
Molecular Insights into the Marine Gastropod Olivancillaria urceus: Transcriptomic and Proteopeptidomic Approaches Reveal Polypeptides with Putative Therapeutic Potential
 , , , , , , and
, , , , , , and 
Abstract
1. Introduction
2. Results
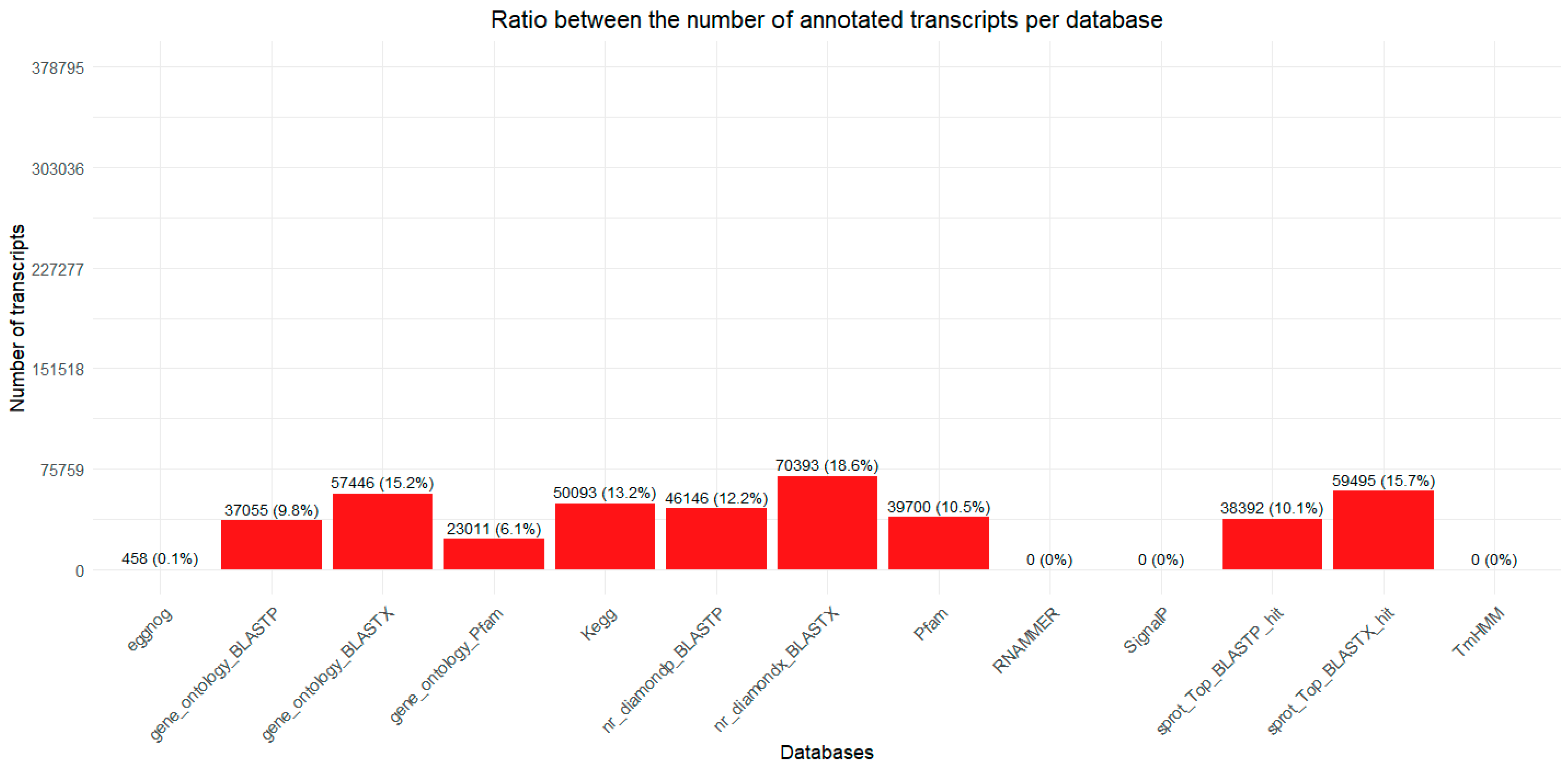
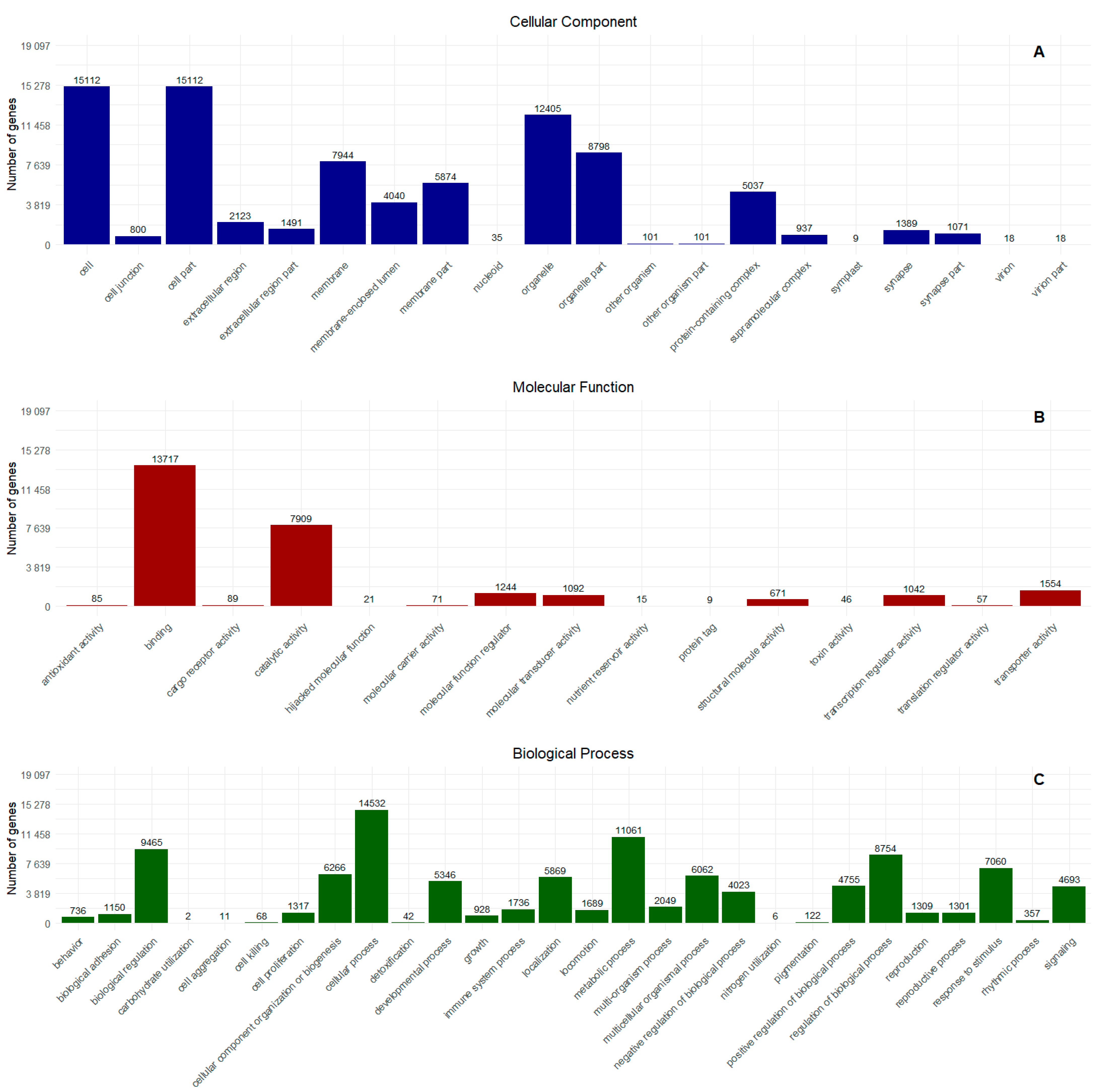
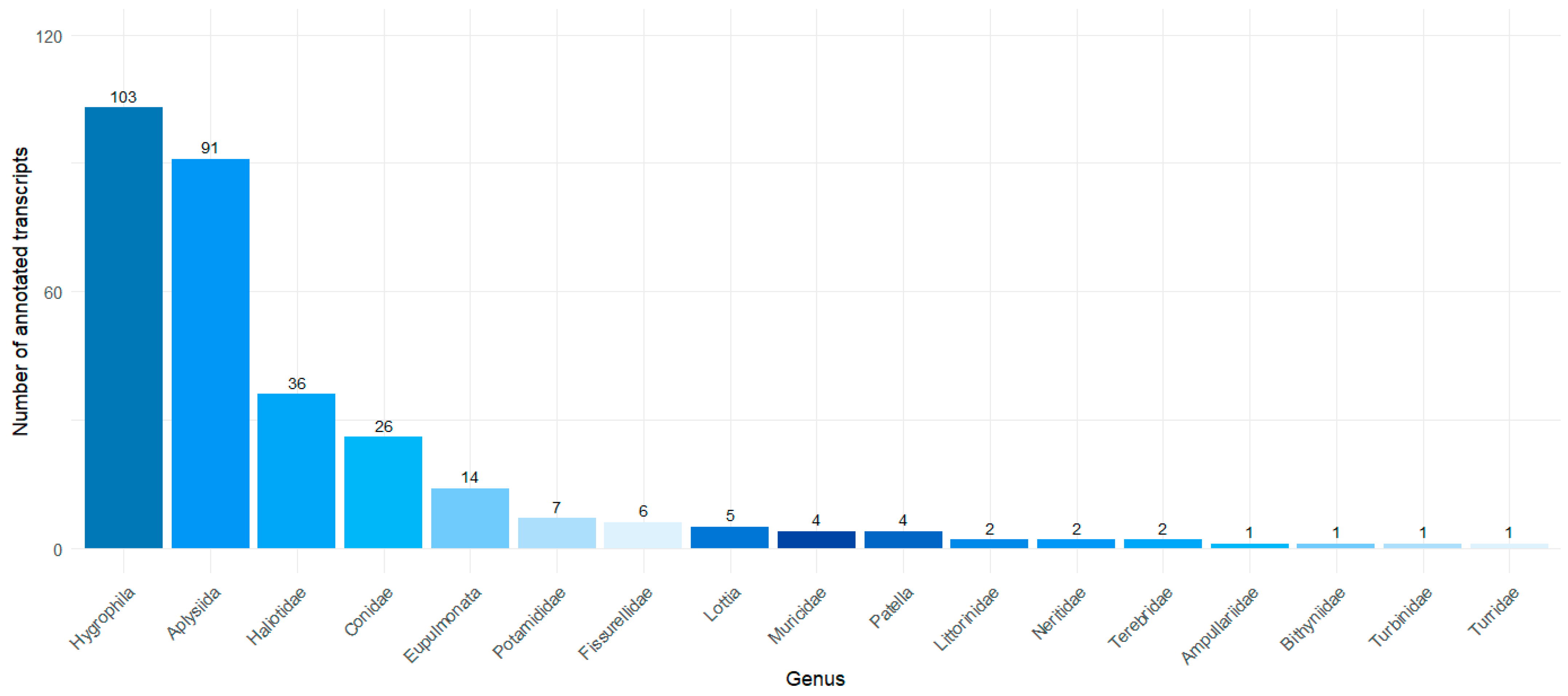
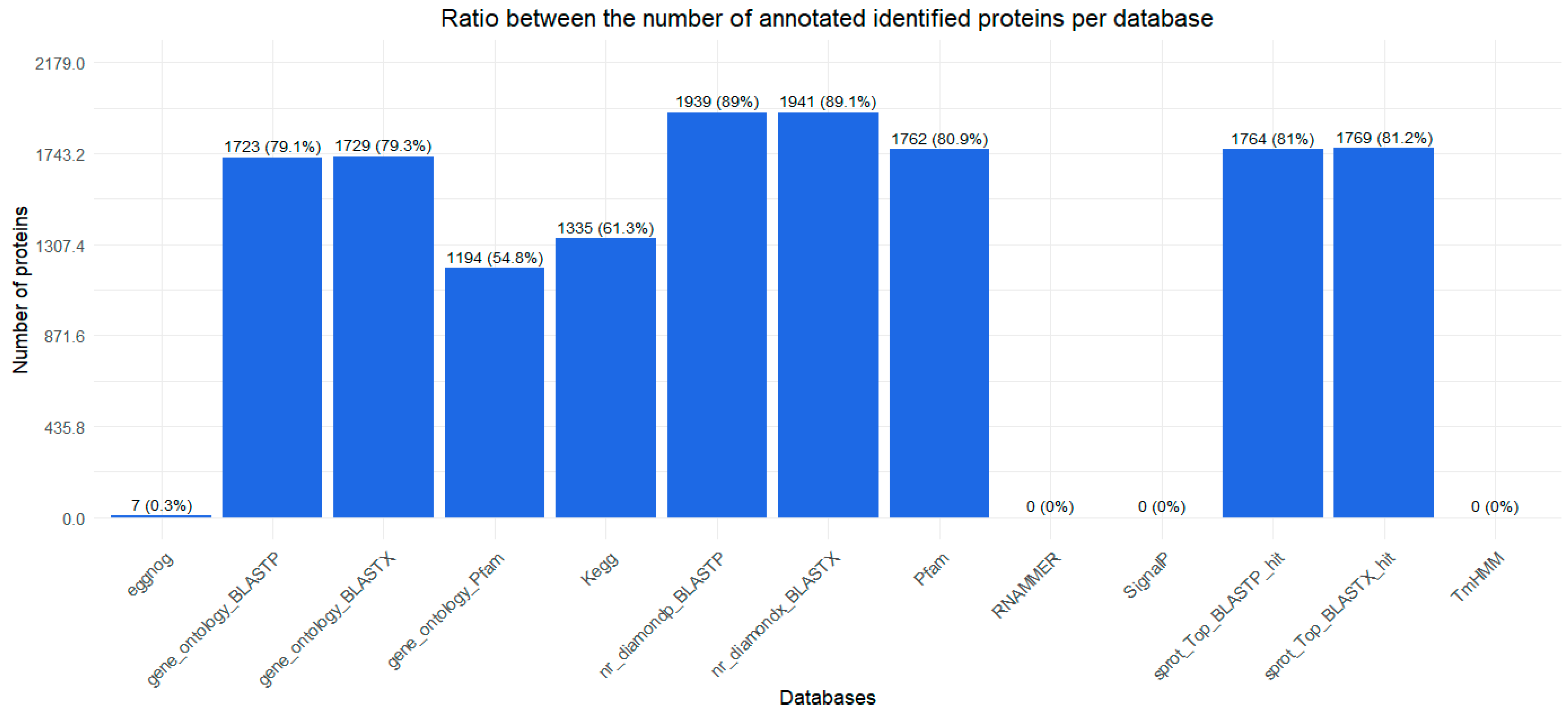
2.1. Transcriptome
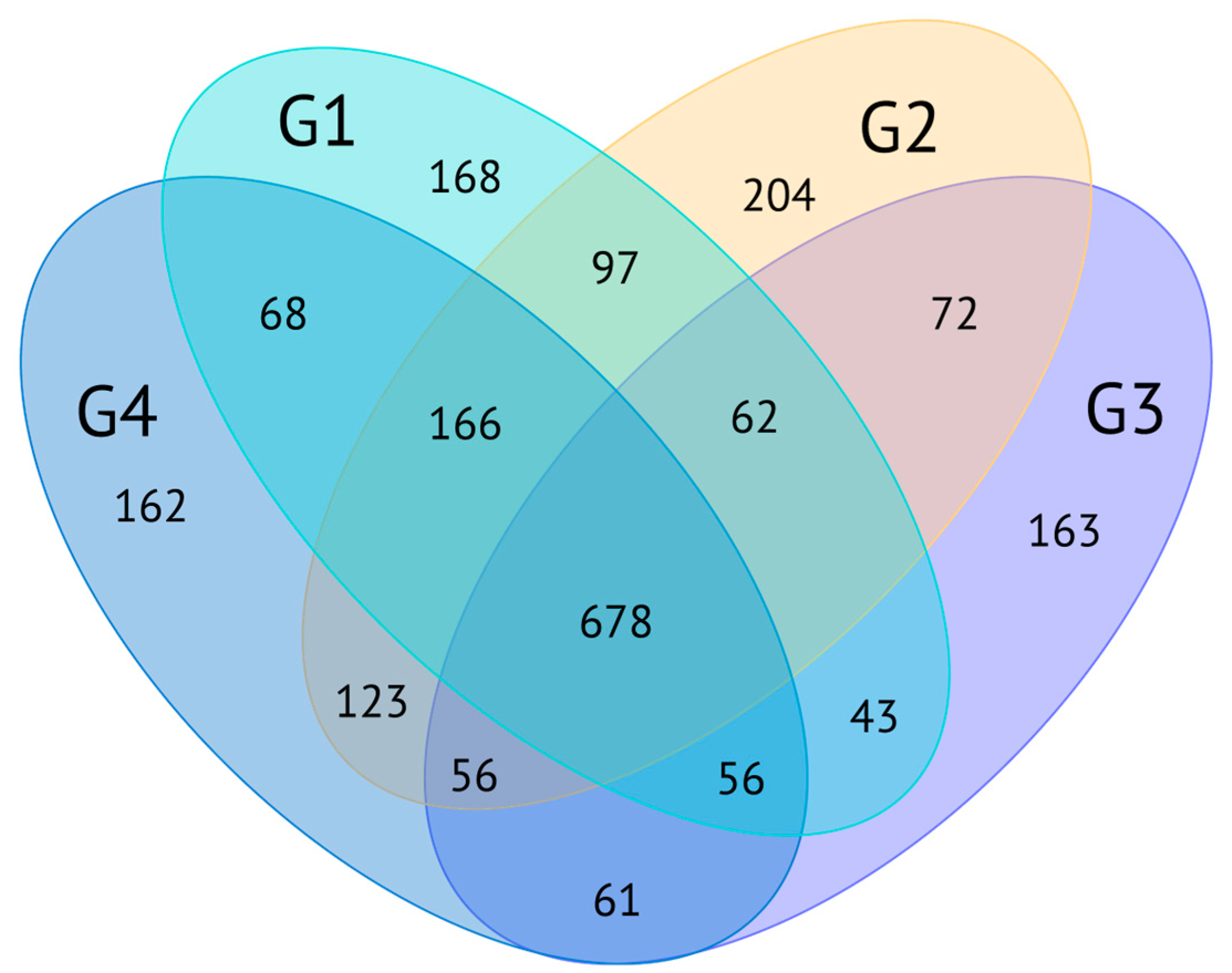
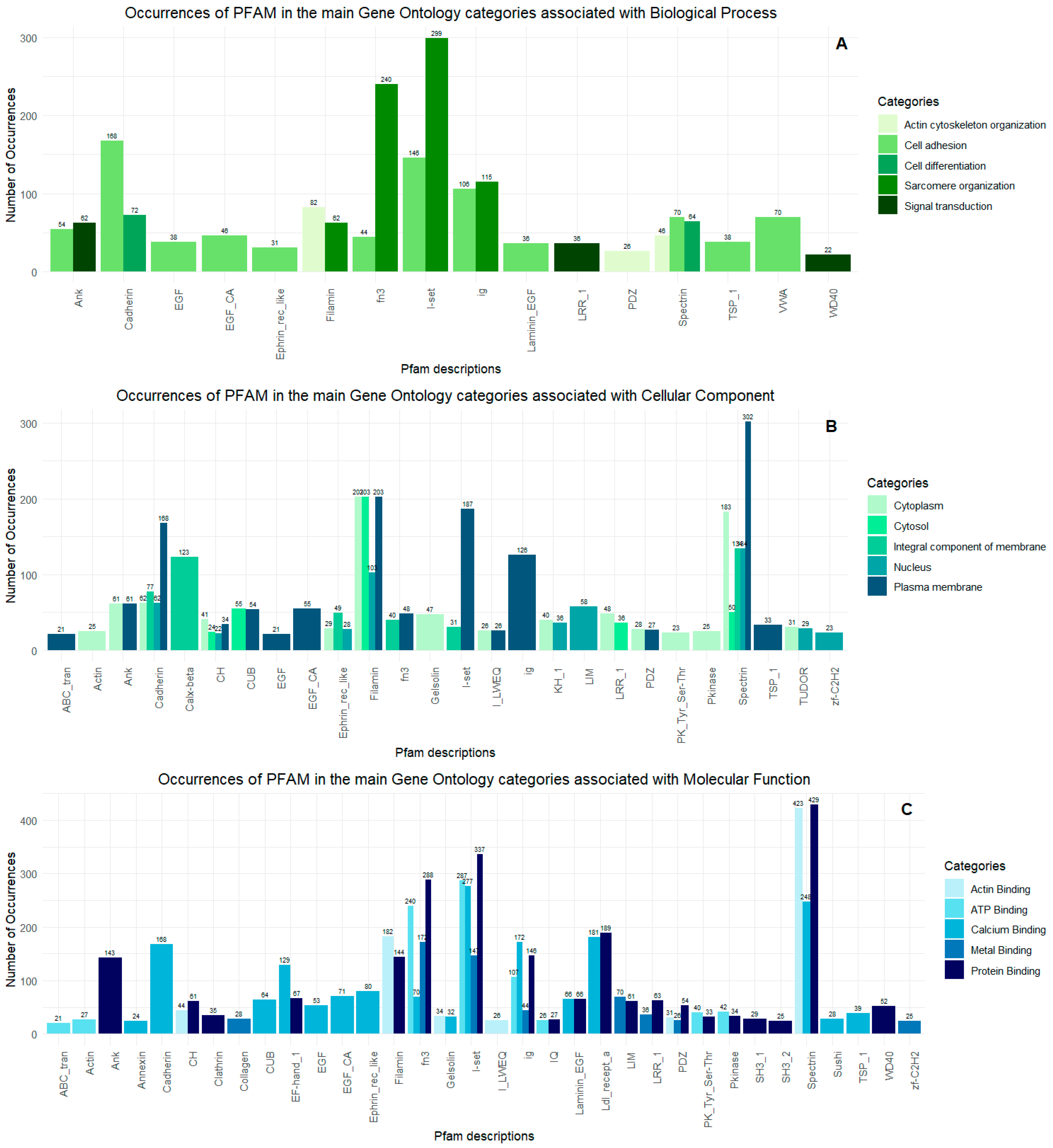
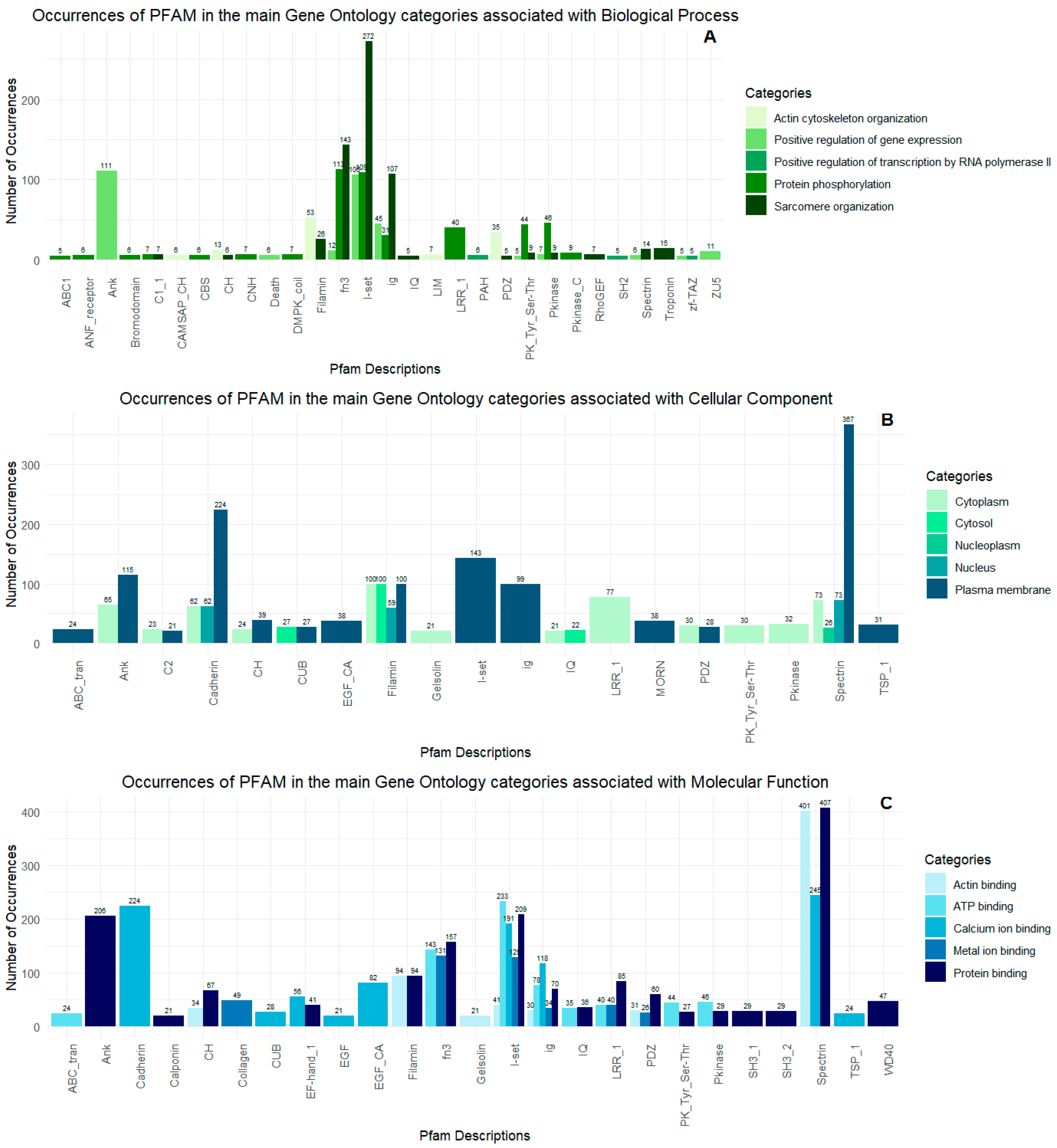
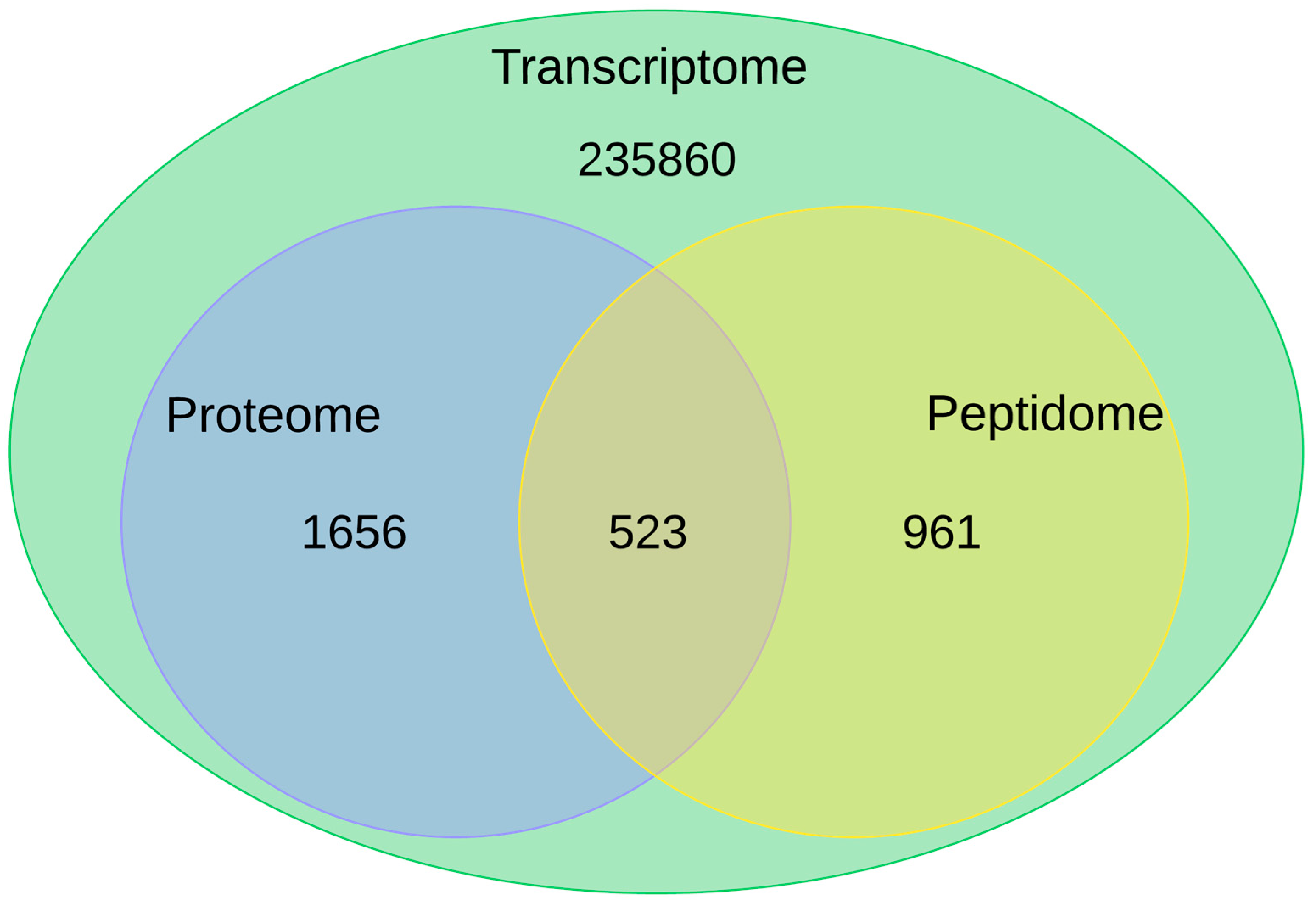
2.2. Proteopeptidome
3. Discussion
3.1. Putative Toxin-Related Transcripts and Polypeptides in O. urceus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript ID—O. urceus | Name of the Toxin in Conus | Probable Toxic Activity | Author |
|---|---|---|---|
| DN16518 c0 g1 i1 | Conodipine-P1 | Phospholipase A2 | [36] |
| DN19184 c2 g1 i1 | |||
| DN19184 c2 g1 i2 | |||
| DN 13158 c3 g1 i1 | Conodipine-P3 | ||
| DN27199 c2 g2 i1 | Conotoxin precursor Pmag02 | Conotoxin | [37,38] |
| DN3301 c0 g1 i1 | Kunitz-type domain | Serine protease inhibitors/toxin | [42,43] |
| DN109957 c0 g1 i1 | Conotoxin Im14.3 | Likely as a neurotoxin | [45] |
| DN126505 c0 g1 i1 | |||
| DN133748 c0 g1 i1 | |||
| DN17279 c3 g1 i1 | |||
| DN132248 c0 g1 i1 | ConoCAP | Decreases heart rate | [46] |
| DN4345 c0 g2 i2 | |||
| DN18169 c0 g1 i3 | Elevenin-Vc1 | Toxin induces hyperactivity | [47] |
| DN18169 c0 g1 i2 | |||
| DN55000 c0 g1 i1 | Tereporin-Ca1 | Function of a pore-forming protein | [48] |
| DN87107 c0 g1 i1 | |||
| DN60625 c0 g1 i1 | Perivitellin-2 protein (31 kDa subunit) | Cytotoxicity | [49] |
| DN46921 c0 g1 i1 | Turripeptide Pal9.2 toxin | Inhibiting ion channels by similarity with other similar toxins | [50] |
| DN12249 c1 g1 i1 | Thyrostimulin alpha-2 subunit | Function of toxin by similarity | [54,55] |
| DN94413 c0 g1 i1 | Thyrostimulin beta-5 subunit |
3.2. Limitations of Omics Analyses
4. Materials and Methods
4.1. Collection and Maintenance of Specimens
4.2. RNA Extraction, mRNA Library Synthesis, and Illumina Sequencing
4.3. Transcriptome Assembly, Annotation and Functional Enrichment
4.4. Proteomics and Peptidomics Procedures
4.4.1. Protein and Peptide Fraction Preparation
4.4.2. NanoLC and Mass Spectrometry
4.4.3. Protein and Peptide Identification
4.4.4. Integrative Data Analyses of the Transcriptome and Proteome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Branco, J.O.; Freitas Júnior, F.; Christoffersen, M.L. Bycatch Fauna of Seabob Shrimp Trawl Fisheries from Santa Catarina State, Southern Brazil. Biota Neotrop. 2015, 15, e20140143. [Google Scholar] [CrossRef]
