
A Survey for Human Tissue-Level Determinants of CAV1 Regulation and Function

 ,
,  , , and
, , and
Abstract

1. Introduction
2. Results
2.1. Enhanced Expression of CAV1 and CAVIN1 in Caveolae-Rich Versus Caveolae-Deficient Tissues
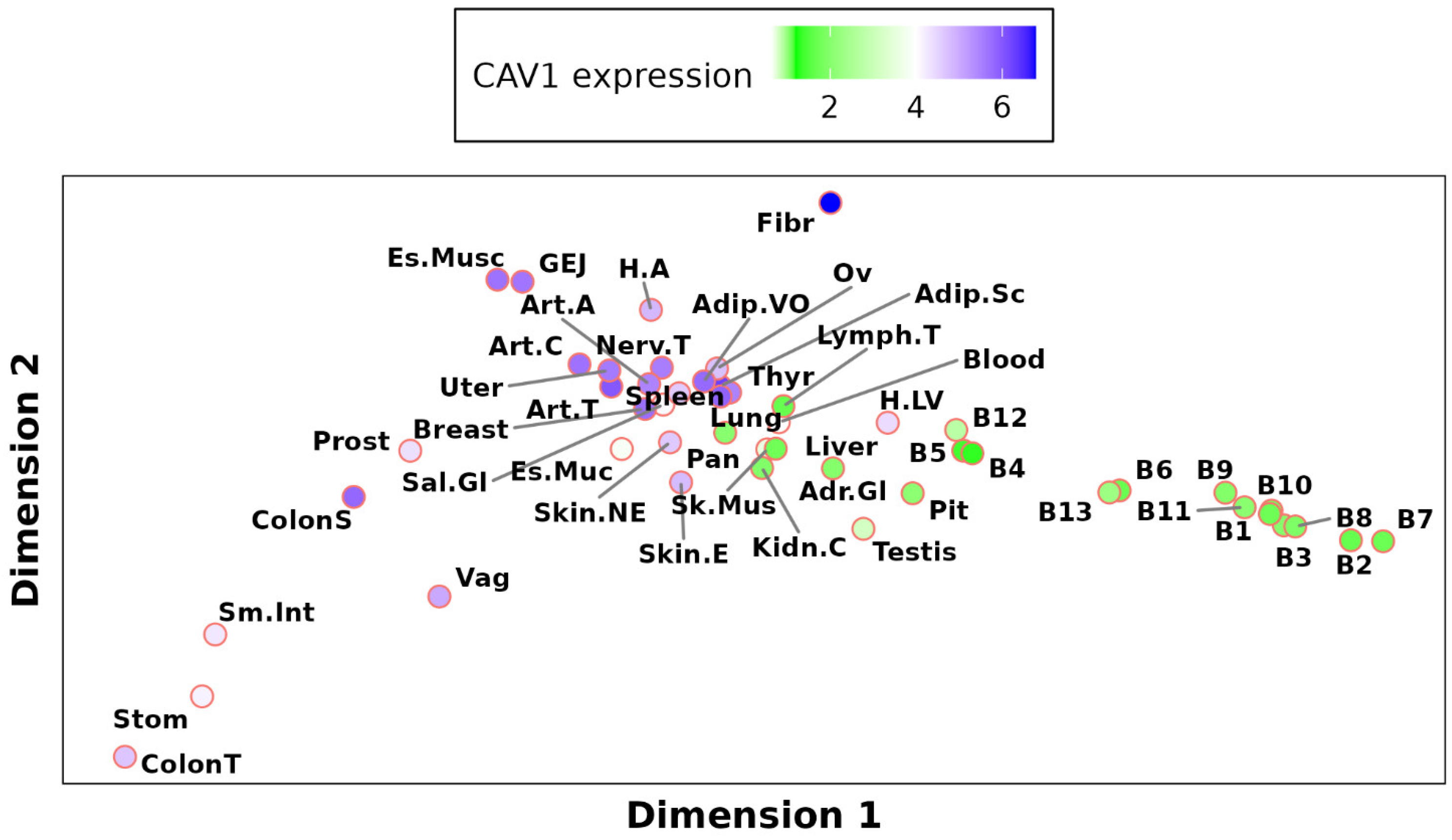
2.2. Understanding the Sources of Variation in CAV1 Expression Across Tissues
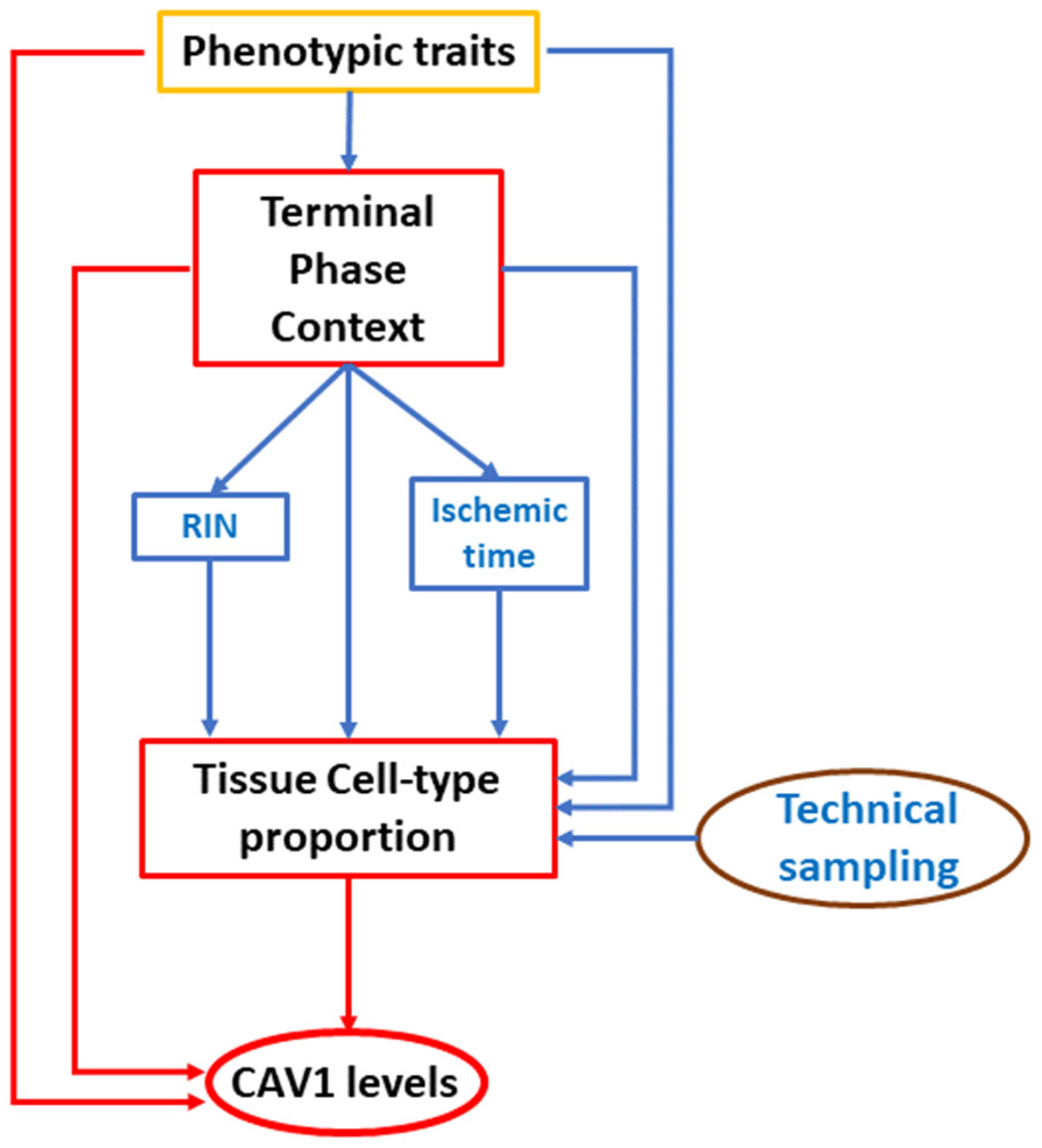
