
Structural Bioinformatics Applied to Acetylcholinesterase Enzyme Inhibition

 ,
,  and
and 
Abstract
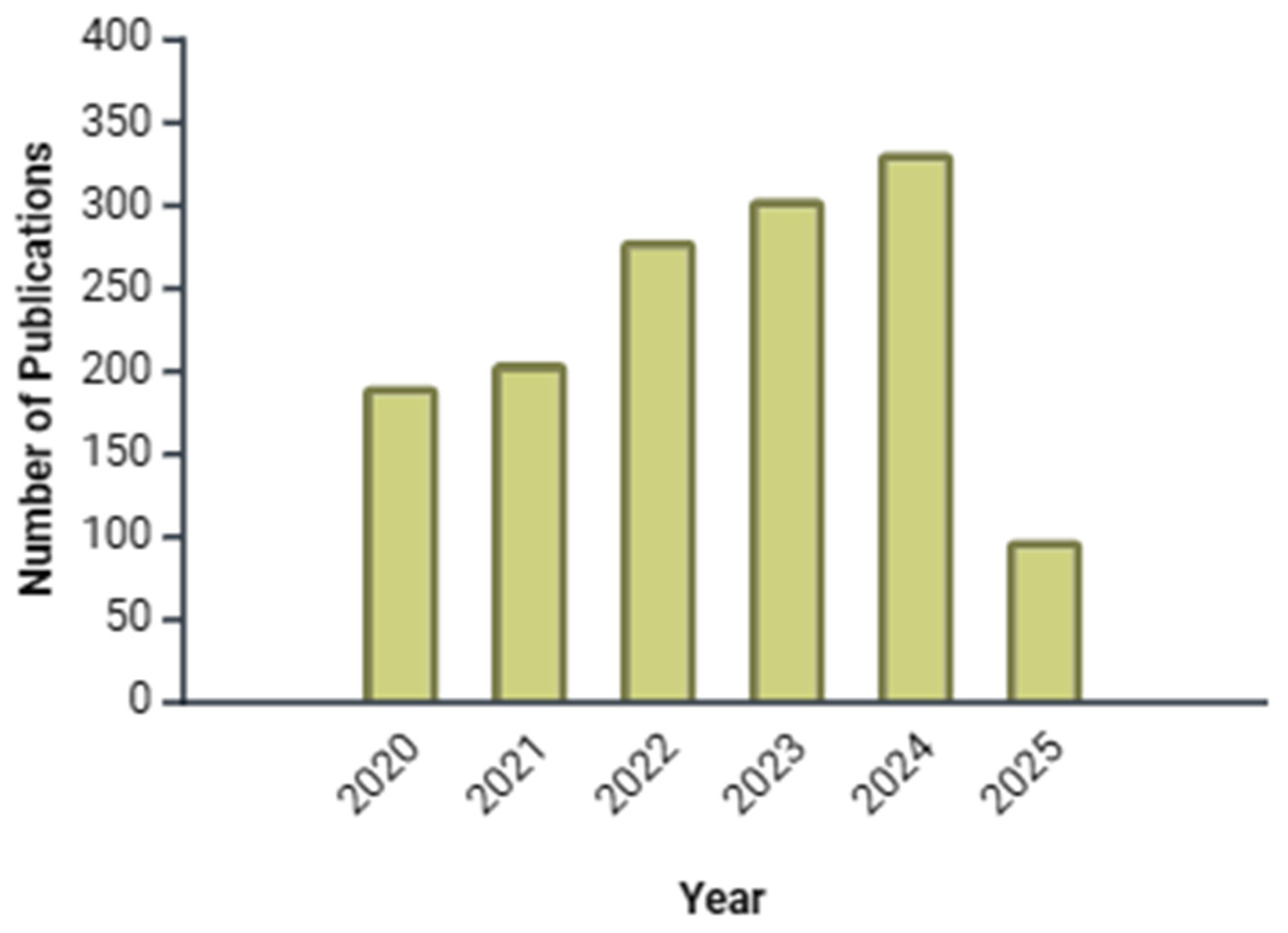
1. Introduction
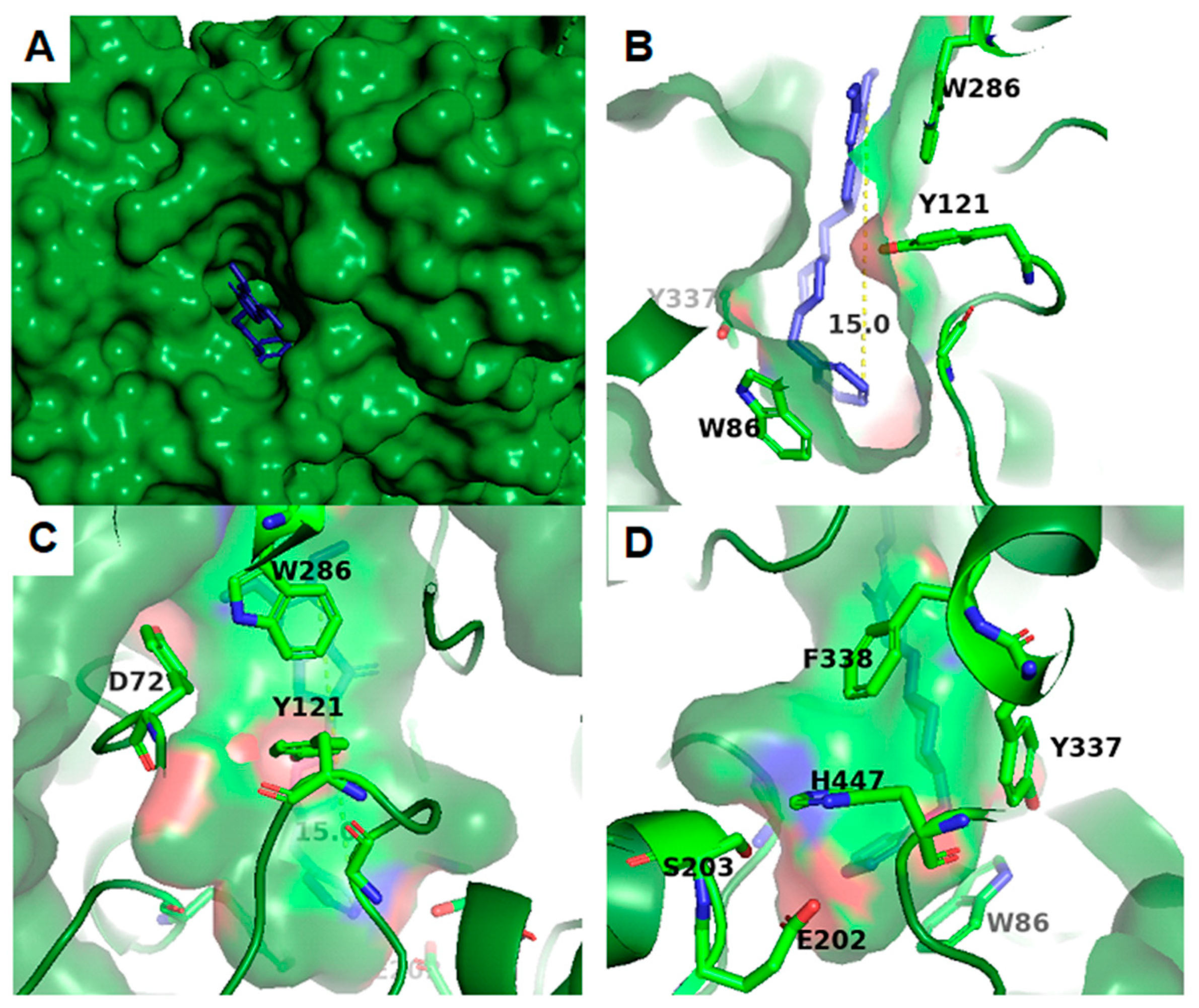
2. Acetylcholinesterase: Structure and Function
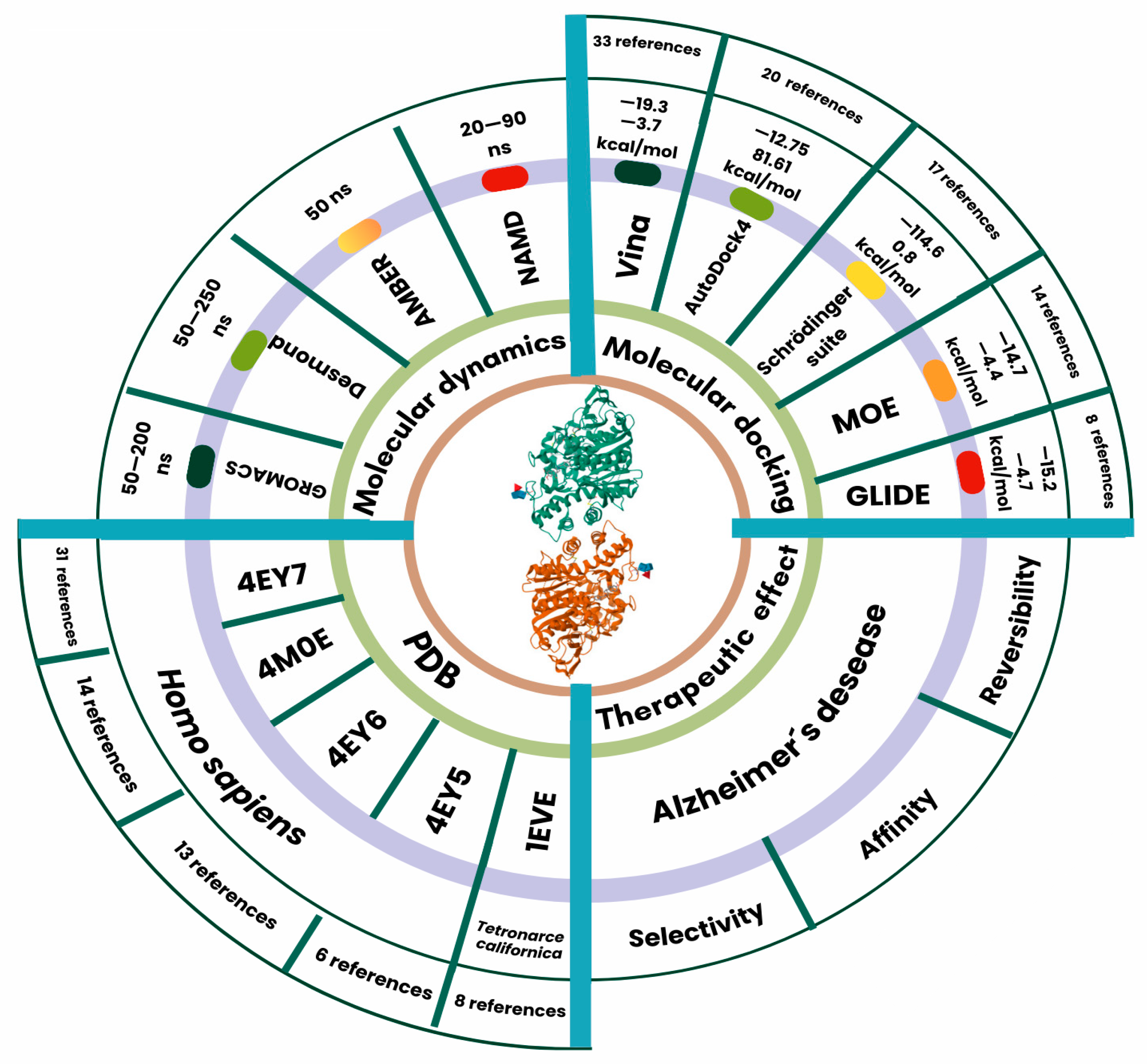
3. Structural Bioinformatics Methods in the Study of AChE
4. Applications of Molecular Docking in AChE
5. Molecular Dynamics Simulations in AChE
6. Discussion
7. Limitations of Computational Tools
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AChE | Acetylcholinesterase |
| MOE | Molecular Operating Environment |
| LD | Linear dichroism |
| PDB | Protein Data Bank |
| NA | Data not available |
References
- Basnet, R.; Khadka, S.; Basnet, B.B.; Gupta, R. Perspective on Acetylcholinesterase: A Potential Target for Alzheimer’s Disease Intervention. Curr. Enzym. Inhib. 2020, 16, 181–188. [Google Scholar] [CrossRef]
- Carvajal, F.J.; Inestrosa, N.C. Interactions of AChE with Aβ Aggregates in Alzheimer’s Brain: Therapeutic Relevance of IDN 5706. Front. Mol. Neurosci. 2011, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Dubey, R. Target Enzyme in Alzheimer’s Disease: Acetylcholinesterase Inhibitors. Curr. Top. Med. Chem. 2019, 19, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Vignaux, P.A.; Minerali, E.; Lane, T.R.; Foil, D.H.; Madrid, P.B.; Puhl, A.C.; Ekins, S. The Antiviral Drug Tilorone Is a Potent and Selective Inhibitor of Acetylcholinesterase. Chem. Res. Toxicol. 2021, 34, 1296–1307. [Google Scholar] [CrossRef]
- García Marín, I.D.; Camarillo López, R.H.; Martínez, O.A.; Padilla-Martínez, I.I.; Correa-Basurto, J.; Rosales-Hernández, M.C. New Compounds from Heterocyclic Amines Scaffold with Multitarget Inhibitory Activity on Aβ Aggregation, AChE, and BACE1 in the Alzheimer Disease. PLoS ONE 2022, 17, e0269129. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, M.; Correa-Basurto, J.; Martínez-Ramos, F.; Padilla-Martínez, I.I.; Benítez-Cardoza, C.G.; Mera-Jiménez, E.; Rosales-Hernández, M.C. Design of Multi-Target Compounds as AChE, BACE1, and Amyloid-Β1-42 Oligomerization Inhibitors: In Silico and In Vitro Studies. J. Alzheimer’s Dis. 2014, 41, 1073–1085. [Google Scholar] [CrossRef]
- Manzoor, S.; Gabr, M.T.; Rasool, B.; Pal, K.; Hoda, N. Dual Targeting of Acetylcholinesterase and Tau Aggregation: Design, Synthesis and Evaluation of Multifunctional Deoxyvasicinone Analogues for Alzheimer’s Disease. Bioorg. Chem. 2021, 116, 105354. [Google Scholar] [CrossRef]
- Baruah, P.; Basumatary, G.; Yesylevskyy, S.O.; Aguan, K.; Bez, G.; Mitra, S. Novel Coumarin Derivatives as Potent Acetylcholinesterase Inhibitors: Insight into Efficacy, Mode and Site of Inhibition. J. Biomol. Struct. Dyn. 2019, 37, 1750–1765. [Google Scholar] [CrossRef]
- Camps, P.; El Achab, R.; Morral, J.; Muñoz-Torrero, D.; Badia, A.; Baños, J.E.; Vivas, N.M.; Barril, X.; Orozco, M.; Luque, F.J. New Tacrine−Huperzine A Hybrids (Huprines): Highly Potent Tight-Binding Acetylcholinesterase Inhibitors of Interest for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2000, 43, 4657–4666. [Google Scholar] [CrossRef]
- Lotfi, S.; Rahmani, T.; Hatami, M.; Pouramiri, B.; Kermani, E.T.; Rezvannejad, E.; Mortazavi, M.; Fathi Hafshejani, S.; Askari, N.; Pourjamali, N.; et al. Design, Synthesis and Biological Assessment of Acridine Derivatives Containing 1,3,4-Thiadiazole Moiety as Novel Selective Acetylcholinesterase Inhibitors. Bioorg. Chem. 2020, 105, 104457. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Kandiah, N.; Pai, M.-C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The Advantages of Dual Inhibition of Acetylcholinesterase and Butyrylcholinesterase and Its Role in Subcortical Vascular Dementia and Parkinson’s Disease Dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef]
- Sharon, N.; Ugale, V.G.; Padmaja, P.; Lokwani, D.; Salunkhe, C.; Shete, P.; Reddy, P.N.; Kulkarni, P.P. Development of Novel 9H-Carbazole-4H-Chromene Hybrids as Dual Cholinesterase Inhibitors for the Treatment of Alzheimer’s Disease. Mol. Divers. 2025, 29, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Ojo, O.A.; Ojo, A.B.; Okolie, C.; Nwakama, M.-A.C.; Iyobhebhe, M.; Evbuomwan, I.O.; Nwonuma, C.O.; Maimako, R.F.; Adegboyega, A.E.; Taiwo, O.A.; et al. Deciphering the Interactions of Bioactive Compounds in Selected Traditional Medicinal Plants against Alzheimer’s Diseases via Pharmacophore Modeling, Auto-QSAR, and Molecular Docking Approaches. Molecules 2021, 26, 1996. [Google Scholar] [CrossRef] [PubMed]
- Thai, Q.M.; Pham, M.Q.; Tran, P.-T.; Nguyen, T.H.; Ngo, S.T. Searching for Potential Acetylcholinesterase Inhibitors: A Combined Approach of Multi-Step Similarity Search, Machine Learning and Molecular Dynamics Simulations. R. Soc. Open Sci. 2024, 11, 240546. [Google Scholar] [CrossRef]
- Son, M.; Park, C.; Rampogu, S.; Zeb, A.; Lee, K.W. Discovery of Novel Acetylcholinesterase Inhibitors as Potential Candidates for the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 1000. [Google Scholar] [CrossRef]
- Onorini, D.; Schoborg, R.; Borel, N.; Leonard, C. Beta Lactamase-Producing Neisseria Gonorrhoeae Alleviates Amoxicillin-Induced Chlamydial Persistence in a Novel in Vitro Co-Infection Model. Curr. Res. Microb. Sci. 2023, 4, 100188. [Google Scholar] [CrossRef]
- Bag, S.; Tulsan, R.; Sood, A.; Datta, S.; Török, M. Pharmacophore Modeling, Virtual and in Vitro Screening for Acetylcholinesterase Inhibitors and Their Effects on Amyloid-β Self- Assembly. Curr. Comput. Aided Drug Des. 2013, 9, 2–14. [Google Scholar]
- Eckert, S.; Eyer, P.; Worek, F. Reversible Inhibition of Acetylcholinesterase by Carbamates or Huperzine a Increases Residual Activity of the Enzyme upon Soman Challenge. Toxicology 2007, 233, 180–186. [Google Scholar] [CrossRef]
- Gebre, T.; Ayele, B.; Zerihun, M.; Genet, A.; Stoller, N.E.; Zhou, Z.; House, J.I.; Yu, S.N.; Ray, K.J.; Emerson, P.M.; et al. Comparison of Annual versus Twice-Yearly Mass Azithromycin Treatment for Hyperendemic Trachoma in Ethiopia: A Cluster-Randomised Trial. Lancet 2012, 379, 143–151. [Google Scholar] [CrossRef]