- De Castro Mendonça, L.M.; Guimarães, C.R.P.; Lima, S.F.B. Mollusk Bycatch in Trawl Fisheries Targeting the Atlantic Seabob Shrimp Xiphopenaeus kroyeri on the Coast of Sergipe, Northeastern Brazil. Pap. Avulsos Zool. 2019, 59, e20195933. [Google Scholar] [CrossRef]
- Herrera-Valdivia, E.; López-Martínez, J.; Vargasmachuca, S.C.; García-Juárez, A.R. Taxonomic and Functional Diversity of the Bycatch Fishes Community of Trawl Fishing from Northern Gulf of California, Mexico. Rev. Biol. Trop. 2016, 64, 587–602. [Google Scholar] [CrossRef]
- Mendo, J.; Mendo, T.; Gil-Kodaka, P.; Martina, J.; Gómez, I.; Delgado, R.; Fernández, J.; Travezaño, A.; Arroyo, R.; Loza, K.; et al. Bycatch and Discards in the Artisanal Shrimp Trawl Fishery in Northern Peru. PLoS ONE 2022, 17, e0268128. [Google Scholar] [CrossRef]
- Rodrigues-Filho, J.L.; Couto, E.d.C.G.; Barbieri, E.; Branco, J.O. Seasonal Cycles of the Carcinofauna Caught as Bycatch in Sea-Bob Shrimp, Xiphopenaeus Kroyeri Fishery on Santa Catarina’s Coast. Bol. Do Inst. Pesca 2016, 42, 648–661. [Google Scholar] [CrossRef]
- Barrilli, G.H.C.; Filho, J.L.R.; do Vale, J.G.; Port, D.; Verani, J.R.; Branco, J.O. Role of the Habitat Condition in Shaping of Epifaunal Macroinvertebrate Bycatch Associated with Small-Scale Shrimp Fisheries on the Southern Brazilian Coast. Reg. Stud. Mar. Sci. 2021, 43, 101695. [Google Scholar] [CrossRef]
- Teso, V.; Pastorino, G. A Revision of the Genus Olivancillaria (Mollusca: Olividae) from the Southwestern Atlantic. Zootaxa 2011, 2889, 1–34. [Google Scholar] [CrossRef]
- Arrighetti, F.; Teso, V.; Brey, T.; Penchaszadeh, P.E. Gastropod Relevance in Predator-Prey Interactions on a Benthic Shallow Sandy Ecosystem at Mar Del Plata, Argentina (38°S). J. Mar. Biol. Assoc. UK 2019, 99, 403–409. [Google Scholar] [CrossRef]
- Tangerina, M.M.P.; Correa, H.; Haltli, B.; Vilegas, W.; Kerr, R.G. Bioprospecting from Cultivable Bacterial Communities of Marine Sediment and Invertebrates from the Underexplored Ubatuba Region of Brazil. Arch. Microbiol. 2017, 199, 155–169. [Google Scholar] [CrossRef]
- Gasu, E.N.; Ahor, H.S.; Borquaye, L.S. Peptide Extract from Olivancillaria hiatula Exhibits Broad-Spectrum Antibacterial Activity. BioMed Res. Int. 2018, 2018, 6010572. [Google Scholar] [CrossRef]
- Gasu, E.N.; Ahor, H.S.; Borquaye, L.S. Peptide Mix from Olivancillaria Hiatula Interferes with Cell-to-Cell Communication in Pseudomonas Aeruginosa. BioMed Res. Int. 2019, 2019, 5313918. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, L. Insights into the Regulation of Algal Proteins and Bioactive Peptides Using Proteomic and Transcriptomic Approaches. Molecules 2019, 24, 1708. [Google Scholar] [CrossRef] [PubMed]