2.2.1. Influence of Public GTEx Variables on CAV1 Expression
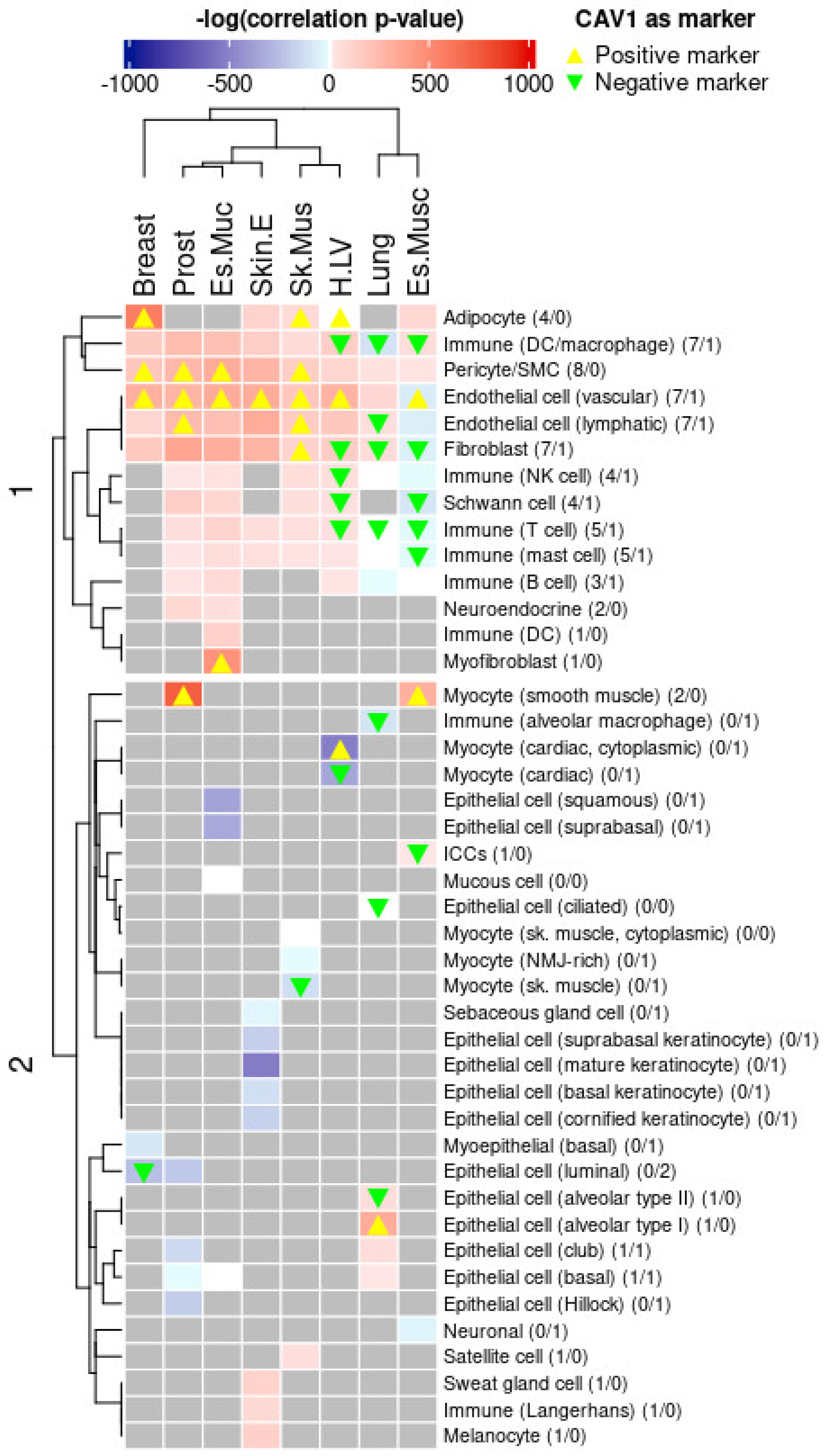
2.2.2. Cell Type Proportions Predict CAV1 Levels
2.2.3. Tissue-Specific CAV1 Expression and Physiological Conditions
2.3. Global Analysis of Tissue-Specific CAV1 Correlation Vectors (TSCVs) Fails to Differentiate Caveolae-Rich from Caveolae-Deficient Tissues