- Mishra, C.B.; Kumari, S.; Manral, A.; Prakash, A.; Saini, V.; Lynn, A.M.; Tiwari, M. Design, Synthesis, in-Silico and Biological Evaluation of Novel Donepezil Derivatives as Multi-Target-Directed Ligands for the Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2017, 125, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Q.; Wang, X.; Liu, Z.; Chen, H.; Su, Z.; Xu, Y.; Zhang, W.; Du, Y.; Tan, Z.; et al. Development of Novel Rivastigmine Derivatives as Selective BuChE Inhibitors for the Treatment of AD. Bioorg. Chem. 2025, 157, 108245. [Google Scholar] [CrossRef]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef]
- Dvir, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D Structure to Function. Chem. Biol. Interact. 2010, 187, 10–22. [Google Scholar] [CrossRef]
- Bourne, Y.; Taylor, P.; Bougis, P.E.; Marchot, P. Crystal Structure of Mouse Acetylcholinesterase. J. Biol. Chem. 1999, 274, 2963–2970. [Google Scholar] [CrossRef]
- Johnson, G.; Moore, S. The Peripheral Anionic Site of Acetylcholinesterase: Structure, Functions and Potential Role in Rational Drug Design. Curr. Pharm. Des. 2006, 12, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Shafferman, A.; Kronman, C.; Flashner, Y.; Leitner, M.; Grosfeld, H.; Ordentlich, A.; Gozes, Y.; Cohen, S.; Ariel, N.; Barak, D. Mutagenesis of Human Acetylcholinesterase. Identification of Residues Involved in Catalytic Activity and in Polypeptide Folding. J. Biol. Chem. 1992, 267, 17640–17648. [Google Scholar] [CrossRef] [PubMed]
- Walczak-Nowicka, Ł.J.; Herbet, M. Acetylcholinesterase Inhibitors in the Treatment of Neurodegenerative Diseases and the Role of Acetylcholinesterase in Their Pathogenesis. Int. J. Mol. Sci. 2021, 22, 9290. [Google Scholar] [CrossRef]
- Fishman, E.B.; Siek, G.C.; MacCallum, R.D.; Bird, E.D.; Volicer, L.; Marquis, J.K. Distribution of the Molecular Forms of Acetylcholinesterase in Human Brain: Alterations in Dementia of the Alzheimer Type. Ann. Neurol. 1986, 19, 246–252. [Google Scholar] [CrossRef]
- Fernandez, H.L.; Moreno, R.D.; Inestrosa, N.C. Tetrameric (G4) Acetylcholinesterase: Structure, Localization, and Physiological Regulation. J. Neurochem. 1996, 66, 1335–1346. [Google Scholar] [CrossRef]
- Coleman, B.A.; Taylor, P. Regulation of Acetylcholinesterase Expression during Neuronal Differentiation. J. Biol. Chem. 1996, 271, 4410–4416. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for Molecular Docking: A Review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a Protein-Small Molecule Docking Web Service Based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef]
- Bermudez-Lugo, J.A.; Rosales-Hernandez, M.C.; Deeb, O.; Trujillo-Ferrara, J.; Correa-Basurto, J. In Silico Methods to Assist Drug Developers in Acetylcholinesterase Inhibitor Design. Curr. Med. Chem. 2011, 18, 1122–1136. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Sánchez, H.; den Haan, H.; Pérez-Garrido, A.; Peña-García, J.; Chakraborty, S.; Erdogan Orhan, I.; Senol Deniz, F.S.; Villalgordo, J.M. Combined Structure and Ligand-Based Design of Selective Acetylcholinesterase Inhibitors. J. Chem. Inf. Model. 2021, 61, 467–480. [Google Scholar] [CrossRef]
- Pereira, G.R.C.; Gonçalves, L.M.; de Azevedo Abrahim-Vieira, B.; De Mesquita, J.F. In Silico Analyses of Acetylcholinesterase (AChE) and Its Genetic Variants in Interaction with the Anti-Alzheimer Drug Rivastigmine. J. Cell Biochem. 2022, 123, 1259–1277. [Google Scholar] [CrossRef]
- Fang, J.; Wu, P.; Yang, R.; Gao, L.; Li, C.; Wang, D.; Wu, S.; Liu, A.-L.; Du, G.-H. Inhibition of Acetylcholinesterase by Two Genistein Derivatives: Kinetic Analysis, Molecular Docking and Molecular Dynamics Simulation. Acta Pharm. Sin. B 2014, 4, 430–437. [Google Scholar] [CrossRef]
- Azmal, M.; Hossen, M.S.; Shohan, M.N.H.; Taqui, R.; Malik, A.; Ghosh, A. A Computational Approach to Identify Phytochemicals as Potential Inhibitor of Acetylcholinesterase: Molecular Docking, ADME Profiling and Molecular Dynamics Simulations. PLoS ONE 2024, 19, e0304490. [Google Scholar] [CrossRef]
- Gupta, M.; Kumar, A.; Prasun, C.; Nair, M.S.; Kini, S.G.; Yadav, D.; Nain, S. Design, Synthesis, Extra-Precision Docking, and Molecular Dynamics Simulation Studies of Pyrrolidin-2-One Derivatives as Potential Acetylcholinesterase Inhibitors. J. Biomol. Struct. Dyn. 2023, 41, 6282–6294. [Google Scholar] [CrossRef] [PubMed]
- Rees, T.M.; Brimijoin, S. The Role of Acetylcholinesterase in the Pathogenesis of Alzheimer’s Disease. Drugs Today 2003, 39, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Scarpini, E. Old and New Acetylcholinesterase Inhibitors for Alzheimer’s Disease. Expert. Opin. Investig. Drugs 2016, 25, 1181–1187. [Google Scholar] [CrossRef]
- El Khatabi, K.; El-Mernissi, R.; Aanouz, I.; Ajana, M.A.; Lakhlifi, T.; Khan, A.; Wei, D.-Q.; Bouachrine, M. Identification of Novel Acetylcholinesterase Inhibitors through 3D-QSAR, Molecular Docking, and Molecular Dynamics Simulation Targeting Alzheimer’s Disease. J. Mol. Model. 2021, 27, 302. [Google Scholar] [CrossRef] [PubMed]
- Sussman, J.L.; Harel, M.; Silman, I. Three-Dimensional Structure of Acetylcholinesterase and of Its Complexes with Anticholinesterase Drugs. Chem. Biol. Interact. 1993, 87, 187–197. [Google Scholar] [CrossRef]
- Nogara, P.A.; de Aquino Saraiva, R.; Caeran Bueno, D.; Lissner, L.J.; Lenz Dalla Corte, C.; Braga, M.M.; Rosemberg, D.B.; Rocha, J.B.T. Virtual Screening of Acetylcholinesterase Inhibitors Using the Lipinski’s Rule of Five and ZINC Databank. Biomed. Res. Int. 2015, 2015, 870389. [Google Scholar] [CrossRef]
- Chen, Z.; Li, H.; Zhang, Q.; Bao, X.; Yu, K.; Luo, X.; Zhu, W.; Jiang, H. Pharmacophore-Based Virtual Screening versus Docking-Based Virtual Screening: A Benchmark Comparison against Eight Targets. Acta Pharmacol. Sin. 2009, 30, 1694–1708. [Google Scholar] [CrossRef]
- Cross, J.B.; Thompson, D.C.; Rai, B.K.; Baber, J.C.; Fan, K.Y.; Hu, Y.; Humblet, C. Comparison of Several Molecular Docking Programs: Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2009, 49, 1455–1474. [Google Scholar] [CrossRef]
- Khan, M.I.; Pathania, S.; Al-Rabia, M.W.; Ethayathulla, A.S.; Khan, M.I.; Allemailem, K.S.; Azam, M.; Hariprasad, G.; Imran, M.A. MolDy: Molecular Dynamics Simulation Made Easy. Bioinformatics 2024, 40, btae313. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Abd El-Karim, S.S.; Anwar, M.M.; Ahmed, N.S.; Syam, Y.M.; Elseginy, S.A.; Aly, H.F.; Younis, E.A.; Khalil, W.K.B.; Ahmed, K.A.; Mohammed, F.F.; et al. Discovery of Novel Benzofuran-Based Derivatives as Acetylcholinesterase Inhibitors for the Treatment of Alzheimer’s Disease: Design, Synthesis, Biological Evaluation, Molecular Docking and 3D-QSAR Investigation. Eur. J. Med. Chem. 2023, 260, 115766. [Google Scholar] [CrossRef] [PubMed]