- Mazzi Esquinca, M.E.; Correa, C.N.; Marques de Barros, G.; Montenegro, H.; Mantovani de Castro, L. Multiomic Approach for Bioprospection: Investigation of Toxins and Peptides of Brazilian Sea Anemone Bunodosoma Caissarum. Mar. Drugs 2023, 21, 197. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.W.; Lu, Y.; Yang, Y.F.; Huang, D.; Li, D.W.; Wang, X.; Gao, Y.; Yang, W.D.; Guan, Y.; Li, H.Y. Systematic Dissection of Genomic Features Determining the Vast Diversity of Conotoxins. BMC Genom. 2023, 24, 598. [Google Scholar] [CrossRef]
- Moutinho Cabral, I.; Madeira, C.; Grosso, A.R.; Costa, P.M. A Drug Discovery Approach Based on Comparative Transcriptomics between Two Toxin-Secreting Marine Annelids: Glycera Alba and Hediste Diversicolor. Mol. Omics 2022, 18, 731–744. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Philip, D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Macmanes, M.D.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq Methods for Transcriptome Analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef]
- Slattery, M.; Ankisetty, S.; Corrales, J.; Marsh-Hunkin, K.E.; Gochfeld, D.J.; Willett, K.L.; Rimoldi, J.M. Marine Proteomics: A Critical Assessment of an Emerging Technology. J. Nat. Prod. 2012, 75, 1833–1877. [Google Scholar] [CrossRef]
- Ahmed, S.; Khan, H.; Fakhri, S.; Aschner, M.; Cheang, W.S. Therapeutic Potential of Marine Peptides in Cervical and Ovarian Cancers. Mol. Cell. Biochem. 2022, 477, 605–619. [Google Scholar] [CrossRef]
- Ribeiro, R.; Pinto, E.; Fernandes, C.; Sousa, E. Marine Cyclic Peptides: Antimicrobial Activity and Synthetic Strategies. Mar. Drugs 2022, 20, 397. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Zhang, L.; Smith, M.D.; Walewska, A.; Vellore, N.A.; Baron, R.; McIntosh, J.M.; White, H.S.; Olivera, B.M.; Bulaj, G. A Marine Analgesic Peptide, Contulakin-G, and Neurotensin Are Distinct Agonists for Neurotensin Receptors: Uncovering Structural Determinants of Desensitization Properties. Front. Pharmacol. 2015, 6, 11. [Google Scholar] [CrossRef]
- Sukmarini, L. Antiviral Peptides (AVPs) of Marine Origin as Propitious Therapeutic Drug Candidates for the Treatment of Human Viruses. Molecules 2022, 27, 2619. [Google Scholar] [CrossRef]
- Jin, A.H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.W.A.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and Biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.H.; Vetter, I.; Himaya, S.W.A.; Alewood, P.F.; Lewis, R.J.; Dutertre, S. Transcriptome and Proteome of Conus Planorbis Identify the Nicotinic Receptors as Primary Target for the Defensive Venom. Proteomics 2015, 15, 4030–4040. [Google Scholar] [CrossRef] [PubMed]