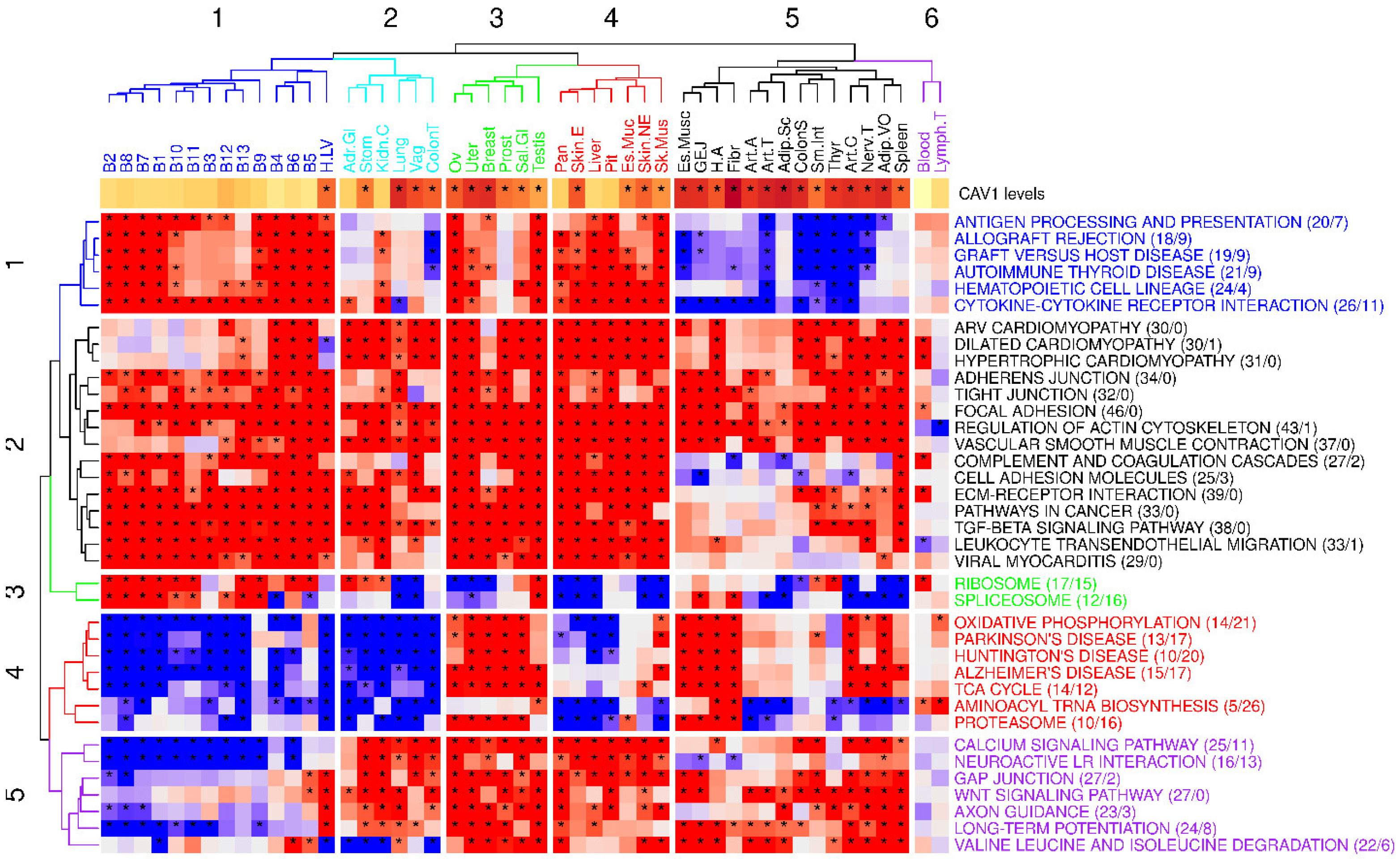
2.3.1. CAV1 Tissue-Specific vs. Ubiquitous Functions
2.3.2. Inferring Transcriptional Regulators in CAV1-TSCVs
2.3.3. CAV1 Tissue Expression and Cell Type-Specific Regulatory Influence
3. Discussion
4. Materials and Methods
4.1. The Genotype-Tissue Expression (GTEx) Project Data
4.2. Defining Cell Type Markers Using GTEx snRNA-Seq Data
4.3. Estimation of Relative Cell Type Proportion for Each Sample
4.4. Linear Modeling of CAV1 Expression: Assessing the Impact of Phenotypic Data and RNA Quality Metrics
4.4.1. Model Set 1: Using Publicly Available GTEx Sample Information
- Essential RNA sample quality metrics:
- RIN (variable SMRIN): The RNA integrity number, a basic measure of the quality of isolated RNA.
