, N.J. Abnormal expression of bHLH3 disrupts a flavonoid homeostasis network, causing differences in pigment composition among mulberry fruits. Hortic. Res. 2020, 7, 83. [Google Scholar] [CrossRef]
- Song, J.L.; Sun, B.M.; Chen, C.M.; Ning, Z.Y.; Zhang, S.L.; Cai, Y.T.; Zheng, X.J.; Cao, B.H.; Chen, G.J.; Jin, D.; et al. An R-R-type MYB transcription factor promotes non-climacteric pepper fruit carotenoid pigment biosynthesis. Plant J. 2023, 115, 724–741. [Google Scholar] [CrossRef]
- Xiao, S.H.; Ming, Y.Q.; Hu, Q.; Ye, Z.X.; Si, H.; Liu, S.M.; Zhang, X.J.; Wang, W.R.; Yu, Y.; Kong, J.; et al. GhWRKY41 forms a positive feedback regulation loop and increases cotton defence response against Verticillium dahliae by regulating phenylpropanoid metabolism. Plant Biotechnol. J. 2023, 21, 961–978. [Google Scholar] [CrossRef]
- Wang, N.; Liu, W.J.; Zhang, T.L.; Jiang, S.H.; Xu, H.F.; Wang, Y.C.; Zhang, Z.Y.; Wang, C.Z.; Chen, X.S. Transcriptomic Analysis of Red-Fleshed Apples Reveals the Novel Role of MdWRKY11 in Flavonoid and Anthocyanin Biosynthesis. J. Agric. Food Chem. 2018, 66, 7076–7086. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222-30. [Google Scholar] [CrossRef]
- Reiser, L.; Bakker, E.; Subramaniam, S.; Chen, X.; Sawant, S.; Khosa, K.; Prithvi, T.; Berardini, T.Z. The Arabidopsis Information Resource in 2024. Genetics 2024, 227, iyae027. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 31–52. [Google Scholar]
- Chen, C.J.; Wu, Y.; Li, J.W.; Wang, X.; Zeng, Z.H.; Xu, J.; Liu, Y.L.; Feng, J.T.; Chen, H.; He, Y.H.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant. 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Chao, J.T.; Kong, Y.Z.; Wang, Q.; Sun, Y.H.; Gong, D.P.; Lv, J.; Liu, G.S. MapGene2Chrom, a tool to draw gene physical map based on Perl and SVG languages. Yi Chuan 2015, 37, 91–97. [Google Scholar] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics. 2015, 31, 1296–1297. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Rombauts, S.; Déhais, P.; Van Montagu, M.; Rouzé, P. PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res. 1999, 27, 295–296. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Peng, Y.; Qu, Y.; Huang, B.; Gong, C.; Wen, Q. Genome-Wide Characterization of WRKY Gene Family in Camellia chekiangoleosa Identifies Potential Regulatory Components in Pigment Biosynthesis Pathways. Int. J. Mol. Sci. 2025, 26, 4622. https://doi.org/10.3390/ijms26104622
Liu Z, Peng Y, Qu Y, Huang B, Gong C, Wen Q. Genome-Wide Characterization of WRKY Gene Family in Camellia chekiangoleosa Identifies Potential Regulatory Components in Pigment Biosynthesis Pathways. International Journal of Molecular Sciences. 2025; 26(10):4622. https://doi.org/10.3390/ijms26104622
Chicago/Turabian StyleLiu, Zhenyu, Yixuan Peng, Yanshu Qu, Bin Huang, Chun Gong, and Qiang Wen. 2025. "Genome-Wide Characterization of WRKY Gene Family in Camellia chekiangoleosa Identifies Potential Regulatory Components in Pigment Biosynthesis Pathways" International Journal of Molecular Sciences 26, no. 10: 4622. https://doi.org/10.3390/ijms26104622
APA StyleLiu, Z., Peng, Y., Qu, Y., Huang, B., Gong, C., & Wen, Q. (2025). Genome-Wide Characterization of WRKY Gene Family in Camellia chekiangoleosa Identifies Potential Regulatory Components in Pigment Biosynthesis Pathways. International Journal of Molecular Sciences, 26(10), 4622. https://doi.org/10.3390/ijms26104622