- Duggan, P.J.; Tuck, K.L. Bioactive Mimetics of Conotoxins and Other Venom Peptides. Toxins 2015, 7, 4175–4198. [Google Scholar] [CrossRef]
- Deer, T.R.; Pope, J.E.; Hanes, M.C.; McDowell, G.C. Intrathecal Therapy for Chronic Pain: A Review of Morphine and Ziconotide as Firstline Options. Pain Med. 2019, 20, 784–798. [Google Scholar] [CrossRef]
- Matis, G.; De Negri, P.; Dupoiron, D.; Likar, R.; Zuidema, X.; Rasche, D. Intrathecal Pain Management with Ziconotide: Time for Consensus? Brain Behav. 2021, 11, e02055. [Google Scholar] [CrossRef]
- McGivern, J.G. Ziconotide: A Review of Its Pharmacology and Use in the Treatment of Pain. Neuropsychiatr. Dis. Treat. 2007, 3, 69–85. [Google Scholar] [CrossRef]
- Seppälä, O.; Walser, J.C.; Cereghetti, T.; Seppälä, K.; Salo, T.; Adema, C.M. Transcriptome Profiling of Lymnaea Stagnalis (Gastropoda) for Ecoimmunological Research. BMC Genom. 2021, 22, 144. [Google Scholar] [CrossRef]
- Santos, C.A.; Sonoda, G.G.; Cortez, T.; Coutinho, L.L.; Andrade, S.C.S. Transcriptome Expression of Biomineralization Genes in Littoraria Flava Gastropod in Brazilian Rocky Shore Reveals Evidence of Local Adaptation. Genome Biol. Evol. 2021, 13, evab050. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yuan, Y.; Meng, M.; Li, S.; Deng, B.; Wang, Y. The Transcriptome Analysis of the Whole-Body of the Gastropod Mollusk Limax Flavus and Screening of Putative Antimicrobial Peptide and Protein Genes. Genomics 2020, 112, 3991–3999. [Google Scholar] [CrossRef]
- Bairati, A.; Comazzi, M.; Gioria, M. An Ultrastructural Study of Connective Tissue in Mollusc Integument: II. Gastropoda. Tissue Cell 2001, 33, 426–438. [Google Scholar] [CrossRef]
- Rigon, F.; Mânica, G.; Guma, F.; Achaval, M.; Faccioni-Heuser, M.C. Ultrastructural Features of the Columellar Muscle and Contractile Protein Analyses in Different Muscle Groups of Megalobulimus abbreviatus (Gastropoda, Pulmonata). Tissue Cell 2010, 42, 53–60. [Google Scholar] [CrossRef]
- Dennis, M.M.; Molnár, K.; Kriska, G.; Lőw, P. Mollusca. In Invertebrate Histology; Wiley: Hoboken, NJ, USA, 2021; pp. 87–132. [Google Scholar] [CrossRef]
- Möller, C.; Clay Davis, W.; Clark, E.; DeCaprio, A.; Marí, F. Conodipine-P1-3, the First Phospholipases A2 Characterized from Injected Cone Snail Venom. Mol. Cell. Proteom. 2019, 18, 876a, 876–891. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Norton, R.S. Conotoxin Gene Superfamilies. Mar. Drugs 2014, 12, 6058. [Google Scholar] [CrossRef] [PubMed]