- Autolysis Score (variable SMATSSCR): Estimation of the destruction of organism cells or tissues by the organisms’ own enzymes or processes. Determined by pathologists based on the visual inspection of histological images.
- Total Ischemic time for a sample (variable SMTSISCH): Interval between actual death, presumed death, or cross-clamp application and final tissue stabilization.
- Phenotypic traits of the individual:
- Age (variable AGE_GROUP): Elapsed time since birth in decades: 20–29, 30–39, 40–49, 50–59, 60–69, and 70–79.
- Gender (variable SEX): Biological gender.
- Terminal Phase Context (variable DTHHRDY): DTHHRDY is a classification of death adapted from the 4-point Hardy Scale, indicating the circumstances and immediacy of the subject’s death, from sudden and violent deaths to slow deaths after a long illness. For more details, see Supplementary Table S2 in the Supplementary Materials.
4.4.2. Model Set 2: Adding Sample Relative Cell Type Proportions
4.4.3. Model Set 3: Using All GTEx Covariates
4.5. Estimation of CAV1 Tissue-Specific Correlation Vectors (TSCVs)
- (1)
- Initial data filtration retained only those transcriptional elements that exhibited at least 4 TPMs across over 5% of the tissue-specific samples.
- (2)
- (3)
- CAV1 TSCVs were estimated by determining the pairwise Pearson correlation coefficient between each transcriptional element (g) expressed in tissue (t) and the CAV1 levels in the same tissue. The formula used is as follows:where rt,g denotes the CAV1 TSCV for tissue t and gene g, represents the log2(TPM) expression of gene g in sample i of tissue t, indicates the log2(TPM) expression of CAV1 sample i of tissue t, is the mean log2(TPM) expression of gene g in tissue t, and is the mean log2(TPM) expression of CAV1 in tissue t.
- (4)
- All the TSCVs were subsequently integrated into a unified matrix; RT:= (rg,t), a transpose of the aforementioned matrix and R:= (rt,g), consisting of 30,385 transcriptional elements expressed in at least one of the 54 tissues evaluated in GTEx. Each column of this matrix corresponds to the CAV1 TSCV from a specific tissue. Genes not expressed in a given tissue were assigned an NA value in this matrix. For gene-level PCA computation, a value of 0 must be assigned to those NA values to allow matrix inversion. It is important to note that the lack of evidence for a correlation due to the lack of expression of a gene does not imply a correlation of 0. Consequently, we employed alternative methods such as Multidimensional Scaling (MDS, see below) to ensure that significant biases were not introduced due to missing data.
- (5)
- Tissues with less than 25 samples were eliminated from the analysis due to insufficient sample size. These included kidney medulla (n = 4), fallopian tube (n = 9), ectocervix (n = 9), endocervix (n = 10), and bladder (n = 21).