- Homoud, Z.A.; Taha, M.; Rahim, F.; Iqbal, N.; Nawaz, M.; Farooq, R.K.; Wadood, A.; Alomari, M.; Islam, I.; Algheribe, S.; et al. Synthesis of Indole Derivatives as Alzheimer Inhibitors and Their Molecular Docking Study. J. Biomol. Struct. Dyn. 2023, 41, 9865–9878. [Google Scholar] [CrossRef] [PubMed]
- Azman, N.A.N.; Alhawarri, M.B.; Rawa, M.S.A.; Dianita, R.; Gazzali, A.M.; Nogawa, T.; Wahab, H.A. Potential Anti-Acetylcholinesterase Activity of Cassia Timorensis DC. Molecules 2020, 25, 4545. [Google Scholar] [CrossRef]
- Tsai, C.-H.; Liou, Y.-L.; Li, S.-M.; Liao, H.-R.; Chen, J.-J. Antioxidant, Anti-α-Glucosidase, Anti-Tyrosinase, and Anti-Acetylcholinesterase Components from Stem of Rhamnus Formosana with Molecular Docking Study. Antioxidants 2024, 14, 8. [Google Scholar] [CrossRef]
- Amir Rawa, M.S.; Nurul Azman, N.A.; Mohamad, S.; Nogawa, T.; Wahab, H.A. In Vitro and In Silico Anti-Acetylcholinesterase Activity from Macaranga Tanarius and Syzygium Jambos. Molecules 2022, 27, 2648. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi-Farani, A.; Moradi, F.; Hosseini, A.; Aliabadi, A. Synthesis, Docking, Pharmacokinetic Prediction, and Acetylcholinesterase Inhibitory Evaluation of N-(2-(Piperidine-1-Yl)Ethyl)Benzamide Derivatives as Potential Anti-Alzheimer Agents. Res. Pharm. Sci. 2024, 19, 698–711. [Google Scholar] [CrossRef]
- Tran, T.S.; Le, M.T.; Nguyen, T.C.V.; Tran, T.H.; Tran, T.D.; Thai, K.M. Synthesis, in Silico and in Vitro Evaluation for Acetylcholinesterase and BACE-1 Inhibitory Activity of Some N-Substituted-4-Phenothiazine-Chalcones. Molecules 2020, 25, 3916. [Google Scholar] [CrossRef]
- Abbas-Mohammadi, M.; Moridi Farimani, M.; Salehi, P.; Ebrahimi, S.N.; Sonboli, A.; Kelso, C.; Skropeta, D. Molecular Networking Based Dereplication of AChE Inhibitory Compounds from the Medicinal Plant Vincetoxicum Funebre (Boiss. & Kotschy). J. Biomol. Struct. Dyn. 2022, 40, 1942–1951. [Google Scholar] [CrossRef]
- Amat-Ur-rasool, H.; Ahmed, M.; Hasnain, S.; Ahmed, A.; Carter, W.G. In Silico Design of Dual-Binding Site Anti-Cholinesterase Phytochemical Heterodimers as Treatment Options for Alzheimer’s Disease. Curr. Issues Mol. Biol. 2022, 44, 152–175. [Google Scholar] [CrossRef]
- Mella, M.; Moraga-Nicolás, F.; Machuca, J.; Quiroz, A.; Mutis, A.; Becerra, J.; Astudillo, Á.; Hormazábal, E. Acetylcholinesterase Inhibitory Activity from Amaryllis belladonna Growing in Chile: Enzymatic and Molecular Docking Studies. Nat. Prod. Res. 2022, 36, 1370–1374. [Google Scholar] [CrossRef]
- Kuzu, B.; Alagoz, M.A.; Demir, Y.; Gulcin, I.; Burmaoglu, S.; Algul, O. Structure-Based Inhibition of Acetylcholinesterase and Butyrylcholinesterase with 2-Aryl-6-Carboxamide Benzoxazole Derivatives: Synthesis, Enzymatic Assay, and in Silico Studies. Mol. Divers. 2025, 29, 671–693. [Google Scholar] [CrossRef]
- Chaichompoo, W.; Rojsitthisak, P.; Pabuprapap, W.; Siriwattanasathien, Y.; Yotmanee, P.; Haritakun, W.; Suksamrarn, A. Stephapierrines A-H, New Tetrahydroprotoberberine and Aporphine Alkaloids from the Tubers of: Stephania Pierrei Diels and Their Anti-Cholinesterase Activities. RSC Adv. 2021, 11, 21153–21169. [Google Scholar] [CrossRef] [PubMed]
- Hamed, A.A.; El-Shiekh, R.A.; Mohamed, O.G.; Aboutabl, E.A.; Fathy, F.I.; Fawzy, G.A.; Al-Taweel, A.M.; Elsayed, T.R.; Tripathi, A.; Al-Karmalawy, A.A. Cholinesterase Inhibitors from an Endophytic Fungus Aspergillus Niveus Fv-Er401: Metabolomics, Isolation and Molecular Docking. Molecules 2023, 28, 2559. [Google Scholar] [CrossRef]
- Silva, L.; Ferreira, E.; Maryam; Espejo-Román, J.; Costa, G.; Cruz, J.; Kimani, N.; Costa, J.; Bittencourt, J.; Cruz, J.; et al. Galantamine Based Novel Acetylcholinesterase Enzyme Inhibitors: A Molecular Modeling Design Approach. Molecules 2023, 28, 1035. [Google Scholar] [CrossRef] [PubMed]
- Haider, R.; Agnello, L.; Shah, S.M.; Sufyan, M.; Khan, N.; Nazir, A.; Ciaccio, M.; Rehman, S. Evaluating the Antioxidant, Anti-inflammatory, and Neuroprotective Potential of Fruiting Body and Mycelium Extracts from Edible Yellow Morel (Morchella esculenta L. Pers.). J. Food Sci. 2025, 90, e17619. [Google Scholar] [CrossRef]
- Mateev, E.; Kondeva-Burdina, M.; Georgieva, M.; Zlatkov, A. Repurposing of FDA-Approved Drugs as Dual-Acting MAO-B and AChE Inhibitors against Alzheimer’s Disease: An in Silico and in Vitro Study. J. Mol. Graph. Model. 2023, 122, 108471. [Google Scholar] [CrossRef] [PubMed]
- Ordoñez, W.O.C.; Palomino, N.V.; Varela, P.E.V.; Martínez, I.B.; Alves, L.B.; Giuliatti, S. Alkaloids from Caliphruria Subedentata (Amaryllidaceae) as Regulators of AChE, BuChE, NMDA and GSK3 Activity: An In Vitro and In Silico Approach for Mimicking Alzheimer’s Disease. Neurochem. Res. 2025, 50, 116. [Google Scholar] [CrossRef]
- Anukanon, S.; Pongpamorn, P.; Tiyabhorn, W.; Chatwichien, J.; Niwetmarin, W.; Sessions, R.B.; Ruchirawat, S.; Thasana, N. In Silico-Guided Rational Drug Design and Semi-Synthesis of C(2)-Functionalized Huperzine A Derivatives as Acetylcholinesterase Inhibitors. ACS Omega 2021, 6, 19924–19939. [Google Scholar] [CrossRef]
- Barakat, A.; Alshahrani, S.; Al-Majid, A.M.; Ali, M.; Altowyan, M.S.; Islam, M.S.; Alamary, A.S.; Ashraf, S.; Ul-Haq, Z. Synthesis of a New Class of Spirooxindole-Benzo[b]Thiophene-Based Molecules as Acetylcholinesterase Inhibitors. Molecules 2020, 25, 4671. [Google Scholar] [CrossRef]
- Chennai, H.Y.; Belaidi, S.; Bourougaa, L.; Ouassaf, M.; Sinha, L.; Samadi, A.; Chtita, S. Identification of Potent Acetylcholinesterase Inhibitors as New Candidates for Alzheimer Disease via Virtual Screening, Molecular Docking, Dynamic Simulation, and Molecular Mechanics–Poisson–Boltzmann Surface Area Calculations. Molecules 2024, 29, 1232. [Google Scholar] [CrossRef]