- Terlau, H.; Olivera, B.M. Conus Venoms: A Rich Source of Novel Ion Channel-Targeted Peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef]
- Bártová, V.; Bárta, J.; Jarošová, M. Antifungal and Antimicrobial Proteins and Peptides of Potato (Solanum tuberosum L.) Tubers and Their Applications. Appl. Microbiol. Biotechnol. 2019, 103, 5533–5547. [Google Scholar] [CrossRef]
- Bonturi, C.R.; Teixeira, A.B.S.; Rocha, V.M.; Valente, P.F.; Oliveira, J.R.; Filho, C.M.B.; Batista, I.d.F.C.; Oliva, M.L.V. Plant Kunitz Inhibitors and Their Interaction with Proteases: Current and Potential Pharmacological Targets. Int. J. Mol. Sci. 2022, 23, 4742. [Google Scholar] [CrossRef]
- Mourão, C.B.F.; Schwartz, E.F. Protease Inhibitors from Marine Venomous Animals and Their Counterparts in Terrestrial Venomous Animals. Mar. Drugs 2013, 11, 2069. [Google Scholar] [CrossRef]
- Mishra, M. Evolutionary Aspects of the Structural Convergence and Functional Diversification of Kunitz-Domain Inhibitors. J. Mol. Evol. 2020, 88, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, S.; McManus, D.P. Structure and Function of Invertebrate Kunitz Serine Protease Inhibitors. Dev. Comp. Immunol. 2013, 39, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Modica, M.V.; Lombardo, F.; Franchini, P.; Oliverio, M. The Venomous Cocktail of the Vampire Snail Colubraria reticulata (Mollusca, Gastropoda). BMC Genom. 2015, 16, 441. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.H.; Dutertre, S.; Dutt, M.; Lavergne, V.; Jones, A.; Lewis, R.J.; Alewood, P.F. Transcriptomic-Proteomic Correlation in the Predation-Evoked Venom of the Cone Snail, Conus Imperialis. Mar. Drugs 2019, 17, 177. [Google Scholar] [CrossRef]
- Möller, C.; Melaun, C.; Castillo, C.; Díaz, M.E.; Renzelman, C.M.; Estrada, O.; Kuch, U.; Lokey, S.; Marí, F. Functional Hypervariability and Gene Diversity of Cardioactive Neuropeptides. J. Biol. Chem. 2010, 285, 40673–40680. [Google Scholar] [CrossRef]
- Krishnarjuna, B.; Sunanda, P.; Seow, J.; Tae, H.S.; Robinson, S.D.; Belgi, A.; Robinson, A.J.; Safavi-Hemami, H.; Adams, D.J.; Norton, R.S. Characterisation of Elevenin-Vc1 from the Venom of Conus Victoriae: A Structural Analogue of α-Conotoxins. Mar. Drugs 2023, 21, 81. [Google Scholar] [CrossRef]
- Gorson, J.; Ramrattan, G.; Verdes, A.; Wright, E.M.; Kantor, Y.; Srinivasan, R.R.; Musunuri, R.; Packer, D.; Albano, G.; Qiu, W.G.; et al. Molecular Diversity and Gene Evolution of the Venom Arsenal of Terebridae Predatory Marine Snails. Genome Biol. Evol. 2015, 7, 1761–1778. [Google Scholar] [CrossRef]