Multidimensional Scaling Analysis (MDS)
4.6. Functional and Upstream Regulator Enrichment of CAV1 TSCVs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Parton, R.G.; del Pozo, M.A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef]
- Palade, G.E. Fine structure of blood capillaries. J. Appl. Phys. 1953, 24, 1424–1448. [Google Scholar]
- Sotodosos-Alonso, L.; Pulgarín-Alfaro, M.; del Pozo, M.A. Caveolae Mechanotransduction at the Interface between Cytoskeleton and Extracellular Matrix. Cells 2023, 12, 942. [Google Scholar] [CrossRef] [PubMed]
- Glenney, J.R. Tyrosine phosphorylation of a 22-kDa protein is correlated with transformation by Rous sarcoma virus. J. Biol. Chem. 1989, 264, 20163–20166. [Google Scholar] [CrossRef] [PubMed]
- Glenney, J.R.; Zokas, L. Novel tyrosine kinase substrates from Rous sarcoma virus-transformed cells are present in the membrane skeleton. J. Cell Biol. 1989, 108, 2401–2408. [Google Scholar] [CrossRef]
- Kurzchalia, T.V.; Dupree, P.; Parton, R.G.; Kellner, R.; Virta, H.; Lehnert, M.; Simons, K. VIP21, a 21-kD membrane protein is an integral component of trans-Golgi-network-derived transport vesicles. J. Cell Biol. 1992, 118, 1003–1024. [Google Scholar] [CrossRef]
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.S.; Glenney, J.R.; Anderson, R.G.W. Caveolin, a protein component of caveolae membrane coats. Cell 1992, 68, 673–682. [Google Scholar] [CrossRef]
- Glenney, J.R.; Soppet, D. Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in Rous sarcoma virus-transformed fibroblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 10517–10521. [Google Scholar] [CrossRef]
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C.; et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001, 293, 2449–2452. Available online: https://www.science.org/doi/abs/10.1126/science.1062688 (accessed on 18 March 2022). [CrossRef]
- Razani, B.; Combs, T.P.; Wang, X.B.; Frank, P.G.; Park, D.S.; Russell, R.G.; Li, M.; Tang, B.; Jelicks, L.A.; Scherer, P.E. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J. Biol. Chem. 2002, 277, 8635–8647. [Google Scholar] [CrossRef]
- Zhao, Y.-Y.; Liu, Y.; Stan, R.-V.; Fan, L.; Gu, Y.; Dalton, N.; Chu, P.-H.; Peterson, K.; Ross, J., Jr.; Chien, K.R. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc. Natl. Acad. Sci. USA 2002, 99, 11375–11380. Available online: https://pubmed.ncbi.nlm.nih.gov/12177436/ (accessed on 14 April 2022). [CrossRef]
- Fra, A.M.; Williamson, E.; Simons, K.; Parton, R.G. De novo formation of caveolae in lymphocytes by expression of VIP21-caveolin. Proc. Natl. Acad. Sci. USA 1995, 92, 8655–8659. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zhang, X.L.; Galbiati, F.; Lisanti, M.P. Chromosomal localization, genomic organization, and developmental expression of the murine caveolin gene family (Cav-1, -2, and -3): Cav-1 and Cav-2 genes map to a known tumor suppressor locus (6-A2/7q31). FEBS Lett. 1998, 429, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, Y.; Sasaoka, T.; Araishi, K.; Imamura, M.; Yorifuji, H.; Nonaka, I.; Ozawa, E.; Kikuchi, T. Caveolin-3 deficiency causes muscle degeneration in mice. Hum. Mol. Genet. 2000, 9, 3047–3054. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, F.; Engelman, J.A.; Volonte, D.; Zhang, X.L.; Minetti, C.; Li, M.; Hou, H.; Kneitz, B.; Edelmann, W.; Lisanti, M.P. Caveolin-3 Null Mice Show a Loss of Caveolae, Changes in the Microdomain Distribution of the Dystrophin-Glycoprotein Complex, and T-tubule Abnormalities. J. Biol. Chem. 2001, 276, 21425–21433. [Google Scholar] [CrossRef]
- Hill, M.M.; Bastiani, M.; Luetterforst, R.; Kirkham, M.; Kirkham, A.; Nixon, S.J.; Walser, P.; Abankwa, D.; Oorschot, V.M.; Martin, S. PTRF-Cavin, a Conserved Cytoplasmic Protein Required for Caveola Formation and Function. Cell 2008, 132, 113–124. [Google Scholar] [CrossRef]