- Saeed, S.; Zahoor, A.F.; Kamal, S.; Raza, Z.; Bhat, M.A. Unfolding the Antibacterial Activity and Acetylcholinesterase Inhibition Potential of Benzofuran-Triazole Hybrids: Synthesis, Antibacterial, Acetylcholinesterase Inhibition, and Molecular Docking Studies. Molecules 2023, 28, 6007. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Shi, H. Study on the Active Ingredients of Shenghui Decoction Inhibiting Acetylcholinesterase Based on Molecular Docking and Molecular Dynamics Simulation. Medicine 2023, 102, e34909. [Google Scholar] [CrossRef]
- Thai, Q.M.; Nguyen, T.H.; Lenon, G.B.; Thu Phung, H.T.; Horng, J.-T.; Tran, P.-T.; Ngo, S.T. Estimating AChE Inhibitors from MCE Database by Machine Learning and Atomistic Calculations. J. Mol. Graph. Model. 2025, 134, 108906. [Google Scholar] [CrossRef] [PubMed]
- SahIn, K.; Durdagi, S. Combined Ligand and Structure-Based Virtual Screening Approaches for Identification of Novel Ache Inhibitors. Turk. J. Chem. 2020, 44, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Makarian, M.; Gonzalez, M.; Salvador, S.M.; Lorzadeh, S.; Hudson, P.K.; Pecic, S. Synthesis, Kinetic Evaluation and Molecular Docking Studies of Donepezil-Based Acetylcholinesterase Inhibitors. J. Mol. Struct. 2022, 1247, 131425. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Li, S.; Liu, C.; Liang, J.; Nong, Y.; Chen, M.; Sun, R. Quaternity Method for Integrated Screening, Separation, Extraction Optimization, and Bioactivity Evaluation of Acetylcholinesterase Inhibitors from Sophora flavescens Aiton. Phytochem. Anal. 2025, 36, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Hemaida, A.Y.; Hassan, G.S.; Maarouf, A.R.; Joubert, J.; El-Emam, A.A. Synthesis and Biological Evaluation of Thiazole-Based Derivatives as Potential Acetylcholinesterase Inhibitors. ACS Omega 2021, 6, 19202–19211. [Google Scholar] [CrossRef]
- Talukder, M.E.K.; Akhter, S.; Ahammad, F.; Aktar, A.; Islam, M.S.; Laboni, A.A.; Afroze, M.; Khan, M.; Uddin, M.J.; Rahman, M.M. Multi-Modal Neuroprotection of Argemone mexicana L. against Alzheimer’s Disease: In Vitro and in Silico Study. Heliyon 2024, 10, e37178. [Google Scholar] [CrossRef]
- El-Hawwary, S.S.; Abd Almaksoud, H.M.; Saber, F.R.; Elimam, H.; Sayed, A.M.; El Raey, M.A.; Abdelmohsen, U.R. Green-Synthesized Zinc Oxide Nanoparticles, Anti-Alzheimer Potential and the Metabolic Profiling of: Sabal Blackburniana Grown in Egypt Supported by Molecular Modelling. RSC Adv. 2021, 11, 18009–18025. [Google Scholar] [CrossRef]
- Banu, Z.; Poduri, R.R.; Bhattamisra, S.K. Phytochemical Profiling, in Silico Molecular Docking and ADMET Prediction of Alkaloid Rich Fraction of Elaeocarpus angustifolius Blume Seeds against Alzheimer’s Disease. Nat. Prod. Res. 2025, 39, 1–9. [Google Scholar] [CrossRef]
- Musa, M.S.; Islam, M.T.; Billah, W.; Hossain, M.S.; Rahat, M.S.S.; Bayil, I.; Munni, Y.A.; Ganguli, S. Structure-Based Virtual Screening of Trachyspermum Ammi Metabolites Targeting Acetylcholinesterase for Alzheimer’s Disease Treatment. PLoS ONE 2024, 19, e0311401. [Google Scholar] [CrossRef]
- de Andrade Medeiros, S.R.; Bezerra, I.C.; Pedroza, L.A.; da Silva, A.J.; Martins, R.M.; Menezes, T.M.; de Melo, A.C.; Neves, J.L.; Gubert, P.; de Melo Filho, A.A. Evaluation of Bauhinia ungulata Essential Oil as a New Acetylcholinesterase Inhibitor from an in silico and in vitro Perspective in the Northern Amazon of Brazil. J. Oleo Sci. 2024, 73, ess23148. [Google Scholar] [CrossRef]
- Moussaoui, S.; Mokrani, E.H.; Kabouche, Z.; Guendouze, A.; Laribi, A.; Bradai, N.; Bensouici, C.; Yilmaz, M.A.; Cakir, O.; Tarhan, A. Evaluation of Polyphenolic Profile, Antioxidant, Anti-Cholinesterase, and Anti-Alpha-Amylase Activities of Pistacia lentiscus L. Leaves. Nat. Prod. Res. 2025, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kadi, I.; Seyhan, G.; Zebbiche, Z.; Sari, S.; Barut, B.; Boumoud, T.; Mermer, A.; Boulebd, H. Novel 2-Alkoxy-3-Cyanopyridine Derivatives as Cholinesterase Inhibitors: Synthesis, Biological Evaluation, and In Silico Investigations. Chem. Biodivers. 2025, 39, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Asgarshamsi, M.H.; Fassihi, A.; Dehkordi, M.M. Design, Synthesis, Molecular Docking, and Molecular Dynamics Simulation Studies of Novel 3-Hydroxypyridine-4-one Derivatives as Potential Acetylcholinesterase Inhibitors. Chem. Biodivers. 2023, 20, e202300325. [Google Scholar] [CrossRef]
- Puopolo, T.; Liu, C.; Ma, H.; Seeram, N.P. Inhibitory Effects of Cannabinoids on Acetylcholinesterase and Butyrylcholinesterase Enzyme Activities. Med. Cannabis Cannabinoids 2022, 5, 85–94. [Google Scholar] [CrossRef]
- Negru, D.C.; Bungau, S.G.; Radu, A.; Tit, D.M.; Radu, A.-F.; Nistor-Cseppento, D.C.; Negru, P.A. Evaluation of the Alkaloids as Inhibitors of Human Acetylcholinesterase by Molecular Docking and ADME Prediction. In Vivo 2025, 39, 236–250. [Google Scholar] [CrossRef]
- Nair, A.C.; Benny, S.; Aneesh, T.P.; Sudheesh, M.S.; Lakshmi, P.K. Comprehensive Profiling of Traditional Herbomineral Formulation Manasamitra Vatakam in Rat Brain Following Oral Administration and In-Silico Screening of the Identified Compound for Anti-Alzheimer’s Activity. J. Ethnopharmacol. 2025, 338, 119024. [Google Scholar] [CrossRef]
- Singh, M.; Jindal, D.; Kumar, R.; Pancham, P.; Haider, S.; Gupta, V.; Mani, S.; R, R.; Tiwari, R.K.; Chanda, S. Molecular Docking and Network Pharmacology Interaction Analysis of Gingko Biloba (EGB761) Extract with Dual Target Inhibitory Mechanism in Alzheimer’s Disease. J. Alzheimer’s Dis. 2023, 93, 705–726. [Google Scholar] [CrossRef]
- Refaay, D.A.; Abdel-Hamid, M.I.; Alyamani, A.A.; Abdel Mougib, M.; Ahmed, D.M.; Negm, A.; Mowafy, A.M.; Ibrahim, A.A.; Mahmoud, R.M. Growth Optimization and Secondary Metabolites Evaluation of Anabaena Variabilis for Acetylcholinesterase Inhibition Activity. Plants 2022, 11, 735. [Google Scholar] [CrossRef]
- Farihi, A.; Bouhrim, M.; Chigr, F.; Elbouzidi, A.; Bencheikh, N.; Zrouri, H.; Nasr, F.A.; Parvez, M.K.; Alahdab, A.; Ahami, A.O.T. Exploring Medicinal Herbs’ Therapeutic Potential and Molecular Docking Analysis for Compounds as Potential Inhibitors of Human Acetylcholinesterase in Alzheimer’s Disease Treatment. Medicina 2023, 59, 1812. [Google Scholar] [CrossRef]
- Reshetnikov, D.V.; Ivanov, I.D.; Baev, D.S.; Rybalova, T.V.; Mozhaitsev, E.S.; Patrushev, S.S.; Vavilin, V.A.; Tolstikova, T.G.; Shults, E.E. Design, Synthesis and Assay of Novel Methylxanthine–Alkynylmethylamine Derivatives as Acetylcholinesterase Inhibitors. Molecules 2022, 27, 8787. [Google Scholar] [CrossRef] [PubMed]