- Giglio, M.L.; Ituarte, S.; Ibañez, A.E.; Dreon, M.S.; Prieto, E.; Fernández, P.E.; Heras, H. Novel Role for Animal Innate Immune Molecules: Enterotoxic Activity of a Snail Egg MACPF-Toxin. Front. Immunol. 2020, 11, 428. [Google Scholar] [CrossRef]
- Olivera, B.M.; Watkins, M.; Bandyopadhyay, P.; Imperial, J.S.; de la Cotera, E.P.H.; Aguilar, M.B.; Vera, E.L.; Concepcion, G.P.; Lluisma, A. Adaptive Radiation of Venomous Marine Snail Lineages and the Accelerated Evolution of Venom Peptide Genes. Ann. N. Y. Acad. Sci. 2012, 1267, 61–70. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Y.; Zhu, S.; Xu, H.; Liu, Y.; Chen, L. The Complete Mitochondrial Genome of Pomacea canaliculata (Gastropoda: Ampullariidae). Mitochondrial DNA 2016, 27, 884–885. [Google Scholar] [CrossRef]
- Liu, Z.; Dai, J.; Chen, Z.; Hu, W.; Xiao, Y.; Liang, S. Isolation and Characterization of Hainantoxin-IV, a Novel Antagonist of Tetrodotoxin-Sensitive Sodium Channels from the Chinese Bird Spider Selenocosmia Hainana. Cell Mol. Life Sci. 2003, 60, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Lewis, R.J. Use of Venom Peptides to Probe Ion Channel Structure and Function. J. Biol. Chem. 2010, 285, 13315–13320. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Safavi-Hemami, H.; McIntosh, L.D.; Purcell, A.W.; Norton, R.S.; Papenfuss, A.T. Diversity of Conotoxin Gene Superfamilies in the Venomous Snail, Conus Victoriae. PLoS ONE 2014, 9, e87648. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Li, Q.; Bandyopadhyay, P.K.; Gajewiak, J.; Yandell, M.; Papenfuss, A.T.; Purcell, A.W.; Norton, R.S.; Safavi-Hemami, H. Hormone-like Peptides in the Venoms of Marine Cone Snails. Gen. Comp. Endocrinol. 2017, 244, 11–18. [Google Scholar] [CrossRef]
- Tian, Y.; Yu, A.M.; Yin, C.; Qian, A. Editorial: Post-Transcriptional Regulation and Its Misregulation: From Molecular Basis to Translational Medicine. Front. Cell Dev. Biol. 2022, 10, 1101576. [Google Scholar] [CrossRef]
- Wu, Q.; Medina, S.G.; Kushawah, G.; Devore, M.L.; Castellano, L.A.; Hand, J.M.; Wright, M.; Bazzini, A.A. Translation Affects MRNA Stability in a Codon-Dependent Manner in Human Cells. eLife 2019, 8, e45396. [Google Scholar] [CrossRef]
- Hawkridge, A.M. Practical Considerations and Current Limitations in Quantitative Mass Spectrometry-Based Proteomics. In Quantitative Proteomics; The Royal Society of Chemistry: London, UK, 2014. [Google Scholar] [CrossRef]
- Li, C.; Chu, S.; Tan, S.; Yin, X.; Jiang, Y.; Dai, X.; Gong, X.; Fang, X.; Tian, D. Towards Higher Sensitivity of Mass Spectrometry: A Perspective From the Mass Analyzers. Front. Chem. 2021, 9, 813359. [Google Scholar] [CrossRef]