- Liu, L.; Pilch, P.F. A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. J. Biol. Chem. 2008, 283, 4314–4322. [Google Scholar] [CrossRef]
- Goetz, J.G.; Lajoie, P.; Wiseman, S.M.; Nabi, I.R. Caveolin-1 in tumor progression: The good, the bad and the ugly. Cancer Metastasis Rev. 2008, 27, 715–735. [Google Scholar] [CrossRef]
- Pol, A.; Morales-Paytuví, F.; Bosch, M.; Parton, R.G. Non-caveolar caveolins—Duties outside the caves. J. Cell Sci. 2020, 133, jcs241562. [Google Scholar] [CrossRef]
- Mehta, N.; Zhang, D.; Li, R.; Wang, T.; Gava, A.; Parthasarathy, P.; Gao, B.; Krepinsky, J.C. Caveolin-1 regulation of Sp1 controls production of the antifibrotic protein follistatin in kidney mesangial cells. Cell Commun. Signal. 2019, 17, 1–16. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Wang, T.; Cai, P. Dihydroartemisinin inhibits the viability of cervical cancer cells by upregulating caveolin 1 and mitochondrial carrier homolog 2: Involvement of p53 activation and NAD(P)H:quinone oxidoreductase 1 downregulation. Int. J. Mol. Med. 2017, 40, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Vicente, R.; Pavón, D.M.; Martín-Padura, I.; Català-Montoro, M.; Díez-Sánchez, A.; Quílez-Álvarez, A.; López, J.A.; Sánchez-Álvarez, M.; Vázquez, J.; Strippoli, R. Caveolin-1 Modulates Mechanotransduction Responses to Substrate Stiffness Through Actin-Dependent Control of YAP. Cell Rep. 2018, 25, 1622–1635. e1626. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Chitnis, S.S.; Higgins, S.P.; Higgins, C.E.; Krepinsky, J.C.; Higgins, P.J. Redox-Induced Src Kinase and Caveolin-1 Signaling in TGF-β1-Initiated SMAD2/3 Activation and PAI-1 Expression. PLoS ONE 2011, 6, e22896. [Google Scholar] [CrossRef]
- Jiao, L.; Wang, S.; Zheng, Y.; Wang, N.; Yang, B.; Wang, D.; Yang, D.; Mei, W.; Zhao, Z.; Wang, Z. Betulinic acid suppresses breast cancer aerobic glycolysis via caveolin-1/NF-κB/c-Myc pathway. Biochem. Pharmacol. 2019, 161, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.H.; Lee, S.-J.; Lee, K.-B.; Lee, J.-H.; Jeong, E.M.; Chung, S.G.; Park, S.-C.; Kim, I.-G. Dexamethasone downregulates caveolin-1 causing muscle atrophy via inhibited insulin signaling. J. Endocrinol. 2015, 225, 27–37. [Google Scholar] [CrossRef]
- Dasari, A.; Bartholomew, J.N.; Volonte, D.; Galbiati, F. Oxidative Stress Induces Premature Senescence by Stimulating Caveolin-1 Gene Transcription through p38 Mitogen-Activated Protein Kinase/Sp1–Mediated Activation of Two GC-Rich Promoter Elements. Cancer Res. 2006, 66, 10805–10814. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Domingues, L.; Hurbain, I.; Gilles-Marsens, F.; Sirés-Campos, J.; André, N.; Dewulf, M.; Romao, M.; Viaris de Lesegno, C.; Macé, A.-S.; Blouin, C. Coupling of melanocyte signaling and mechanics by caveolae is required for human skin pigmentation. Nat. Commun. 2020, 11, 2988. [Google Scholar] [CrossRef]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Silva, C.; Rotellar, F.; Gil, M.J.; Cienfuegos, J.A.; Salvador, J.; Frühbeck, G. Expression of caveolin-1 in human adipose tissue is upregulated in obesity and obesity-associated type 2 diabetes mellitus and related to inflammation. Clin. Endocrinol. 2008, 68, 213–219. [Google Scholar] [CrossRef]
- Al Madhoun, A.; Kochumon, S.; Haddad, D.; Thomas, R.; Nizam, R.; Miranda, L.; Sindhu, S.; Bitar, M.S.; Ahmad, R.; Al-Mulla, F. Adipose Tissue Caveolin-1 Upregulation in Obesity Involves TNF-α/NF-κB Mediated Signaling. Cells 2023, 12, 1019. [Google Scholar] [CrossRef]
- Puente-Santamaria, L.; Wasserman, W.W.; Del Peso, L. TFEA.ChIP: A tool kit for transcription factor binding site enrichment analysis capitalizing on ChIP-seq datasets. Bioinformatics 2019, 35, 5339–5340. [Google Scholar] [CrossRef]