- Grodner, B.; Napiórkowska, M.; Pisklak, D.M. In Vitro and in Silico Kinetic Studies of Patented 1,7-diethyl and 1,7-dimethyl Aminoalkanol Derivatives as New Inhibitors of Acetylcholinesterase. Int. J. Mol. Sci. 2022, 23, 270. [Google Scholar] [CrossRef] [PubMed]
- Drozdowska, D.; Maliszewski, D.; Wróbel, A.; Ratkiewicz, A.; Sienkiewicz, M. New Benzamides as Multi-Targeted Compounds: A Study on Synthesis, AChE and BACE1 Inhibitory Activity and Molecular Docking. Int. J. Mol. Sci. 2023, 24, 14901. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- El Khatabi, K.; Aanouz, I.; El-Mernissi, R.; Singh, A.K.; Ajana, M.A.; Lakhlifi, T.; Kumar, S.; Bouachrine, M. Integrated 3D-QSAR, Molecular Docking, and Molecular Dynamics Simulation Studies on 1,2,3-Triazole Based Derivatives for Designing New Acetylcholinesterase Inhibitors. Turk. J. Chem. 2021, 45, 647–660. [Google Scholar] [CrossRef]
- Bilginer, S.; Anil, B.; Koca, M.; Demir, Y.; Gülçin, I. Novel Mannich Bases with Strong Carbonic Anhydrases and Acetylcholinesterase Inhibition Effects: 3-(Aminomethyl)-6-{3-[4-(Trifluoromethyl)Phenyl]Acryloyl}-2(3H)-Benzoxazolones. Turk. J. Chem. 2021, 45, 805–818. [Google Scholar] [CrossRef]
- Suwanhom, P.; Saetang, J.; Khongkow, P.; Nualnoi, T.; Tipmanee, V.; Lomlim, L. Synthesis, Biological Evaluation, and in Silico Studies of New Acetylcholinesterase Inhibitors Based on Quinoxaline Scaffold. Molecules 2021, 26, 4895. [Google Scholar] [CrossRef]
- Zarei, S.; Shafiei, M.; Firouzi, M.; Firoozpour, L.; Divsalar, K.; Asadipour, A.; Akbarzadeh, T.; Foroumadi, A. Design, Synthesis and Biological Assessment of New 1-Benzyl-4-((4-Oxoquinazolin-3(4H)-Yl)Methyl) Pyridin-1-Ium Derivatives (BOPs) as Potential Dual Inhibitors of Acetylcholinesterase and Butyrylcholinesterase. Heliyon 2021, 7, e06683. [Google Scholar] [CrossRef]
- Chowdhury, S.; Rahman, A.; Hussain, F.; Rahman, S.M.A. Synthesis, characterization and in vitro, in vivo, in silico biological evaluations of substituted benzimidazole derivatives. Saudi J. Biol. Sci. 2022, 29, 239–250. [Google Scholar] [CrossRef]
- Ramos, A.S.F.; Techert, S. Influence of the Water Structure on the Acetylcholinesterase Efficiency. Biophys. J. 2005, 89, 1990–2003. [Google Scholar] [CrossRef]
- Bondžić, A.M.; Lazarević-Pašti, T.D.; Leskovac, A.R.; Petrović, S.Ž.; Čolović, M.B.; Parac-Vogt, T.N.; Janjić, G.V. A New Acetylcholinesterase Allosteric Site Responsible for Binding Voluminous Negatively Charged Molecules—The Role in the Mechanism of AChE Inhibition. Eur. J. Pharm. Sci. 2020, 151, 105376. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.J.; Rupprecht, L.E.; Hayes, M.R.; Blendy, J.A.; Schmidt, H.D. Galantamine, an Acetylcholinesterase Inhibitor and Positive Allosteric Modulator of Nicotinic Acetylcholine Receptors, Attenuates Nicotine Taking and Seeking in Rats. Neuropsychopharmacology 2012, 37, 2310–2321. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In Silico Pharmacology for Drug Discovery: Methods for Virtual Ligand Screening and Profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Cao, Y.; Zhang, L. Exploring the Computational Methods for Protein-Ligand Binding Site Prediction. Comput. Struct. Biotechnol. J. 2020, 18, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Dutkiewicz, Z. Computational Methods for Calculation of Protein-Ligand Binding Affinities in Structure-Based Drug Design. Phys. Sci. Rev. 2022, 7, 933–968. [Google Scholar] [CrossRef]
- Agu, P.C.; Afiukwa, C.A.; Orji, O.U.; Ezeh, E.M.; Ofoke, I.H.; Ogbu, C.O.; Ugwuja, E.I.; Aja, P.M. Molecular Docking as a Tool for the Discovery of Molecular Targets of Nutraceuticals in Diseases Management. Sci. Rep. 2023, 13, 13398. [Google Scholar] [CrossRef]
- Khare, N.; Maheshwari, S.K.; Jha, A.K. Screening and Identification of Secondary Metabolites in the Bark of Bauhinia variegata to Treat Alzheimer’s Disease by Using Molecular Docking and Molecular Dynamics Simulations. J. Biomol. Struct. Dyn. 2021, 39, 5988–5998. [Google Scholar] [CrossRef]
- Yi, P.; Zhang, Z.; Huang, S.; Huang, J.; Peng, W.; Yang, J. Integrated Meta-Analysis, Network Pharmacology, and Molecular Docking to Investigate the Efficacy and Potential Pharmacological Mechanism of Kai-Xin-San on Alzheimer’s Disease. Pharm. Biol. 2020, 58, 932–943. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, B.; Pang, X.; Yang, R.; Chen, M.; Zhao, J.; Wang, J.; Wang, Z.; Yu, Z.; Wang, Y.; et al. Network Pharmacology-Based Analysis of Xiao-Xu-Ming Decoction on the Treatment of Alzheimer’s Disease. Front. Pharmacol. 2020, 11, 595254. [Google Scholar] [CrossRef]
- Hussein, R.A.; Afifi, A.H.; Soliman, A.A.F.; El Shahid, Z.A.; Zoheir, K.M.A.; Mahmoud, K.M. Neuroprotective Activity of Ulmus pumila L. in Alzheimer’s Disease in Rats; Role of Neurotrophic Factors. Heliyon 2020, 6, e05678. [Google Scholar] [CrossRef]
- Kareti, S.R.; Pharm, S.M. In Silico Molecular Docking Analysis of Potential Anti-Alzheimer’s Compounds Present in Chloroform Extract of Carissa Carandas Leaf Using Gas Chromatography MS/MS. Curr. Ther. Res. Clin. Exp. 2020, 93, 100615. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.P.; Ren, H.Q.; Liu, E.Y.L.; Leung, K.W.; Guo, S.C.; Duan, R.; Dong, T.T.X.; Tsim, K.W.K. The Cholinesterase Inhibitory Properties of Stephaniae Tetrandrae Radix. Molecules 2020, 25, 5914. [Google Scholar] [CrossRef]
- Heo, J.H.; Eom, B.H.; Ryu, H.W.; Kang, M.G.; Park, J.E.; Kim, D.Y.; Kim, J.H.; Park, D.; Oh, S.R.; Kim, H. Acetylcholinesterase and Butyrylcholinesterase Inhibitory Activities of Khellactone Coumarin Derivatives Isolated from Peucedanum Japonicum Thurnberg. Sci. Rep. 2020, 10, 21695. [Google Scholar] [CrossRef]
- Azevedo, R.D.S.; Falcão, K.V.G.; Assis, C.R.D.; Martins, R.M.G.; Araújo, M.C.; Yogui, G.T.; Neves, J.L.; Seabra, G.M.; Maia, M.B.S.; Amaral, I.P.G.; et al. Effects of Pyriproxyfen on Zebrafish Brain Mitochondria and Acetylcholinesterase. Chemosphere 2021, 263, 128029. [Google Scholar] [CrossRef] [PubMed]
- Ogata, N.; Tagishi, H.; Tsuji, M. Inhibition of Acetylcholinesterase by Wood Creosote and Simple Phenolic Compounds. Chem. Pharm. Bull. 2020, 68, 1193–1200. [Google Scholar] [CrossRef]