- Venkataramanan, K.P.; Min, L.; Hou, S.; Jones, S.W.; Ralston, M.T.; Lee, K.H.; Papoutsakis, E.T. Complex and Extensive Post-Transcriptional Regulation Revealed by Integrative Proteomic and Transcriptomic Analysis of Metabolite Stress Response in Clostridium Acetobutylicum. Biotechnol. Biofuels 2015, 8, 81. [Google Scholar] [CrossRef]
- Caburatan, L.; Park, J. Differential Expression, Tissue-Specific Distribution, and Posttranslational Controls of Phosphoenolpyruvate Carboxylase. Plants 2021, 10, 1887. [Google Scholar] [CrossRef]
- Campos, B.; Fletcher, D.; Piña, B.; Tauler, R.; Barata, C. Differential Gene Transcription across the Life Cycle in Daphnia Magna Using a New All Genome Custom-Made Microarray. BMC Genom. 2018, 19, 370. [Google Scholar] [CrossRef]
- Andrews, S. FastQC. In Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A Fast, Lock-Free Approach for Efficient Parallel Counting of Occurrences of k-Mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: A Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Wheeler, T.J.; Eddy, S.R. Nhmmer: DNA Homology Search with Profile HMMs. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. Pfam Protein Families Database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Farrell, C.M.; Feldgarden, M.; Fine, A.M.; Funk, K.; et al. Database Resources of the National Center for Biotechnology Information in 2023. Nucleic Acids Res. 2023, 51, D29–D38. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F.A.; Zdobnov, E.M. OrthoDB V10: Sampling the Diversity of Animal, Plant, Fungal, Protist, Bacterial and Viral Genomes for Evolutionary and Functional Annotations of Orthologs. Nucleic Acids Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef]
- Ma, B.; Zhang, K.; Hendrie, C.; Liang, C.; Li, M.; Doherty-Kirby, A.; Lajoie, G. PEAKS: Powerful Software for Peptide de Novo Sequencing by Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. RCM 2003, 17, 2337–2342. [Google Scholar] [CrossRef]
- Zhang, J.; Xin, L.; Shan, B.; Chen, W.; Xie, M.; Yuen, D.; Zhang, W.; Zhang, Z.; Lajoie, G.A.; Ma, B. PEAKS DB: De Novo Sequencing Assisted Database Search for Sensitive and Accurate Peptide Identification. Mol. Cell. Proteom. 2012, 11, M111.010587. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2. Available online: https://link.springer.com/book/10.1007/978-3-319-24277-4 (accessed on 13 March 2024). [CrossRef]
- Wickham, H.; François, R.; Henry, L.; Müller, K. dplyr: A Grammar of Data Manipulation. 2019. Available online: https://dplyr.tidyverse.org/ (accessed on 3 March 2024).