- Hammal, F.; De Langen, P.; Bergon, A.; Lopez, F.; Ballester, B. ReMap 2022: A database of Human, Mouse, Drosophila and Arabidopsis regulatory regions from an integrative analysis of DNA-binding sequencing experiments. Nucleic Acids Res. 2022, 50, D316–D325. [Google Scholar] [CrossRef] [PubMed]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef]
- Sun, Z.-W.; Wang, X.; Zhao, Y.; Sun, Z.-X.; Wu, Y.-H.; Hu, H.; Zhang, L.; Wang, S.-D.; Li, F.; Wei, A.-J. Blood-brain barrier dysfunction mediated by the EZH2-Claudin-5 axis drives stress-induced TNF-α infiltration and depression-like behaviors. Brain Behav. Immun. 2024, 115, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Ihezie, S.A.; Mathew, I.E.; McBride, D.W.; Dienel, A.; Blackburn, S.L.; Pandit, P.K.T. Epigenetics in blood–brain barrier disruption. Fluids Barriers CNS 2021, 18, 1–16. [Google Scholar] [CrossRef]
- Goetz, J.G.; Minguet, S.; Navarro-Lérida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibáñez, T.; Pellinen, T.; Echarri, A. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Strippoli, R.; Sandoval, P.; Moreno-Vicente, R.; Rossi, L.; Battistelli, C.; Terri, M.; Pascual-Antón, L.; Loureiro, M.; Matteini, F.; Calvo, E. Caveolin1 and YAP drive mechanically induced mesothelial to mesenchymal transition and fibrosis. Cell Death Dis. 2020, 11, 647. [Google Scholar] [CrossRef]
- Cao, J.; Navis, A.; Cox, B.D.; Dickson, A.L.; Gemberling, M.; Karra, R.; Bagnat, M.; Poss, K.D. Single epicardial cell transcriptome sequencing identifies Caveolin 1 as an essential factor in zebrafish heart regeneration. Development 2016, 143, 232–243. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Eraslan, G.; Drokhlyansky, E.; Anand, S.; Fiskin, E.; Subramanian, A.; Slyper, M.; Wang, J.; Van Wittenberghe, N.; Rouhana, J.M.; Waldman, J. Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science 2022, 376, eabl4290. [Google Scholar] [CrossRef]
- van Buuren, S.; Groothuis-Oudshoorn, K. mice: Multivariate Imputation by Chained Equations in R. J. Stat. Softw. 2011, 45, 1–67. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. Available online: https://academic.oup.com/bioinformatics/article/26/1/139/182458 (accessed on 30 May 2022). [CrossRef] [PubMed]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. Biorxiv 2016, 060012. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Set 1 | Model Set 2 | Model Set 3 | |
|---|---|---|---|
| Death-related variables | 17 | 15 | 16 |
| RIN | 15 | 4 | 4 |
| Gender | 5 | 2 | 1 |
| Age | 5 | 2 | 1 |
| Ischemic time | 3 | 0 | 1 |
| Autolysis | 2 | 0 | 0 |
| PAXgene fixation time | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-Jiménez, V.; Sánchez-Cabo, F.; Schwartz, M.A.; Sánchez-Álvarez, M.; del Pozo, M.Á. A Survey for Human Tissue-Level Determinants of CAV1 Regulation and Function. Int. J. Mol. Sci. 2025, 26, 3789. https://doi.org/10.3390/ijms26083789
Jiménez-Jiménez V, Sánchez-Cabo F, Schwartz MA, Sánchez-Álvarez M, del Pozo MÁ. A Survey for Human Tissue-Level Determinants of CAV1 Regulation and Function. International Journal of Molecular Sciences. 2025; 26(8):3789. https://doi.org/10.3390/ijms26083789
Chicago/Turabian StyleJiménez-Jiménez, Víctor, Fátima Sánchez-Cabo, Martin A. Schwartz, Miguel Sánchez-Álvarez, and Miguel Ángel del Pozo. 2025. "A Survey for Human Tissue-Level Determinants of CAV1 Regulation and Function" International Journal of Molecular Sciences 26, no. 8: 3789. https://doi.org/10.3390/ijms26083789
APA StyleJiménez-Jiménez, V., Sánchez-Cabo, F., Schwartz, M. A., Sánchez-Álvarez, M., & del Pozo, M. Á. (2025). A Survey for Human Tissue-Level Determinants of CAV1 Regulation and Function. International Journal of Molecular Sciences, 26(8), 3789. https://doi.org/10.3390/ijms26083789