- Kocakaya, S.O.; Ertas, A.; Yener, I.; Ercan, B.; Oral, E.V.; Akdeniz, M.; Kaplaner, E.; Topcu, G.; Kolak, U. Selective In-Vitro Enzymes’ Inhibitory Activities of Fingerprints Compounds of Salvia Species and Molecular Docking Simulations. Iran. J. Pharm. Res. 2020, 19, 187–198. [Google Scholar] [CrossRef]
- Karasova, J.Z.; Mzik, M.; Kucera, T.; Vecera, Z.; Kassa, J.; Sestak, V. Interaction of Cucurbit [7]Uril with Oxime K027, Atropine, and Paraoxon: Risky or Advantageous Delivery System? Int. J. Mol. Sci. 2020, 21, 7883. [Google Scholar] [CrossRef] [PubMed]
- Adalat, B.; Rahim, F.; Taha, M.; Alshamrani, F.J.; Anouar, E.H.; Uddin, N.; Shah, S.A.A.; Ali, Z.; Zakaria, Z.A. Synthesis of Benzimidazole–Based Analogs as Anti Alzheimer’s Disease Compounds and Their Molecular Docking Studies. Molecules 2020, 25, 4828. [Google Scholar] [CrossRef]
- Pitchai, A.; Rajaretinam, R.K.; Mani, R.; Nagarajan, N. Molecular Interaction of Human Acetylcholinesterase with Trans-Tephrostachin and Derivatives for Alzheimer’s Disease. Heliyon 2020, 6, e04930. [Google Scholar] [CrossRef]
- Tran, T.S.; Tran, T.D.; Tran, T.H.; Mai, T.T.; Nguyen, N.L.; Thai, K.M.; Le, M.T. Synthesis, in Silico and in Vitro Evaluation of Some Flavone Derivatives for Acetylcholinesterase and BACE-1 Inhibitory Activity. Molecules 2020, 25, 4064. [Google Scholar] [CrossRef]
- Jusril, N.A.; Juhari, A.N.N.M.; Bakar, S.I.A.; Saad, W.M.M.; Adenan, M.I. Combining in Silico and in Vitro Studies to Evaluate the Acetylcholinesterase Inhibitory Profile of Different Accessions and the Biomarker Triterpenes of Centella Asiatica. Molecules 2020, 25, 3353. [Google Scholar] [CrossRef] [PubMed]
- Kausar, N.; Murtaza, S.; Arshad, M.N.; Zaib Saleem, R.S.; Asiri, A.M.; Kausar, S.; Altaf, A.A.; Tatheer, A.; Elnaggar, A.Y.; El-Bahy, S.M. Design, Synthesis, Crystal Structure, in Vitro Cytotoxicity Evaluation, Density Functional Theory Calculations and Docking Studies of 2-(Benzamido) Benzohydrazide Derivatives as Potent AChE and BChE Inhibitors. RSC Adv. 2021, 12, 154–167. [Google Scholar] [CrossRef]
- Krátký, M.; Štěpánková, Š.; Konečná, K.; Svrčková, K.; Maixnerová, J.; Švarcová, M.; Jand’ourek, O.; Trejtnar, F.; Vinšová, J. Novel Aminoguanidine Hydrazone Analogues: From Potential Antimicrobial Agents to Potent Cholinesterase Inhibitors. Pharmaceuticals 2021, 14, 1229. [Google Scholar] [CrossRef]
- Iqbal, D.; Khan, M.S.; Waiz, M.; Rehman, M.T.; Alaidarous, M.; Jamal, A.; Alothaim, A.S.; AlAjmi, M.F.; Alshehri, B.M.; Banawas, S.; et al. Exploring the Binding Pattern of Geraniol with Acetylcholinesterase through In Silico Docking, Molecular Dynamics Simulation, and In Vitro Enzyme Inhibition Kinetics Studies. Cells 2021, 10, 3533. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, N.M.; Mostafa, A.M.; Ashour, M.L.; Elhady, S.S. Neuroprotective Effects of Black Pepper Cold-Pressed Oil on Scopolamine-Induced Oxidative Stress and Memory Impairment in Rats. Antioxidants 2021, 10, 1993. [Google Scholar] [CrossRef] [PubMed]
- Onikanni, A.S.; Lawal, B.; Olusola, A.O.; Olugbodi, J.O.; Sani, S.; Ajiboye, B.O.; Ilesanmi, O.B.; Alqarni, M.; Mostafahedeab, G.; Obaidullah, A.J.; et al. Sterculia Tragacantha Lindl Leaf Extract Ameliorates STZ-Induced Diabetes, Oxidative Stress, Inflammation and Neuronal Impairment. J. Inflamm. Res. 2021, 14, 6749–6764. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Lin, M.; Zhuo, W.; Li, Y. Chemical Constituents from the Wild Atractylodes Macrocephala Koidz and Acetylcholinesterase Inhibitory Activity Evaluation as Well as Molecular Docking Study. Molecules 2021, 26, 7299. [Google Scholar] [CrossRef]
- Johnson, T.O.; Ojo, O.A.; Ikiriko, S.; Ogunkua, J.; Akinyemi, G.O.; Rotimi, D.E.; Oche, J.R.; Adegboyega, A.E. Biochemical Evaluation and Molecular Docking Assessment of Cymbopogon Citratus as a Natural Source of Acetylcholine Esterase (AChE)- Targeting Insecticides. Biochem. Biophys. Rep. 2021, 28, 101175. [Google Scholar] [CrossRef]
- Upadhyay, S.P.; Singh, V.; Sharma, R.; Zhou, J.; Thapa, P.; Johnson, D.K.; Keightley, A.; Chen, M.; Suo, W.; Sharma, M. Influence of Ligand Geometry on Cholinesterase Enzyme—A Comparison of 1-Isoindolinone Based Structural Analog with Donepezil. J. Mol. Struct. 2022, 1247, 131385. [Google Scholar] [CrossRef]
- Kiziltas, H.; Goren, A.C.; Alwasel, S.H.; Gulcin, İ. Sahlep (Dactylorhiza Osmanica): Phytochemical Analyses by LC-HRMS, Molecular Docking, Antioxidant Activity, and Enzyme Inhibition Profiles. Molecules 2022, 27, 6907. [Google Scholar] [CrossRef]
- Lolak, N.; Akocak, S.; Durgun, M.; Duran, H.E.; Necip, A.; Türkeş, C.; Işık, M.; Beydemir, Ş. Novel Bis-Ureido-Substituted Sulfaguanidines and Sulfisoxazoles as Carbonic Anhydrase and Acetylcholinesterase Inhibitors. Mol. Divers. 2023, 27, 1735–1749. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Rahim, F.; Ullah, H.; Khan, S.; Sarfraz, M.; Iqbal, R.; Suleman, F.; Al-Sadoon, M.K. Design, Synthesis, In Vitro Biological Evaluation and In Silico Molecular Docking Study of Benzimidazole-Based Oxazole Analogues: A Promising Acetylcholinesterase and Butyrylcholinesterase Inhibitors. Molecules 2023, 28, 7015. [Google Scholar] [CrossRef]
- Jaśkiewicz, A.; Budryn, G.; Carmena-Bargueño, M.; Pérez-Sánchez, H. Evaluation of Activity of Sesquiterpene Lactones and Chicory Extracts as Acetylcholinesterase Inhibitors Assayed in Calorimetric and Docking Simulation Studies. Nutrients 2022, 14, 3633. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, M.; Dimitrov, I.; Ivanov, S.; Georgiev, B.; Berkov, S.; Zheleva-Dimitrova, D.; Doytchinova, I. Virtual Screening and Hit Selection of Natural Compounds as Acetylcholinesterase Inhibitors. Molecules 2022, 27, 3139. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Lu, T.; Wang, J.; Nie, X.; Xiong, W.; Yin, Z.; Peng, D. Design, Synthesis and Bioactivity Evaluation of Coumarin–BMT Hybrids as New Acetylcholinesterase Inhibitors. Molecules 2022, 27, 2142. [Google Scholar] [CrossRef]
- Xiao, Y.; Liang, W.; Liu, D.; Zhang, Z.; Chang, J.; Zhu, D. Isolation and Acetylcholinesterase Inhibitory Activity of Asterric Acid Derivatives Produced by Talaromyces Aurantiacus FL15, an Endophytic Fungus from Huperzia Serrata. 3 Biotech 2022, 12, 60. [Google Scholar] [CrossRef]
- Rahim, F.; Ullah, H.; Taha, M.; Hussain, R.; Sarfraz, M.; Iqbal, R.; Iqbal, N.; Khan, S.; Ali Shah, S.A.; Albalawi, M.A.; et al. Synthesis of New Triazole-Based Thiosemicarbazone Derivatives as Anti-Alzheimer’s Disease Candidates: Evidence-Based In Vitro Study. Molecules 2022, 28, 21. [Google Scholar] [CrossRef]