- Wickham, H. Package stringr: Simple, Consistent Wrappers for Common String Operations. Available online: https://stringr.tidyverse.org/ (accessed on 13 March 2024).
- Wickham, H.; Grolemund, G. R for Data Science: Visualize, Model, Transform, Tidy, and Import Data; O’Reilly: Sebastopol, CA, USA, 2023. [Google Scholar]



| Transcript ID (Trinity ID) | Uniprot/Swiss-Prot ID | Alignment Region in the Sequence | Similarity | p-Value | Description | TPM * |
|---|---|---|---|---|---|---|
| DN27199 c2 g2 i1 | DAZ86947.1 | Q:6-98, H:3-86 | 33.1% | 1 × 10−6 | Conotoxin precursor Pmag02 | 371,809 |
| DN16518 c0 g1 i1 | COP3_CONPU | Q:37-94, H:33-86 | 44.8% | 1.3 × 10−6 | Conodipine-P3 | 36,110 |
| DN3301 c0 g1 i1 | KCP_HALAI | Q:132-479, H:5-120 | 49.1% | 5.53 × 10−41 | BPTI/Kunitz domain | 10,069 |
| DN126505 c0 g1 i1 | CUE3_CONIM | Q:9-44, H:38-73 | 47.2% | 1.38 × 10−6 | Conotoxin Im14.3 | 29 |
| DN13158 c3 g1 i1 | COP1_CONPU | Q:27-93, H:28-90 | 44.8% | 1.47 × 10−12 | Conodipine-P1 | 1679 |
| DN132248 c0 g1 i1 | CCAP_CONVL | Q:46-124, H:22-96 | 50.6% | 1.15 × 10−17 | ConoCAP | 28 |
| DN133748 c0 g1 i1 | CUE3_CONIM | Q:13-42, H:41-70 | 56.7% | 5.69 × 10−7 | Conotoxin Im14.3 | 42 |
| DN17279 c3 g1 i1 | CUE3_CONIM | Q:21-50, H:44-73 | 56.7% | 8.29 × 10−7 | Conotoxin Im14.3 | 27 |
| DN109957 c0 g1 i1 | CUE3_CONIM | Q:147-177, H:44-73 | 64.5% | 6.39 × 10−7 | Conotoxin Im14.3 | 641 |
| DN18169 c0 g1 i3 | CELE_CONVC | Q:21-125, H:1-98 | 60% | 1.33 × 10−27 | Elevenin-Vc1 | 100 |
| DN18169 c0 g1 i2 | CELE_CONVC | Q:21-125, H:1-98 | 60% | 1.33 × 10−27 | Elevenin-Vc1 | 91 |
| DN19184 c2 g1 i2 | COP1_CONPU | Q:7-183, H:8-176 | 31% | 7.61 × 10−26 | Conodipine-P1 | 68 |
| DN19184 c2 g1 i1 | COP1_CONPU | Q:7-183, H:8-176 | 31% | 7.61 × 10−26 | Conodipine-P1 | 19 |
| DN4345 c0 g2 i2 | CCAP_CONVL | Q:14-48, H:155-189 | 71.4% | 1.11 × 10−12 | ConoCAP | 78 |
| DN46921 c0 g1 i1 | TU92_POLAB | Q:1-74, H:1-70 | 55.4% | 2.78 × 10−23 | Turripeptide Pal9.2 | 25 |
| DN55000 c0 g1 i1 | ACTP1_TERAN | Q:43-230, H:1-190 | 70% | 3.68 × 10−98 | Tereporin-Ca1 | 38 |
| DN60625 c0 g1 i1 | PV22_POMMA | Q:4-113, H:179-283 | 33.3% | 6.28 × 10−11 | Perivitellin-2 31 kDa sub. | 21 |
| DN87107 c0 g1 i1 | ACTP1_TERAN | Q:1-62, H:127-190 | 65.6% | 248 × 10−11 | Tereporin-Ca1 | 34 |
| DN94413 c0 g1 i1 | CTHB5_CONVC | Q:55-149, H:1-96 | 80.2% | 5.1 × 10−51 | Thyrostimulin beta-5 sub. | 22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barros, G.M.d.; Gama, L.F.; Mello, F.R.d.; Corrêa, C.N.; Fiametti, L.O.; Montenegro, H.; Ottoni, C.A.; Castro, L.M.d. Molecular Insights into the Marine Gastropod Olivancillaria urceus: Transcriptomic and Proteopeptidomic Approaches Reveal Polypeptides with Putative Therapeutic Potential. Int. J. Mol. Sci. 2025, 26, 3751. https://doi.org/10.3390/ijms26083751
Barros GMd, Gama LF, Mello FRd, Corrêa CN, Fiametti LO, Montenegro H, Ottoni CA, Castro LMd. Molecular Insights into the Marine Gastropod Olivancillaria urceus: Transcriptomic and Proteopeptidomic Approaches Reveal Polypeptides with Putative Therapeutic Potential. International Journal of Molecular Sciences. 2025; 26(8):3751. https://doi.org/10.3390/ijms26083751
Chicago/Turabian StyleBarros, Gabriel Marques de, Letícia Fontes Gama, Felipe Ricardo de Mello, Claudia Neves Corrêa, Louise Oliveira Fiametti, Horácio Montenegro, Cristiane Angélica Ottoni, and Leandro Mantovani de Castro. 2025. "Molecular Insights into the Marine Gastropod Olivancillaria urceus: Transcriptomic and Proteopeptidomic Approaches Reveal Polypeptides with Putative Therapeutic Potential" International Journal of Molecular Sciences 26, no. 8: 3751. https://doi.org/10.3390/ijms26083751
APA StyleBarros, G. M. d., Gama, L. F., Mello, F. R. d., Corrêa, C. N., Fiametti, L. O., Montenegro, H., Ottoni, C. A., & Castro, L. M. d. (2025). Molecular Insights into the Marine Gastropod Olivancillaria urceus: Transcriptomic and Proteopeptidomic Approaches Reveal Polypeptides with Putative Therapeutic Potential. International Journal of Molecular Sciences, 26(8), 3751. https://doi.org/10.3390/ijms26083751