- Liao, Y.; Hu, X.; Pan, J.; Zhang, G. Inhibitory Mechanism of Baicalein on Acetylcholinesterase: Inhibitory Interaction, Conformational Change, and Computational Simulation. Foods 2022, 11, 168. [Google Scholar] [CrossRef]
- Tarabasz, D.; Szczeblewski, P.; Laskowski, T.; Płaziński, W.; Baranowska-Wójcik, E.; Szwajgier, D.; Kukula-Koch, W.; Meissner, H.O. The Distribution of Glucosinolates in Different Phenotypes of Lepidium Peruvianum and Their Role as Acetyl- and Butyrylcholinesterase Inhibitors—In Silico and In Vitro Studies. Int. J. Mol. Sci. 2022, 23, 4858. [Google Scholar] [CrossRef]
- da Silva Mesquita, R.; Kyrylchuk, A.; Cherednichenko, A.; Costa Sá, I.S.; Macedo Bastos, L.; Moura Araújo da Silva, F.; Saraiva Nunomura, R.d.C.; Grafov, A. In Vitro and In Silico Evaluation of Cholinesterase Inhibition by Alkaloids Obtained from Branches of Abuta Panurensis Eichler. Molecules 2022, 27, 3138. [Google Scholar] [CrossRef]
- Wu, T.; Hou, W.; Liu, C.; Li, S.; Zhang, Y. Efficient Combination of Complex Chromatography, Molecular Docking and Enzyme Kinetics for Exploration of Acetylcholinesterase Inhibitors from Poria Cocos. Molecules 2023, 28, 1228. [Google Scholar] [CrossRef] [PubMed]
- Eltahawy, N.A.; Ali, A.I.; Ibrahim, S.A.; Nafie, M.S.; Sindi, A.M.; Alkharobi, H.; Almalki, A.J.; Badr, J.M.; Elhady, S.S.; Abdelhameed, R.F.A. Analysis of Marrubiin in Marrubium alysson L. Extract Using Advanced HPTLC: Chemical Profiling, Acetylcholinesterase Inhibitory Activity, and Molecular Docking. Metabolites 2023, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Jamal, Q.M.S.; Khan, M.I.; Alharbi, A.H.; Ahmad, V.; Yadav, B.S. Identification of Natural Compounds of the Apple as Inhibitors against Cholinesterase for the Treatment of Alzheimer’s Disease: An In Silico Molecular Docking Simulation and ADMET Study. Nutrients 2023, 15, 1579. [Google Scholar] [CrossRef]
- Pishgouii, F.; Lotfi, S.; Sedaghati, E. Anti-AChE and Anti-BuChE Screening of the Fermentation Broth Extracts from Twelve Aspergillus Isolates and GC-MS and Molecular Docking Studies of the Most Active Extracts. Appl. Biochem. Biotechnol. 2023, 195, 5199–5216. [Google Scholar] [CrossRef]
- Belaiba, M.; Aldulaijan, S.; Messaoudi, S.; Abedrabba, M.; Dhouib, A.; Bouajila, J. Evaluation of Biological Activities of Twenty Flavones and In Silico Docking Study. Molecules 2023, 28, 2419. [Google Scholar] [CrossRef]
- Laghchioua, F.E.; da Silva, C.F.M.; Pinto, D.C.G.A.; Cavaleiro, J.A.S.; Mendes, R.F.; Paz, F.A.A.; Faustino, M.A.F.; Rakib, E.M.; Neves, M.G.P.M.S.; Pereira, F.; et al. Design of Promising Thiazoloindazole-Based Acetylcholinesterase Inhibitors Guided by Molecular Docking and Experimental Insights. ACS Chem. Neurosci. 2024, 15, 2853–2869. [Google Scholar] [CrossRef]
- Raturi, A.; Yadav, V.; Hoda, N.; Subbarao, N.; Chaudhry, S.A. In Silico Identification of Colchicine Derivatives as Novel and Potential Inhibitors Based on Molecular Docking and Dynamic Simulations Targeting Multifactorial Drug Targets Involved in Alzheimer’s Disease. J. Biomol. Struct. Dyn. 2024, 42, 11555–11573. [Google Scholar] [CrossRef] [PubMed]
- Faloye, K.O.; Mahmud, S.; Fakola, E.G.; Oyetunde, Y.M.; Fajobi, S.J.; Ugwo, J.P.; Olusola, A.J.; Famuyiwa, S.O.; Olajubutu, O.G.; Oguntade, T.I.; et al. Revealing the Acetylcholinesterase Inhibitory Potential of Phyllanthus amarus and Its Phytoconstituents: In Vitro and in Silico Approach. Bioinform. Biol. Insights 2022, 16, 1–11. [Google Scholar] [CrossRef]
- Kurbanova, M.; Maharramov, A.; Safarova, A.; Ahmad, S.; El Bakri, Y. Molecular Docking Study and Molecular Dynamics Simulation of Ethyl 3,5-diphenyl-1 H. -pyrrole-2-carboxylate and (Z)-ethyl-2-(3-oxo-1,3-diphenylprop-1-enylamino)Acetate. J. Biochem. Mol. Toxicol. 2022, 36, e23013. [Google Scholar] [CrossRef]
- Gholami, A.; Minai-Tehrani, D.; Eriksson, L.A. In Silico and in Vitro Studies Confirm Ondansetron as a Novel Acetylcholinesterase and Butyrylcholinesterase Inhibitor. Sci. Rep. 2023, 13, 643. [Google Scholar] [CrossRef]
- de Almeida, R.B.M.; Barbosa, D.B.; do Bomfim, M.R.; Amparo, J.A.O.; Andrade, B.S.; Costa, S.L.; Campos, J.M.; Cruz, J.N.; Santos, C.B.R.; Leite, F.H.A.; et al. Identification of a Novel Dual Inhibitor of Acetylcholinesterase and Butyrylcholinesterase: In Vitro and In Silico Studies. Pharmaceuticals 2023, 16, 95. [Google Scholar] [CrossRef] [PubMed]
- Mendes, G.O.; Pita, S.S.d.R.; Carvalho, P.B.d.; Silva, M.P.d.; Taranto, A.G.; Leite, F.H.A. Molecular Multi-Target Approach for Human Acetylcholinesterase, Butyrylcholinesterase and β-Secretase 1: Next Generation for Alzheimer’s Disease Treatment. Pharmaceuticals 2023, 16, 880. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, N.; Darshan, V.M.D.; Venketesh, V.; Pradhan, S.S.; Garg, A.; Sivaramakrishnan, V.; Kanchi, S.; Mahalingam, S.M. Synthesis, Computational Docking and Molecular Dynamics Studies of a New Class of Spiroquinoxalinopyrrolidine Embedded Chromanone Hybrids as Potent Anti-Cholinesterase Agents. RSC Adv. 2024, 14, 18815–18831. [Google Scholar] [CrossRef]
- Saha, B.; Das, A.; Jangid, K.; Kumar, A.; Kumar, V.; Jaitak, V. Identification of Coumarin Derivatives Targeting Acetylcholinesterase for Alzheimer’s Disease by Field-Based 3D-QSAR, Pharmacophore Model-Based Virtual Screening, Molecular Docking, MM/GBSA, ADME and MD Simulation Study. Curr. Res. Struct. Biol. 2024, 7, 100124. [Google Scholar] [CrossRef]
- Keçeci Sarıkaya, M.; Yıldırım, Ş.; Kocyigit, U.M.; Ceylan, M.; Yırtıcı, Ü.; Eyüpoğlu, V. Novel Aminothiazole–Chalcone Analogs: Synthesis, Evaluation Acetylcholinesterase Activity, In Silico Analysis. Chem. Biodivers. 2025, e202402777. [Google Scholar] [CrossRef]
- Rawat, K.; Tewari, D.; Bisht, A.; Chandra, S.; Tiruneh, Y.K.; Hassan, H.M.; Al-Emam, A.; Sindi, E.R.; Al-Dies, A.-A.M. Identification of AChE Targeted Therapeutic Compounds for Alzheimer’s Disease: An in-Silico Study with DFT Integration. Sci. Rep. 2024, 14, 30356. [Google Scholar] [CrossRef]
- Emam, M.; El-Newary, S.A.; Aati, H.Y.; Wei, B.; Seif, M.; Ibrahim, A.Y. Anti-Alzheimer’s Potency of Rich Phenylethanoid Glycosides Extract from Marrubium vulgare L.: In Vitro and In Silico Studies. Pharmaceuticals 2024, 17, 1282. [Google Scholar] [CrossRef]
- Al-Maqtari, H.M.; Hasan, A.H.; Suleiman, M.; Ahmad Zahidi, M.A.; Noamaan, M.A.; Alexyuk, P.; Alexyuk, M.; Bogoyavlenskiy, A.; Jamalis, J. Benzyloxychalcone Hybrids as Prospective Acetylcholinesterase Inhibitors against Alzheimer’s Disease: Rational Design, Synthesis, In Silico ADMET Prediction, QSAR, Molecular Docking, DFT, and Molecular Dynamic Simulation Studies. ACS Omega 2024, 9, 32901–32919. [Google Scholar] [CrossRef]
- Szeleszczuk, Ł.; Pisklak, D.M.; Grodner, B. Thiamine and Thiamine Pyrophosphate as Non-Competitive Inhibitors of Acetylcholinesterase—Experimental and Theoretical Investigations. Molecules 2025, 30, 412. [Google Scholar] [CrossRef]
- Zhu, J.; Xu, Z.; Liu, X. Chemical Composition, Antioxidant Activities, and Enzyme Inhibitory Effects of Lespedeza bicolour Turcz. Essential Oil. J. Enzym. Inhib. Med. Chem. 2025, 40, 2460053. [Google Scholar] [CrossRef]
- Khedraoui, M.; Abchir, O.; Nour, H.; Yamari, I.; Errougui, A.; Samadi, A.; Chtita, S. An In Silico Study Based on QSAR and Molecular Docking and Molecular Dynamics Simulation for the Discovery of Novel Potent Inhibitor against AChE. Pharmaceuticals 2024, 17, 830. [Google Scholar] [CrossRef] [PubMed]
- Tabbiche, A.; Bouchama, A.; Fadli, K.; Ahmad, B.; Kumar, N.; Chiter, C.; Yahiaoui, M.; Zaidi, F.; Boudjemaa, K.; Dege, N.; et al. Development of New Benzil-Hydrazone Derivatives as Anticholinesterase Inhibitors: Synthesis, X-Ray Analysis, DFT Study and in Vitro/in Silico Evaluation. J. Biomol. Struct. Dyn. 2025, 43, 2518–2533. [Google Scholar] [CrossRef] [PubMed]
- Żołek, T.; Purgatorio, R.; Kłopotowski, Ł.; Catto, M.; Ostrowska, K. Coumarin Derivative Hybrids: Novel Dual Inhibitors Targeting Acetylcholinesterase and Monoamine Oxidases for Alzheimer’s Therapy. Int. J. Mol. Sci. 2024, 25, 12803. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Software | No. Refs. | Binding Affinity [Kcal/mol] | |||
|---|---|---|---|---|---|
| Mean | Min | Max | STDV | ||
| Vina | 33 | −9.76 | −19.3 | −3.7 | −2.45 |
| AutoDock4 | 20 | −7.68 | −12.75 | 81.61 | 11.47 |
| Schrödinger Suite | 17 | −13.40 | −114.6 | 0.8 | 18.10 |
| MOE | 14 | −8.25 | −14.7 | −4.4 | 2.36 |
| Glide | 8 | −9.92 | −15.2 | −4.7 | 2.44 |
| Biovia Discovery Studio | 6 | −13.04 | −32.2 | −4.6 | 8.40 |
| CDOCKER | 4 | −3.51 | −46.3 | 84.5 | 45.53 |
| GOLD | 3 | 14.27 | −65.8 | 60.1 | 54.95 |
| DSHC | 2 | −28.08 | −54.5 | −16.6 | 10.90 |
| FlexX | 3 | −24.63 | −36.5 | −11.8 | 4.52 |
| Smina | 2 | −10.92 | −15.0 | −8.6 | 1.70 |
| Achilles Docking Server | 1 | −7.79 | −8.5 | −6.9 | 0.68 |
| ArgusLab 4.0 | 1 | −9.72 | −12.7 | −8.9 | 0.98 |
| ClusPro y Pymol | 1 | −883.10 | −974.0 | −792.2 | 128.55 |
| DOCK | 1 | −30.71 | −52.4 | −14.0 | 12.42 |
| ICM Pro Molsoft | 1 | −15.79 | −18.4 | −12.7 | 1.68 |
| iGEMDOCK | 1 | −87.49 | −92.4 | −81.1 | 3.73 |
| PyVSvina | 1 | −10.60 | −10.60 | −10.60 | 0.0 |
| Surflex-Dock | 1 | −8.96 | −114.6 | 18.1 | 12.847 |
| Vina + Umbrella Sampling simulation | 1 | −37.14 | −53.3 | −18.5 | 14.53 |
| Donepezil | Galantamine | ||||||
|---|---|---|---|---|---|---|---|
| PDB | Affinity Energy (kcal/mol) | Software | Cite | PDB | Affinity Energy (kcal/mol) | Software | Cite |
| 1ACL | −6.3226 | MOE | [52] | 1W6R | −9.63 | AutoDock4 | [53] |
| 1C2B | −11.7 | Vina | [54] | −8.68 | [55] | ||
| 1EVE | −12.74 | ArgusLab 4.0 | [56] | 1DX6 | −28.53 | Vina | [57] |
| −9.81 | Glide | [58] | 4EY5 | −7.7 | Vina | [59] | |
| −9.81 | 1C2B | −7.9 | Blind docking | [60] | |||
| −6.49 | Schrödinger suite | [61] | 4EY6 | −7.91 | AutoDock4 | [62] | |
| 1OCE | −8.04 | MOE | [63] | −9.9 | [64] | ||
| 4BDT | −8.5 | [65] | −11.54 | Glide | [66] | ||
| 4EY5 | −10.5 | Vina | [59] | 59.74 | GOLD | [67] | |
| −12.42 | AutoDock4 | [68] | −9.28 | MOE | [69] | ||
| 4EY7 | −10.8 | Biovia Discovery studio | [70] | −7.07 | [71] | ||
| 4EY7 | −31.26 | CDOCKER | [72] | −14.2 | Smina | [73] | |
| −5.552 | Glide | [74] | −9.61 | Vina | [15] | ||
| −17.7 | ICM Pro Molsoft | [75] | −9.1 | [76] | |||
| −15.5 | MOE | [77] | 4EY7 | −9.268 | Glide | [74] | |
| −10.171 | Schrodinger suite | [78] | −9 | Vina | [79] | ||
| −18.909 | [80] | −8.9 | [81] | ||||
| −8.7 | Vina | [79] | −10.5 | [82] | |||
| −11.94 | [15] | 4M0E | −21.2 | FlexX | [83] | ||
| −11.8 | [84] | −7 | Glide | [84] | |||
| −10.5 | [85] | 5HFA | −8.2 | Biovia Discovery Studio | [86] | ||
| −11.7 | [87] | 6O4W | −10.4 | Vina | [88] | ||
| −18.1 | [73] | 6O4X | −8.02 | Glide | [89] | ||
| 4M0E | −45.18 | CDOCKER | [90] | NA | |||
| −8.271 | Schrödinger suite | [81] | |||||
| 4PQE | −8.6 | Biovia Discovery Studio | [91] | ||||
| 6O4W | −14.817 | Glide | [92] | ||||
| 6O4X | −11.1 | Vina | [93] | ||||
| 73EH | −11.6 | [94] | |||||
| No. References | PDB ID |
|---|---|
| 31 | 4EY7 |
| 14 | 4M0E |
| 13 | 4EY6 |
| 8 | 1EVE |
| 6 | 4EY5 |
| 4 | 4PQE, 6O4W |
| 3 | 1C2B, 4BDT, 6O4X, 7D9P |
| 2 | 1ACJ, 1DX6, 1OCE, 1W6R, 3LII, 4EY4, 6H12, 7D90, 7D9Q, 7XN1 |
| 1 | 1C2O, 1EA5, 1EEA, 1F8U, 1GQS, 1H23, 1O86, 1QON, 2ACK, 3I6M, 3I6Z, 5FPQ, 5FUM, 5HF5, 5HFA, 6CQV, 6CQZ, 6EUC, 6EYF, 6NTL, 6NTO, 6O50, 6O69, 6U37, 6WO4, 6WUZ, 6WVO, 6WVQ, 6XYU, 73EH, 7E3H |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reynoso-García, M.F.; Nicolás-Álvarez, D.E.; Tenorio-Barajas, A.Y.; Reyes-Chaparro, A. Structural Bioinformatics Applied to Acetylcholinesterase Enzyme Inhibition. Int. J. Mol. Sci. 2025, 26, 3781. https://doi.org/10.3390/ijms26083781
Reynoso-García MF, Nicolás-Álvarez DE, Tenorio-Barajas AY, Reyes-Chaparro A. Structural Bioinformatics Applied to Acetylcholinesterase Enzyme Inhibition. International Journal of Molecular Sciences. 2025; 26(8):3781. https://doi.org/10.3390/ijms26083781
Chicago/Turabian StyleReynoso-García, María Fernanda, Dulce E. Nicolás-Álvarez, A. Yair Tenorio-Barajas, and Andrés Reyes-Chaparro. 2025. "Structural Bioinformatics Applied to Acetylcholinesterase Enzyme Inhibition" International Journal of Molecular Sciences 26, no. 8: 3781. https://doi.org/10.3390/ijms26083781
APA StyleReynoso-García, M. F., Nicolás-Álvarez, D. E., Tenorio-Barajas, A. Y., & Reyes-Chaparro, A. (2025). Structural Bioinformatics Applied to Acetylcholinesterase Enzyme Inhibition. International Journal of Molecular Sciences, 26(8), 3781. https://doi.org/10.3390/ijms26083781